

Abstract
ATP-binding cassette transporter A1 (ABCA1) plays a crucial role in exporting cholesterol from macrophages, a function relevant to its involvement in the prevention of atherosclerosis. Quercetin, one of flavonoids, has been described to reduce atherosclerotic lesion formation. This study is aimed to investigate the effect of quercetin on regulation of ABCA1 expression and to explore its underlying mechanisms in macrophages. The results show that quercetin markedly enhanced cholesterol efflux from macrophages in a concentration-dependent manner, which was associated with an increase in ABCA1 mRNA and protein expression. Remarkably, quercetin is able to stimulate the phosphorylation of p38 by up to 234-fold at 6 h via an activation of the transforming growth factor β-activated kinase 1 (TAK1) and mitogen-activated kinase kinase 3/6 (MKK3/6). Inhibition of p38 with a pharmacological inhibitor or small hairpin RNA (shRNA) suppressed the stimulatory effects of quercetin on ABCA1 expression and cholesterol efflux. Moreover, knockdown of p38 reduced quercetin-enhanced ABCA1 promoter activity and the binding of specificity protein 1 (Sp1) and liver X receptor α (LXRα) to the ABCA1 promoter using chromatin immunoprecipitation assays. These findings provide evidence that p38 signaling is essential for the regulation of quercetin-induced ABCA1 expression and cholesterol efflux in macrophages.
Keywords: ATP-binding cassette transporter A1, cell signaling, nuclear receptors, promoter activity
Quercetin, one of widely distributed flavonoids in plants, is found abundantly in onions, apples, berries, broccoli, Ginkgo biloba, and red wine (1). Like other members of flavonoids, quercetin has wide range of biological functions associated with the modulation of oxidative stress and inflammatory processes (2–4). In addition to these properties, it has been reported that quercetin is able to reduce plasma cholesterol levels in hyperlipidemia animals (5–7). Hypercholesterolemia is one of the major risk factors for the development and progression of atherosclerosis (8, 9). It seems that quercetin may play an essential role in the prevention of atherosclerosis. Hypercholesterolemia promotes the entry and retention of cholesterol-rich low-density lipoprotein (LDL) within the arterial wall. The accumulated oxidized LDL (oxLDL) in the subendothelial space of vessels is taken up by macrophages through scavenger receptors. However, unlimited uptake of oxLDL results in foam cell formation, a pathological hallmark of early atherosclerosis. Cholesterol efflux from macrophages is a key mechanism to prevent the development of atherosclerosis (10–13). Despite the importance of cholesterol accumulation in the development of atherosclerosis, little is known about the biological effects and molecular mechanisms of quercetin on cholesterol transportation in macrophages.
ATP-binding cassette transporter A1 (ABCA1), a member of the ATP-binding cassette transporter family, is involved in the control of apolipoprotein AI (apoAI)-mediated cholesterol efflux from macrophages (14, 15). A previous study has shown that overexpression of ABCA1 in LDL receptor-deficient mice reduces lipid accumulation in the arterial wall (16). Thus, induction of ABCA1 expression is considered an attractive approach to preventing excessive cholesterol accumulation and atherosclerosis. The expression of ABCA1 is transcriptionally regulated by nuclear transcription factor liver X receptor (LXR) and retinoid X receptor (RXR) (17, 18). When LXR is activated by its natural ligand, oxysterols, it forms heterodimers with RXR and binds to the ABCA1 promoter to activate gene expression. Thymiakou et al. found that transcription factor specificity protein 1 (Sp1) regulated ABCA1 expression via physical interaction with the LXR/RXR heterodimer in human hepatoma cells treated with oxysterols and retinoids (19). In addition, Sp1 has been reported to be involved in quercetin-regulated gene expression (20, 21); however, the involvement of Sp1 and LXR in quercetin-induced ABCA1 transcription is still to be elucidated.
In addition to transcription factor-mediated ABCA1 gene regulation, mitogen-activated protein kinase (MAPK) signaling cascades have been reported to modulate ABCA1 expression (22–24). The MAPK family comprises extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinase 1/2 (JNK1/2), and p38 (25). Inhibition of ERK activation leads to increase ABCA1 mRNA and protein stabilities, which results in the activation of ABCA1 expression (22). Activation of p38 has been reported to modulate ABCA1 expression in vascular smooth muscle cells and macrophages (23, 24). Nevertheless, little is known about the contribution of the MAPK signaling cascades to quercetin-mediated ABCA1 gene regulation.
The aim of the study was to investigate whether quercetin stimulates cholesterol efflux via the induction of ABCA1 expression in macrophages, as well as to explore its molecular mechanisms. Our results show that quercetin markedly increases apoAI-mediated cholesterol efflux via elevation of ABCA1 gene expression. Furthermore, quercetin activates the p38 signaling cascade via stimulating phosphorylation of transforming growth factor β-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase 3/6 (MKK3/6), the upstream kinases of p38. Moreover, quercetin enhances p38-mediated binding of Sp1 and LXRα to the corresponding binding sites located within −250/−1 region of the ABCA1 promoter. Our results provide a new insight into the mechanisms underlying the biological effect of quercetin on cholesterol efflux.
MATERIALS AND METHODS
Materials
SB203580 (p38 inhibitor) and 5Z-7-Oxozeaenol (TAK1 inhibitor) were obtained from Calbiochem (San Diego, CA). Antibodies recognizing ABCA1, α-tubulin, LXRα, B23, and rabbit control IgG were purchased from Abcam (Cambridge, UK). Antibodies against phospho-p38, phospho-ERK1/2, phospho-JNK1/2, phospho-MKK3/6, phospho-TAK1, total p38, total ERK1/2, total JNK1/2, and total MKK3 were obtained from Cell Signaling Technology (Beverly, MA). Total TAK1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Sp1 antibody was obtained from Millipore (Temecula, CA).
Cell culture
Murine macrophage cell line RAW264.7 was originally obtained from the American Type Culture Collection (Manassas, VA). Cells were grown in Dulbecco's modified Eagle's medium (DMEM; Hyclone Laboratories, Logan, UT) supplemented with 10% fetal bovine serum (FBS; PAA Laboratories, Linz, Austria), 100 units/ml penicillin, and 100 μg/ml streptomycin. All cell experiments were performed in a humidified atmosphere at 37°C with 5% CO2. Bone marrow-derived macrophages (BMDM) were isolated and cultured from 6- to 12-week-old C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) as previously described (26). The animal experiments conformed to the PHS policy and approved by the Animal Care and Utilization committee of National Yang-Ming University. Briefly, bone marrow cells isolated from femurs were cultured for 7 days in Petri dishes with DMEM containing 20% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml macrophage colony-stimulating factor (M-CSF; R and D Systems, Minneapolis, MN) at 37°C in a 5% CO2 atmosphere. On day 7, adherent BMDM were obtained and cultured in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml M-CSF. Quercetin dihydrate (Sigma-Aldrich, St. Louis, MO) was first dissolved in dimethyl sulfoxide (DMSO) and diluted by medium containing 2% FBS to achieve the final concentration. Medium containing 0.05% DMSO was used as the control group.
Cholesterol efflux
RAW264.7 macrophages or BMDM plated on 24-well plates were radiolabeled with 0.5 μCi/ml [1,2-3H]cholesterol (PerkinElmer, Boston, MA) in DMEM containing 0.2% BSA (BSA; Sigma-Aldrich) for 24 h. To deliver macrophages with more cholesterol, cells were also treated with cholesterol (Wako Pure Chemical Industries, Osaka, Japan) dissolved in ethanol and diluted into serum-free medium at the appropriate concentrations. RAW264.7 macrophages were incubated in 0.2% BSA-containing DMEM with 10 μg/ml of acetylated low-density lipoprotein (AcLDL; Molecular Probes, Eugene, OR) or 30 μg/ml of cholesterol in the presence of 0.5 μCi/ml [1,2-3H]cholesterol for 24 h. Subsequently, cells were washed and incubated with quercetin in indicated concentrations for another 24 h. To perform apoAI-mediated cholesterol efflux, the medium was replaced with fresh medium containing 0.2% BSA with or without 10 μg/ml lipid-free human apoAI (Sigma-Aldrich) for 24 h. The efflux medium was then collected and centrifuged to remove cell debris. Cells were lysed in 0.1 N NaOH, and radioactivities of the medium and the cell lysate were measured by liquid scintillation counting. Cholesterol efflux was expressed as the percentage of counts in the medium relative to the total counts (medium and cells). ApoAI-specific cholesterol efflux was obtained by subtracting the nonspecific efflux that occurred in apoAI-free medium. For TAK1 inhibition study, RAW264.7 macrophages or BMDM were pretreated with 5 μM 5Z-7-Oxozeaenol for 1 h and then treated with 100 μM quercetin for 24 h.
RNA extraction and quantitative real-time PCR analysis
Total cellular RNA was extracted by TRI Reagent (Sigma-Aldrich) following the manufacturer's protocol. Reverse transcription was carried out with 2 μg total RNA using moloney murine leukemia virus reverse transcriptase (MMLV; Invitrogen, Carlsbad, CA). Quantitative real-time PCR was assessed using SYBR Green PCR Master Mix (Finnzymes, Espoo, Finland) on a Roche LightCycler system (Roche Diagnostics, Mannheim, Germany). The primer sequences were as follows: ABCA1 (forward 5′-GGTTTGGAGATGGTTATACAATAGTTGT-3′ and reverse 5′-CCCGGAAACGCAAGTCC-3′) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, forward 5′-GTATGACTCCACTCACGGCAAA-3′ and reverse 5′-GGTCTCGCTCCTGGAAGATG-3′). GAPDH was used for the internal normalization.
Western blot analysis
Cells were lysed with lysis buffer [20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 50 mM dithiothreitol, complete protease inhibitor cocktail (Roche Diagnostics) and phosphatase inhibitor cocktail I and II (Sigma-Aldrich)]. The cell lysates were centrifuged and the supernatants were collected. The protein concentration was assayed using Bradford reagent (Bio-Rad, Hercules, CA) with BSA as the standard. Equal amounts of protein were analyzed by SDS-polyacrylamide gel, and transferred to nitrocellulose membranes (Pall, Glen Cove, NY). The immunoblots were blocked with 5% nonfat milk in PBST (5.8 mM Na2HPO4·7H2O, 130 mM NaCl and 0.05% Tween-20) for 1 h at room temperature, and then incubated with first antibodies overnight at 4°C. After PBST wash, the blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma-Aldrich), and the target protein bands were visualized by using enhanced chemiluminescence system (ECL; PerkinElmer). The blots were then stripped for further probing with α-tubulin or B23 antibody as internal controls for the total cellular or nuclear proteins, respectively. Relative intensities of protein bands were quantified by densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA).
Lentivirus-mediated RNA interference
Knockdown of p38 expression was performed by the lentivirus-packaged small hairpin RNA (shRNA) approach. Two different clones expressing p38-targeting shRNAs were obtained from the National RNAi Core Facility of Academia Sinica (Taiwan). The targeting sequences of these two clones were as follows: p38 shRNA-1 (5′-CCTCTTGTTGAAAGATTCCTT-3′) and p38 shRNA-2 (5′-GCTGAATTGGATGCACTATAA-3′). Luciferase shRNA was used as a control shRNA. Briefly, lentiviral-containing shRNAs at an MOI of 3 were used to infect RAW264.7 macrophages in the presence of 8 μg/ml polybrene (Sigma-Aldrich). After 24 h, the infected cells were selected with 2 μg/ml puromycin (Sigma-Aldrich) for two weeks to generate stable-knockdown cells. The knockdown efficiency of p38 was examined by Western blot analysis using anti-total p38 antibody.
Construction of ABCA1 promoter-driven luciferase reporter plasmids
Various lengths of the mouse ABCA1 promoter sequences were amplified by PCR using the primers as indicated (Table 1). Each PCR fragment was subsequently cloned into the XhoI/HindIII restriction sites of the luciferase reporter plasmid pGL3-basic vector (Promega, Madison, WI). To introduce site-specific mutations in putative transcription factor binding sites within −250/−1 region of the ABCA1 promoter, site-directed mutagenesis was performed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. The DNA sequences of the constructs were verified by automated DNA sequencing. The mouse p38 expression plasmid (pRcCMV-p38) was a gift from Professor Fung-Fang Wang's lab (National Yang-Ming University, Taipei, Taiwan).
TABLE 1.
Primers used for the plasmid constructions
| Primer Set | Sequence |
| WT-1550 | 5′-CCG CTCGAG ATTTAGTAGGAGTACTCTTGG-3′ (XhoI) |
| 5′-CCG AAGCTT GGTTTTTGCCGCGACTAGTTC-3′ (HindIII) | |
| WT-1100 | 5′-CCG CTCGAG AATGCTTGCCTGCTATGCAAG-3′ (XhoI) |
| 5′-CCG AAGCTT GGTTTTTGCCGCGACTAGTTC-3′ (HindIII) | |
| WT-700 | 5′-CCG CTCGAG AGGTCCCTGGAAGGTAGAGAT-3′ (XhoI) |
| 5′-CCG AAGCTT GGTTTTTGCCGCGACTAGTTC-3′ (HindIII) | |
| WT-400 | 5′-CCG CTCGAG TCAAAAAGCAACACCCACAAA-3′ (XhoI) |
| 5′-CCG AAGCTT GGTTTTTGCCGCGACTAGTTC-3′ (HindIII) | |
| WT-250 | 5′-TGC CTCGAG GGCCAGGGCTACAGAAAGCGG-3′ (XhoI) |
| 5′-CCG AAGCTT GGTTTTTGCCGCGACTAGTTC-3′ (HindIII) | |
| mSp1a | 5′-CACTGTCGCCGGTTTAAGGttttGGCCATGTCTCCACGTGCT-3′ |
| 5′-AGCACGTGGAGACATGGCCaaaaCCTTAAACCGGCGACAGTG-3′ | |
| mE-box | 5′-GGGCGGGCCATGTCTCCAaaTGCTTTCTGCTGAGTGAC-3′ |
| 5′-GTCACTCAGCAGAAAGCAttTGGAGACATGGCCCGCCC-3′ | |
| mAP1 | 5′-CGTGCTTTCTGCTGAGTtgtTGAACTACATAAACAGAGGCCG-3′ |
| 5′-CGGCCTCTGTTTATGTAGTTCAacaACTCAGCAGAAAGCACG-3′ | |
| mSp1b | 5′-GGCCGGGAAGGtttCGGGGGAAAGAGGGAGAGAACAGCGTTTGACCG-3′ |
| 5′-CGGTCAAACGCTGTTCTCTCCCTCTTTCCCCCGaaaCCTTCCCGGCC-3′ | |
| mLXR | 5′-GGGAGAGAACAGCGTTTGtgtGGTAGTAACtaCGGCGCTCGGCAC-3′ |
| 5′-GTGCCGAGCGCCGtaGTTACTACCacaCAAACGCTGTTCTCTCCC-3′ |
Restriction enzyme cutting sites are underlined. Mutations are indicated by lowercase letters.
Transient transfection and luciferase assay
Transient transfection was performed using Lipofectamine LTX and PLUS reagents (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Cells were cotransfected with 1 μg of ABCA1 promoter-driven luciferase reporter plasmid and 0.5 μg of pCMV-β-galactosidase expression plasmid in serum-free DMEM. After 6 h of transfection, the medium was replaced with fresh DMEM containing 10% FBS and incubated for further 18 h at 37°C. Cells were then treated with 100 μM quercetin for 24 h and lysed with lysis buffer (70 mM K2HPO4, 2.1 mM MgCl2, 55 mM Tris-HCl, pH 7.8, 0.7 mM dithiothreitol, and 1% Triton X-100). Cell lysates were harvested by centrifugation. Luciferase activity was quantified with a VICTOR2 Multilabel Reader (PerkinElmer). β-galactosidase activity was measured by incubating cell extracts with o-nitrophenyl-β-D-galactopyranoside (Sigma-Aldrich). Relative luciferase activity was presented as firefly luciferase values normalized to β-galactosidase activity.
Nuclear protein extraction
Cells were lysed by hypotonic lysis buffer [20 mM HEPES, pH 7.4, 10 mM KCl, 1 mM MgCl2, 0.5% NP-40, 0.5 mM dithiothreitol, and complete protease inhibitor cocktail (Roche Diagnostics)]. Nuclei were pelleted by centrifugation and lysed by high salt nuclear extraction buffer [20 mM HEPES, pH 7.4, 10 mM KCl, 1 mM MgCl2, 400 mM NaCl, 0.5 mM dithiothreitol, and complete protease inhibitor cocktail (Roche Diagnostics)]. The nuclear proteins were harvested after centrifugation.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) experiments were performed according to the manufacturer's instructions (Cell Signaling Technology, Beverly, MA) with some modifications. RAW264.7 macrophages were treated with or without 100 μM quercetin for 3 h, harvested, and crosslinked with 1% formaldehyde at 37°C for 10 min. Crosslinking was stopped by adding glycine to 125 mM for 5 min at room temperature. Cells were washed twice with ice-cold PBS, and then lysed with hypotonic cell lysis buffer [5 mM HEPES, pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM dithiothreitol, and complete protease inhibitor cocktail (Roche Diagnostics)]. After centrifugation, nuclear pellets were collected and resuspended in micrococcal nuclease (MNase) digestion buffer [50 mM Tris-HCl, pH 7.4, 0.32 M sucrose, 4 mM MgCl2, 1 mM CaCl2, and complete protease inhibitor cocktail (Roche Diagnostics)]. Chromatin was digested into fragments of 150–900 bp by MNase (New England Biolabs, Beverly, MA), and this was followed by sonication to disrupt nuclear membrane. For immunoprecipitation, 15–25 μg of chromatin was incubated with 1 μg of antibodies against Sp1 or LXRα. Nonimmune IgG was used as a negative control. Antibody-protein-DNA complexes were pulled down with 50% protein A agarose beads/salmon sperm DNA slurry (Millipore, Temecula, CA). Precipitated chromatin complexes were eluted with elution buffer (1% SDS and 0.1 M NaHCO3). Crosslinking was reversed with 250 mM NaCl and proteinase K overnight at 65°C. DNA was purified by phenol/chloroform extraction and isopropanol precipitation in the presence of glycogen. The fragments of the ABCA1 promoter containing the predicted Sp1 and LXRα binding sites were detected by PCR using the primers as follows: forward 5′-GGCGGGCCATGTCTCCACG-3′ and reverse 5′-GCTCGCTCGCCCTCGGAATT-3′. The PCR products were separated on a 2% agarose gel and visualized by ethidium bromide staining.
Statistical analysis
Data are expressed as mean ± SEM of at least three independent experiments. Statistical analysis was performed using Student t-test or one-way ANOVA with the Tukey's method as a posthoc test. A value of P < 0.05 was considered statistically significant.
RESULTS
Quercetin enhances cholesterol efflux and ABCA1 expression in macrophages
To determine whether quercetin may influence cholesterol homeostasis, we first investigated the role of quercetin in the control of apoAI-mediated cholesterol efflux from macrophages. The results in Fig. 1A showed that quercetin concentration-dependently enhanced apoAI-mediated cholesterol efflux from RAW264.7 macrophages and primary macrophages, BMDM. We also treated RAW264.7 macrophages with AcLDL or cholesterol to examine the effect of quercetin on cholesterol efflux from the cholesterol-loaded macrophages. As shown in Fig. 1B, quercetin increased apoAI-mediated cholesterol efflux from AcLDL- or cholesterol-loaded macrophages in a dose-dependent manner. As the importance of ABCA1 in apoAI-mediated cholesterol efflux has been established (14, 15), we next examined the effect of quercetin on the mRNA level of ABCA1 by quantitative real-time PCR. The results revealed that quercetin also upregulated the ABCA1 mRNA level in a dose-dependent manner up to 6-fold at 100 μM quercetin treatment (Fig. 1C). We then investigated whether the elevated ABCA1 mRNA level resulted in an increased expression of ABCA1 protein. As shown in Fig. 1D, ABCA1 protein level was increased in a concentration-dependent manner up to 8-fold when cells were subjected to 100 μM quercetin treatment. These results suggest that quercetin-induced ABCA1 is a facilitating factor in the stimulation of cholesterol efflux from macrophages.
Fig. 1.

Quercetin enhances cholesterol efflux and expression of ABCA1 in macrophages. A: [3H]cholesterol-labeled RAW264.7 macrophages or BMDM were treated with quercetin as indicated concentrations for 24 h. ApoAI-dependent cholesterol efflux was measured by incubating [3H]cholesterol-labeled macrophages with or without 10 μg/ml apoAI for 24 h. Cholesterol efflux was expressed as the percentage of radioactivity in the medium relative to the total radioactivity (medium and cells). B: RAW264.7 macrophages were loaded with 10 μg/ml of acetylated low-density lipoprotein (AcLDL) or 30 μg/ml cholesterol in the presence of [3H]cholesterol for 24 h. Cholesterol efflux was measured as described in Materials and Methods. C: After cells were treated with quercetin for 18 h, mRNA levels of ABCA1 were detected by quantitative real-time PCR and normalized to GAPDH. D: Cells were treated with quercetin for 24 h, and then total cell extracts were harvested. ABCA1 protein expressions were measured by Western blot analysis and α-tubulin was utilized as a loading control. The normalized level of mRNA or protein from cells without quercetin treatment was set as 1. Results are expressed as mean ± SEM (n = 3–5). *P < 0.05, **P < 0.01 versus control group.
Quercetin induces phosphorylation of p38, TAK1, and MKK3/6
To investigate the mechanisms underlying the regulation of ABCA1 expression by quercetin, we assessed the effect of quercetin on the phosphorylation of p38, ERK, and JNK. Quercetin time-dependently induced the phosphorylation of p38 by 168-fold to 234-fold at 2 to 6 h (Fig. 2A). However, there were no significant differences in the levels of phosphorylated ERK and JNK among the groups at various times compared with the 0 h group. As TAK1 and MKK3/6 are upstream kinases of p38 (27), we next examined the effect of quercetin on the activation of TAK1 and MKK3/6. The levels of phosphorylated TAK1 and MKK3/6 were significantly increased after quercetin treatment for 0.5 to 6 h (Fig. 2B). Therefore, we next assessed the effect of 5Z-7-Oxozeazenol (5Z-7-Oxo), a specific TAK1 inhibitor, on the quercetin-induced phosphorylation of p38, TAK1, and MKK3/6. As shown in Fig. 2B, the phosphorylation levels of p38, TAK1, and MKK3/6 were markedly suppressed by 5Z-7-Oxo. Moreover, we investigated the effect of 5Z-7-Oxo on quercetin-induced cholesterol efflux from RAW264.7 macrophages or BMDM. 5Z-7-Oxo significantly reversed the induction of cholesterol efflux by quercetin (Fig. 2C, D). These results indicate that TAK1-MKK3/6-p38 signaling cascade is essential for the regulation of quercetin-stimulated cholesterol efflux from macrophages.
Fig. 2.

Quercetin activates p38 via the TAK1-MKK3/6 signaling cascade. A: The proteins from total cell lysates were separated by SDS-PAGE and immunoblotted with anti-phospho-MAPK antibodies. Immunoblots were reprobed with total MAPK antibodies for internal normalization. B: RAW264.7 macrophages were pretreated with 5 μM 5Z-7-Oxo for 1 h, and then treated with 100 μM quercetin as indicated time to determine whether the p38 activation was via phosphorylation of TAK1 and MKK3/6. An arrow indicates MKK3 band. Histograms show the relative intensity of normalized phospho-MAPK, phospho-TAK1, and phospho-MKK3/6 over the 0 h group. Data represent mean ± SEM (n = 3–5). **P < 0.01 versus 0 h group, #P < 0.05 versus 2 h group. C, D: Analyses were performed to determine the effect of 5Z-7-Oxo on cholesterol efflux in RAW264.7 macrophages (C) or BMDM (D) as described in Materials and Methods. Bars are mean ± SEM (n = 3–4). **P < 0.01, ##P < 0.01.
p38 activation is crucial for the regulation of quercetin-induced ABCA1 expression
To understand the mechanisms of quercetin-induced regulation of ABCA1 expression, we addressed whether the p38 signaling pathway was necessary for ABCA1 induction. RAW264.7 macrophages were pretreated with SB203580, a pharmacological inhibitor of p38, followed by treatment with quercetin. Quercetin-induced ABCA1 mRNA and protein expression were remarkably attenuated in cells treated with SB203580 (Fig. 3A, B). Moreover, a significant reduction in p38 was observed when p38 shRNA-1- and p38 shRNA-2-expressing cells (RAW264.7 macrophages were infected with lentivirus containing p38-targeting shRNAs) were examined (Fig. 3C). Luciferase shRNA was used as a control shRNA. Importantly, both mRNA and protein levels of ABCA1 induced by quercetin were significantly suppressed in p38 shRNA-expressing cells (Fig. 3D, E). In line with the inhibitory effect of p38 knockdown on ABCA1 expression, quercetin-induced cholesterol efflux was also suppressed in p38 shRNA-expressing cells (Fig. 3F). Together, these results support the critical role of p38 in the induction of ABCA1 expression by quercetin and the subsequent changes in cholesterol efflux in macrophages.
Fig. 3.

p38 is involved in the quercetin-induced ABCA1 expression and cholesterol efflux. RAW264.7 macrophages were pretreated with 20 μM SB203580 (p38 inhibitor), and then treated with 100 μM quercetin. A: The ABCA1 mRNA levels were measured by quantitative real-time PCR and normalized to GAPDH. B: The ABCA1 protein levels were detected by Western blot analysis and α-tubulin was utilized as a loading control. The normalized level of mRNA or protein from cells without quercetin treatment was set as 1. RAW264.7 macrophages were infected with lentivirus expressing p38 shRNA-1 or p38 shRNA-2 to confirm the effects of p38 on quercetin-induced ABCA1 expression and cholesterol efflux. Luciferase shRNA (Luc. shRNA) was used as a control shRNA. C: The knockdown efficiency of p38 was checked by Western blot analysis. D, E: The effect of p38 knockdown on quercetin-induced ABCA1 mRNA (D) and protein (E) expression was examined by quantitative real-time PCR and Western blot analysis, respectively. The normalized level of mRNA or protein from parental cells without quercetin treatment was given as 1. F: Cholesterol efflux from p38 knockdown cells to media was measured in the presence of 10 μg/ml apoAI. Cholesterol efflux was expressed as the percentage of radioactivity in the medium relative to the total radioactivity (medium and cells). Values are mean ± SEM (n = 3–6). *P < 0.05, **P < 0.01, #P < 0.05, ##P < 0.01.
Identification of the quercetin-responsive region within the ABCA1 promoter
The results in Fig. 1B implies that quercetin may regulate ABCA1 expression at the transcription level. To define the quercetin-responsive region within the ABCA1 promoter, we generated five 5′-serial deletion constructs, wild-type (WT)-1550, WT-1100, WT-700, WT-400, and WT-250, that contained the ABCA1 promoter region between −1550 and −1 bp. All fragments of the ABCA1 promoter constructs were analyzed for luciferase activity in RAW264.7 macrophages treated in the absence or presence of 100 μM quercetin. As shown in Fig. 4, the luciferase activity of the constructs from −1550 to −250 sites showed significantly higher luciferase activity (WT-250, 6.1 ± 0.5-fold; WT-400, 6.6 ± 0.6-fold; WT-700, 6.5 ± 0.6-fold; WT-1100, 5.9 ± 0.4-fold; and WT-1550, 4.8 ± 0.2-fold) compared with the promoter activity in the transfected cells without treatment of quercetin (WT-250, 2.3 ± 0.2-fold; WT-400, 2.5 ± 0.2-fold; WT-700, 2.8 ± 0.2-fold; WT-1100, 2.4 ± 0.1-fold; and WT-1550, 1.9 ± 0.2-fold). These observations imply that the main quercetin-responsive region required for ABCA1 promoter activity is located in the region between −250 and −1 bp.
Fig. 4.

Characterization of quercetin-responsive domains in the ABCA1 promoter. Schematic diagram of putative binding sites for transcription factors and serial deletion constructs of ABCA1 promoter are shown. RAW264.7 macrophages were transiently cotransfected with 1 μg of indicated constructs and 0.5 μg of pCMV β-galactosidase expression plasmid, and then treated with 100 μM quercetin (Q100) for 24 h. Luciferase activity was normalized with β-galactosidase activity. Results are shown as fold changes in luciferase activity relative to the untreated pGL3-basic vector group. The level of luciferase activity without quercetin treatment in pGL3-basic vector group was given the value of 1. Bars are mean ± SEM (n = 3). **P < 0.01 versus quercetin-treated pGL3-basic vector group.
The Sp1 and LXR binding sites are critical for the regulation of quercetin-induced ABCA1 promoter activity
The potential binding sites for Sp1, E-box, AP1, and LXR/RXR within the −250/−1 region of the ABCA1 promoter were predicted by using the TRANSFAC program (28) (Fig. 4). To address the importance of these transcription factor binding sites in quercetin-induced ABCA1 promoter activities, we generated several mutated constructs (mSp1a, mE-box, mAP1, mSp1b, and mlXR) by site-directed mutagenesis as shown in Fig. 5A. The results showed that mutagenesis of the Sp1b or LXR sites attenuated quercetin-induced ABCA1 promoter activity compared with the level of WT-250 promoter. Moreover, a construct containing double mutations of Sp1b and LXR sites (mSp1b/mLXR) was generated to validate whether these two sites were essential for the regulation of quercetin-induced ABCA1 promoter activity. It is clear that quercetin-induced promoter activity of mSp1b/mLXR showed a more significant decrease compared with that of Sp1b or LXR mutation alone (Fig. 5A). We thus assume that Sp1b and LXR sites are both required for the regulation of ABCA1 gene expression in cells under quercetin treatment.
Fig. 5.

Sp1 and LXR binding sites are quercetin-responsive elements within the ABCA1 promoter. A: Six mutation constructs of ABCA1 promoter in the potential Sp1a, E-box, AP1, Sp1b, and LXR recognition sites were generated using site-directed mutatgenesis. RAW264.7 macrophages were transiently cotransfected with 1 μg of indicated mutant constructs and 0.5 μg of pCMV β-galactosidase expression plasmid, and then treated with 100 μM quercetin (Q100) for 24 h. Luciferase activity was normalized with β-galactosidase activity and expressed as fold changes in luciferase activity compared with those of the control. The level of luciferase activity without quercetin treatment was given the value of 1. WT-250, −250/−1 region of ABCA1 promoter. mSp1b/mLXR, double mutations at Sp1b and LXR sites. B: Cells were treated with quercetin for 3 h, and nuclear extracts were harvested. The nuclear expression levels of Sp1 and LXR were analyzed by Western blot analysis. The arrow indicates Sp1 band. B23 was used as nuclear loading control, and α-tubulin was used to exclude cytosolic contamination. The level of Sp1 or LXR without quercetin treatment was given the value of 1. C: ChIP assays were performed to observe the binding of Sp1 and LXRα to the ABCA1 promoter in quercetin-treated cells as described in Materials and Methods. Nonimmune IgG was used as the negative control. The results of the ChIP assays were evaluated by PCR and gel electrophoresis. Values are quantified by densitometer and expressed as ratio relative to inputs. Results are mean ± SEM (n = 3–4). **P < 0.01, #P < 0.05, ##P < 0.01.
As depicted in Fig. 5B, the nuclear levels of Sp1 and LXR were increased in cells treated with quercetin, suggesting that both Sp1 and LXR may be involved in the regulation of quercetin-induced ABCA1 gene expression. The binding properties of Sp1 and LXRα to the ABCA1 promoter were also examined by ChIP assays. Antibodies against Sp1 or LXRα were used to immunoprecipitate the protein-chromatin complex. It was found that the interaction of Sp1 and LXRα with the −250/−1 region of ABCA1 promoter was significantly increased (Fig. 5C). These results are consistent with those of the mutagenesis study, indicating that quercetin stimulates ABCA1 promoter activity via the binding of Sp1 and LXRα to the ABCA1 promoter region between −250 and −1 bp. These observations confirm the essential role of Sp1 and LXRα activation in quercetin-induced ABCA1 gene expression.
The binding of Sp1 and LXRalpha to the ABCA1 promoter is mediated through p38 activation
We also investigated whether the stimulatory effect of quercetin on ABCA1 gene expression was mediated through p38 signaling in p38 shRNA-expressing cells. As shown in Fig. 6A, attenuation of quercetin-induced ABCA1 promoter activity was observed in the p38 knockdown cells. In addition, we conducted rescue experiments using macrophages that overexpressed p38. The results showed that overexpression of p38 rescued the promoter activity when compared with the p38 knockdown cells (Fig. 6A). Together, these findings suggest that p38 plays an essential role in the quercetin-induced ABCA1 promoter activity. Moreover, the binding of Sp1 and LXRα to the ABCA1 promoter induced by quercetin was greatly reduced in p38 knockdown cells using ChIP assays (Fig. 6B). These observations confirm that p38 is required for the binding of Sp1 and LXRα to the ABCA1 promoter, suggesting that p38-mediated Sp1 and LXRα activation is involved in the regulation of quercetin-induced ABCA1 gene expression.
Fig. 6.
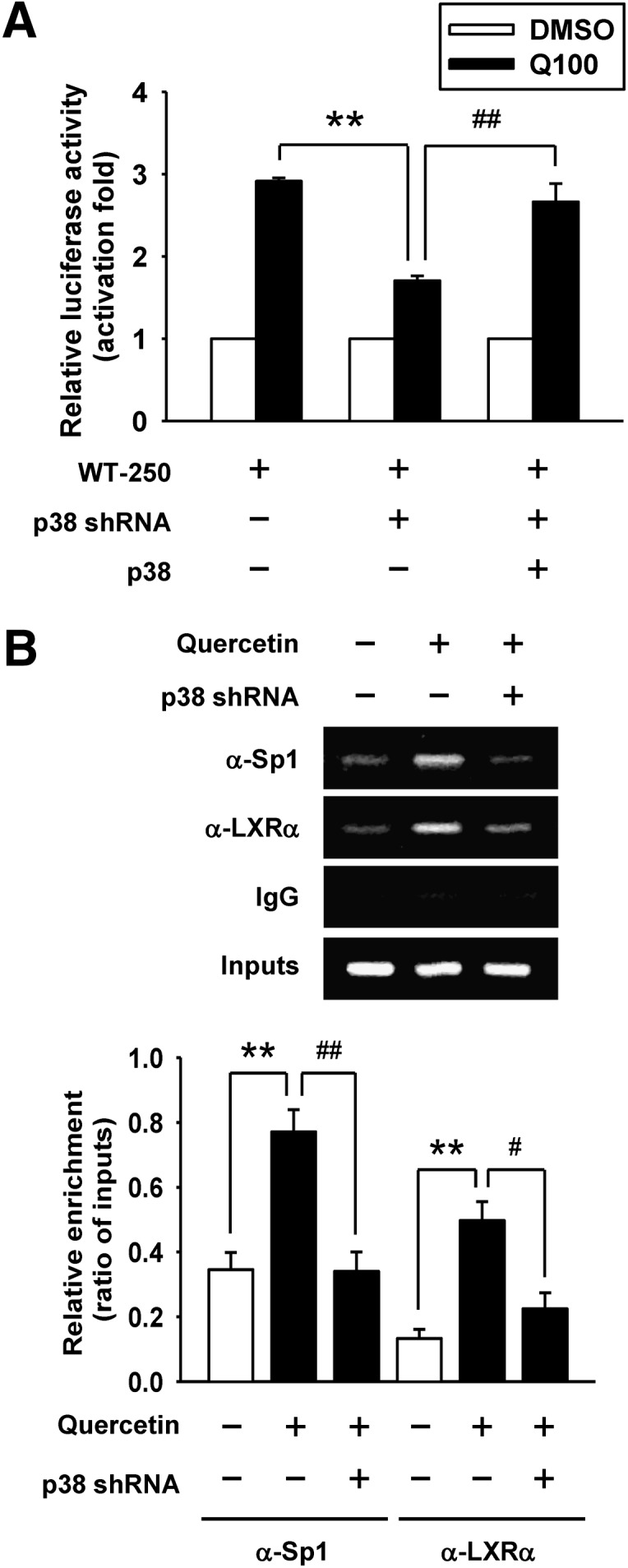
Effect of p38 knockdown by shRNA on the attenuation of Sp1 and LXRα binding to the ABCA1 promoter. A: Parental cells or p38 knockdown cells were transiently cotransfected with 0.5 μg of WT-250 construct, 0.5 μg of pRcCMV vector, and 0.5 μg of pCMV β-galactosidase plasmid. For p38 overexpression, p38 knockdown cells were transiently cotransfected with 0.5 μg of WT-250 construct, 0.5 μg of pRcCMV-p38 plasmid, and 0.5 μg of pCMV β-galactosidase plasmid. After transfection, cells were treated with 100 μM quercetin (Q100) for 24 h. Luciferase activity was normalized with β-galactosidase activity and expressed as fold changes in luciferase activity compared with respective control groups. The level of luciferase activity without quercetin treatment was given the value of 1. WT-250, −250/−1 region of ABCA1 promoter. B: ChIP assays were performed to observe the binding of Sp1 and LXRα to the ABCA1 promoter in p38 knockdown cells as described in Materials and Methods. Nonimmune IgG was used as the negative control. The results of ChIP assays were evaluated by PCR and gel electrophoresis. Values are quantified by densitometer and expressed as ratio relative to inputs. Bars are mean ± SEM (n = 3). **P < 0.01, #P < 0.05, ##P < 0.01.
DISCUSSION
In this study, we investigated the molecular mechanisms underlying quercetin-regulated ABCA1 expression and cholesterol efflux in macrophages. As illustrated in Fig. 7, our results show that quercetin induces apoAI-mediated cholesterol efflux through activation of ABCA1 mRNA and protein expression in RAW264.7 macrophages. It is clear that quercetin-induced ABCA1 expression and cholesterol efflux are mediated by activating the TAK1-MKK3/6-p38 signaling cascade. Moreover, activated p38 signaling increases the binding of Sp1 and LXRα to their corresponding cis-elements within the −250/−1 region of the ABCA1 promoter, which in turn increases ABCA1 gene expression in macrophages.
Fig. 7.

A model describing the mechanisms of quercetin-induced cholesterol efflux in macrophages. Quercetin elicits the TAK1-MKK3/6 signaling cascade to activate p38. Activated p38 subsequently increases the binding of Sp1 and LXRα to the ABCA1 promoter, which in turn enhances the expression of ABCA1 as well as cholesterol efflux from macrophages.
Over the past decade, some in vitro and in vivo studies have shown that quercetin has anti-oxidative and anti-inflammatory effects (2, 3, 29). Other researchers have indicated that quercetin reduces plasma cholesterol levels and atherosclerotic progression in rabbits and hamsters fed with a high-fat diet, which implies that quercetin may also have anti-lipidemia and anti-atherosclerotic properties (6, 7). Cholesterol efflux from cholesterol-loaded macrophages is a key atheroprotective mechanism that counteracts cholesterol uptake (10, 11). Several lines of evidence indicate that induction of cholesterol efflux retards the development of atherosclerosis in apolipoprotein E (apoE) knockout mice and LDL receptor knockout mice (30, 31). Our results showed that quercetin treatment markedly induced apoAI-mediated cholesterol efflux in primary macrophages and cholesterol-loaded macrophages, which seems to provide a new explanation for the effect of quercetin on the amelioration of atherosclerotic plaques. In this study, we also explored the molecular mechanisms that affect cholesterol transportation in macrophages. The concentration of quercetin used in our present study was similar to that used in other studies (32, 33). A previous study showed that rats, fed 0.2% quercetin diet for 10 days, maintain 100 μM concentration of quercetin in the plasma (34). Moreover, the concentrations of quercetin in plasma are positively correlated with the intake of quercetin in diet. In the animal study, it seems that the intake of quercetin between 40 and 1,900 mg/kg/day is a safe range for clinical use (35–38).
The importance of ABCA1 in apoAI-mediated cholesterol efflux has been well established (14). Transplantation of bone marrow containing ABCA1-overexpressing macrophages leads to an inhibition of atherosclerotic lesion progression (16). On the other hand, macrophages lacking ABCA1 are defective in cholesterol efflux in vivo (39, 40). Increasing evidence points to induced expression of ABCA1 or promoted function of cholesterol efflux, which is responsible for the reduction of cholesterol accumulation in macrophages treated with dietary flavonoids, such as wogonin and procyanidin (41, 42). Our results suggest that the upregulation of ABCA1 expression, both at mRNA and protein levels, by quercetin may be beneficial because it increases the cholesterol efflux from macrophages, thereby resulting in atherosclerotic protection. Moreover, this study is the first to show molecular mechanisms involved in quercetin-mediated ABCA1 gene regulation and cholesterol efflux.
The sequence required for human ABCA1 promoter activity is located in the −175/+224 region (43). This region includes one LXR binding site (position −62) and two Sp1 binding sites (positions −90 and −156). Additionally, a number of other transcription factor binding sites have been predicted in this region and are conserved in both humans and mice (44). Thymiakou et al. reported that both the LXR site and the two Sp1 binding sites were required for the induction of ABCA1 gene transcription in human hepatoma cells treated with oxysterols and retinoids (19). We observed that only the proximal Sp1 binding site (Sp1b, position −99) was involved in quercetin-stimulated ABCA1 promoter activity. This observation is in agreement with those of Zhao et al. and Chen et al., who found that this Sp1 binding motif (position −99) is essential for ABCA1 promoter activity in apoE- or LDL-treated RAW264.7 macrophages (45, 46). It has been reported that LXR also participates in the regulation of ABCA1 gene expression (17). Similar results were reported by Sevov et al. showing that resveratrol upregulates LXRα expression to induce ABCA1 expression in human macrophages (47). Our results indicated that quercetin increased the binding of both LXRα and Sp1 to the ABCA1 promoter in murine cell line RAW264.7 macrophages.
We also found that quercetin potently induced p38 phosphorylation in RAW264.7 macrophages. This result is consistent with the previous finding that quercetin induces p38 activation and Sp1 binding to the promoters of tissue-type plasminogen activator (21) and filamin A (48). These observations suggest that p38 can increase Sp1 promoter binding activity and induce gene transcription. We therefore tested hypothesis that quercetin modulates ABCA1 promoter activity through p38 signaling pathway. This hypothesis was confirmed by a promoter assay showing that quercetin-induced ABCA1 promoter activity was suppressed in p38 knockdown RAW264.7 macrophages. Kaplan et al. reported that lipopolysaccharide-induced ABCA1 expression was through the p38 signaling pathway in human monocytic leukemia cells (24). In contrast, a high concentration of glucose induces p38 activation, but it suppresses ABCA1 expression in vascular smooth muscle cells (23). In this study, we found that quercetin induced the TAK1-MKK3/6-p38 signaling cascade and upregulated ABCA1 expression in RAW264.7 macrophages. Moreover, we showed that the quercetin-induced binding of Sp1 to the ABCA1 promoter is suppressed in the p38 knockdown cells. A recent study has shown that a p38 inhibitor, SB202190, inhibits the induction of ABCA1 mRNA expression by LXR ligand in prostate cancer epithelial cells (49), implying that p38 could regulate LXR-mediated gene transcription. We also found that the quercetin-induced binding of LXRα to the ABCA1 promoter was attenuated in p38 knockdown RAW264.7 macrophages. Collectively, our results indicate that quercetin enhances p38 signaling and subsequently this potentiates the binding of Sp1 and LXRα to the ABCA1 promoter, which in turn increases ABCA1 expression and cholesterol efflux in macrophages.
In conclusion, this study provides novel insights into the protective effect of quercetin on enhancing cholesterol efflux via upregulating ABCA1 expression, which is mediated by increasing p38-dependent Sp1 and LXRα binding to the ABCA1 promoter in macrophages. Quercetin may therefore be a promising therapeutic agent for the prevention of atherosclerotic progression.
Acknowledgments
The authors thank Professor Fung-Fang Wang's lab (National Yang-Ming University, Taipei, Taiwan) for providing the mouse p38 expression plasmid, and Professor Hsing-Jien Kung (University of California at Davis, Sacramento, CA) for editing the manuscript.
Footnotes
Abbreviations:
- AcLDL
- acetylated LDL
- BMDM
- bone marrow-derived macrophages
- ERK1/2
- extracellular signal-regulated kinase 1/2
- JNK1/2
- c-Jun N-terminal kinase 1/2
- LXRα
- liver X receptor α
- MAPK
- mitogen-activated protein kinase
- MKK3/6
- mitogen-activated protein kinase kinase 3/6
- oxLDL
- oxidized LDL
- RXR
- retinoid X receptor
- Sp1
- specificity protein 1
- shRNA
- small hairpin RNA
- TAK1
- transforming growth factor β-activated kinase 1
- WT
- wild-type
- 5Z-7-Oxo
- 5Z-7-Oxozeaenol
This work was supported by grants from Ministry of Education, Aim for the Top University Plan (99AC-P504 and 100AC-P504), Taiwan.
REFERENCES
- 1.Peluso M. R. 2006. Flavonoids attenuate cardiovascular disease, inhibit phosphodiesterase, and modulate lipid homeostasis in adipose tissue and liver. Exp. Biol. Med. (Maywood). 231: 1287–1299. [DOI] [PubMed] [Google Scholar]
- 2.Boots A. W., Haenen G. R., Bast A. 2008. Health effects of quercetin: from antioxidant to nutraceutical. Eur. J. Pharmacol. 585: 325–337. [DOI] [PubMed] [Google Scholar]
- 3.Bischoff S. C. 2008. Quercetin: potentials in the prevention and therapy of disease. Curr. Opin. Clin. Nutr. Metab. Care. 11: 733–740. [DOI] [PubMed] [Google Scholar]
- 4.Kleemann R., Verschuren L., Morrison M., Zadelaar S., van Erk M. J., Wielinga P. Y., Kooistra T. 2011. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis. 218: 44–52. [DOI] [PubMed] [Google Scholar]
- 5.Rivera L., Moron R., Sanchez M., Zarzuelo A., Galisteo M. 2008. Quercetin ameliorates metabolic syndrome and improves the inflammatory status in obese Zucker rats. Obesity (Silver Spring). 16: 2081–2087. [DOI] [PubMed] [Google Scholar]
- 6.Juz´wiak S., Wójcicki J., Mokrzycki K., Marchlewicz M., Bialecka M., Wenda-Rózewicka L., Gawron´ska-Szklarz B., Droz´dzik M. 2005. Effect of quercetin on experimental hyperlipidemia and atherosclerosis in rabbits. Pharmacol. Rep. 57: 604–609. [PubMed] [Google Scholar]
- 7.Auger C., Teissedre P. L., Gerain P., Lequeux N., Bornet A., Serisier S., Besancon P., Caporiccio B., Cristol J. P., Rouanet J. M. 2005. Dietary wine phenolics catechin, quercetin, and resveratrol efficiently protect hypercholesterolemic hamsters against aortic fatty streak accumulation. J. Agric. Food Chem. 53: 2015–2021. [DOI] [PubMed] [Google Scholar]
- 8.Steinberg D. 2008. The statins in preventive cardiology. N. Engl. J. Med. 359: 1426–1427. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg D., Glass C. K., Witztum J. L. 2008. Evidence mandating earlier and more aggressive treatment of hypercholesterolemia. Circulation. 118: 672–677. [DOI] [PubMed] [Google Scholar]
- 10.Moore K. J., Tabas I. 2011. Macrophages in the pathogenesis of atherosclerosis. Cell. 145: 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libby P., Ridker P. M., Hansson G. K. 2011. Progress and challenges in translating the biology of atherosclerosis. Nature. 473: 317–325. [DOI] [PubMed] [Google Scholar]
- 12.Tabas I., Williams K. J., Boren J. 2007. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 116: 1832–1844. [DOI] [PubMed] [Google Scholar]
- 13.Kolodgie F. D., Burke A. P., Nakazawa G., Virmani R. 2007. Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler. Thromb. Vasc. Biol. 27: 986–989. [DOI] [PubMed] [Google Scholar]
- 14.Kang M. H., Singaraja R., Hayden M. R. 2010. Adenosine-triphosphate-binding cassette transporter-1 trafficking and function. Trends Cardiovasc. Med. 20: 41–49. [DOI] [PubMed] [Google Scholar]
- 15.Oram J. F., Heinecke J. W. 2005. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 85: 1343–1372. [DOI] [PubMed] [Google Scholar]
- 16.Van Eck M., Singaraja R. R., Ye D., Hildebrand R. B., James E. R., Hayden M. R., Van Berkel T. J. 2006. Macrophage ATP-binding cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 26: 929–934. [DOI] [PubMed] [Google Scholar]
- 17.Zhao C., Dahlman-Wright K. 2010. Liver X receptor in cholesterol metabolism. J. Endocrinol. 204: 233–240. [DOI] [PubMed] [Google Scholar]
- 18.Schmitz G., Langmann T. 2005. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim. Biophys. Acta. 1735: 1–19. [DOI] [PubMed] [Google Scholar]
- 19.Thymiakou E., Zannis V. I., Kardassis D. 2007. Physical and functional interactions between liver X receptor/retinoid X receptor and Sp1 modulate the transcriptional induction of the human ATP binding cassette transporter A1 gene by oxysterols and retinoids. Biochemistry. 46: 11473–11483. [DOI] [PubMed] [Google Scholar]
- 20.Yuan H., Young C. Y., Tian Y., Liu Z., Zhang M., Lou H. 2010. Suppression of the androgen receptor function by quercetin through protein-protein interactions of Sp1, c-Jun, and the androgen receptor in human prostate cancer cells. Mol. Cell. Biochem. 339: 253–262. [DOI] [PubMed] [Google Scholar]
- 21.Pan W., Chang M. J., Booyse F. M., Grenett H. E., Bradley K. M., Wolkowicz P. E., Shang Q., Tabengwa E. M. 2008. Quercetin induced tissue-type plasminogen activator expression is mediated through Sp1 and p38 mitogen-activated protein kinase in human endothelial cells. J. Thromb. Haemost. 6: 976–985. [DOI] [PubMed] [Google Scholar]
- 22.Zhou X., Yin Z., Guo X., Hajjar D. P., Han J. 2010. Inhibition of ERK1/2 and activation of liver X receptor synergistically induce macrophage ABCA1 expression and cholesterol efflux. J. Biol. Chem. 285: 6316–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu X., Murao K., Imachi H., Li J., Nishiuchi T., Hosomi N., Masugata H., Zhang G. X., Iwama H., Ishida T. 2010. Hyperglycemia suppresses ABCA1 expression in vascular smooth muscle cells. Horm. Metab. Res. 42: 241–246. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan R., Gan X., Menke J. G., Wright S. D., Cai T. Q. 2002. Bacterial lipopolysaccharide induces expression of ABCA1 but not ABCG1 via an LXR-independent pathway. J. Lipid Res. 43: 952–959. [PubMed] [Google Scholar]
- 25.Cargnello M., Roux P. P. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75: 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marim F. M., Silveira T. N., Lima D. S., Jr., Zamboni D. S. 2010. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE. 5: e15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuadrado A., Nebreda A. R. 2010. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429: 403–417. [DOI] [PubMed] [Google Scholar]
- 28.Matys V., Kel-Margoulis O. V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K., et al. 2006. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34: D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Pascual-Teresa S., Moreno D. A., Garcia-Viguera C. 2010. Flavanols and anthocyanins in cardiovascular health: a review of current evidence. Int. J. Mol. Sci. 11: 1679–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amar M. J., D'Souza W., Turner S., Demosky S., Sviridov D., Stonik J., Luchoomun J., Voogt J., Hellerstein M., Remaley A. T. 2010. 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J. Pharmacol. Exp. Ther. 334: 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bielicki J. K., Zhang H., Cortez Y., Zheng Y., Narayanaswami V., Patel A., Johansson J., Azhar S. 2010. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J. Lipid Res. 51: 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan H., Gong A., Young C. Y. 2005. Involvement of transcription factor Sp1 in quercetin-mediated inhibitory effect on the androgen receptor in human prostate cancer cells. Carcinogenesis. 26: 793–801. [DOI] [PubMed] [Google Scholar]
- 33.Chow J. M., Shen S. C., Huan S. K., Lin H. Y., Chen Y. C. 2005. Quercetin, but not rutin and quercitrin, prevention of H2O2-induced apoptosis via anti-oxidant activity and heme oxygenase 1 gene expression in macrophages. Biochem. Pharmacol. 69: 1839–1851. [DOI] [PubMed] [Google Scholar]
- 34.Manach C., Morand C., Demigne C., Texier O., Regerat F., Remesy C. 1997. Bioavailability of rutin and quercetin in rats. FEBS Lett. 409: 12–16. [DOI] [PubMed] [Google Scholar]
- 35.Egert S., Wolffram S., Bosy-Westphal A., Boesch-Saadatmandi C., Wagner A. E., Frank J., Rimbach G., Mueller M. J. 2008. Daily quercetin supplementation dose-dependently increases plasma quercetin concentrations in healthy humans. J. Nutr. 138: 1615–1621. [DOI] [PubMed] [Google Scholar]
- 36.de Boer V. C., Dihal A. A., van der Woude H., Arts I. C., Wolffram S., Alink G. M., Rietjens I. M., Keijer J., Hollman P. C. 2005. Tissue distribution of quercetin in rats and pigs. J. Nutr. 135: 1718–1725. [DOI] [PubMed] [Google Scholar]
- 37.Ma Z., Hung Nguyen T., Hoa Huynh T., Tien Do P., Huynh H. 2004. Reduction of rat prostate weight by combined quercetin-finasteride treatment is associated with cell cycle deregulation. J. Endocrinol. 181: 493–507. [DOI] [PubMed] [Google Scholar]
- 38.Dunnick J. K., Hailey J. R. 1992. Toxicity and carcinogenicity studies of quercetin, a natural component of foods. Fundam. Appl. Toxicol. 19: 423–431. [DOI] [PubMed] [Google Scholar]
- 39.Wang X., Collins H. L., Ranalletta M., Fuki I. V., Billheimer J. T., Rothblat G. H., Tall A. R., Rader D. J. 2007. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 117: 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang M. D., Franklin V., Marcel Y. L. 2007. In vivo reverse cholesterol transport from macrophages lacking ABCA1 expression is impaired. Arterioscler. Thromb. Vasc. Biol. 27: 1837–1842. [DOI] [PubMed] [Google Scholar]
- 41.Chen C. Y., Shyue S. K., Ching L. C., Su K. H., Wu Y. L., Kou Y. R., Chiang A. N., Pan C. C., Lee T. S. 2011. Wogonin promotes cholesterol efflux by increasing protein phosphatase 2B-dependent dephosphorylation at ATP-binding cassette transporter-A1 in macrophages. J. Nutr. Biochem. 22: 1015–1021. [DOI] [PubMed] [Google Scholar]
- 42.Terra X., Fernandez-Larrea J., Pujadas G., Ardevol A., Blade C., Salvado J., Arola L., Blay M. 2009. Inhibitory effects of grape seed procyanidins on foam cell formation in vitro. J. Agric. Food Chem. 57: 2588–2594. [DOI] [PubMed] [Google Scholar]
- 43.Langmann T., Porsch-Ozcurumez M., Heimerl S., Probst M., Moehle C., Taher M., Borsukova H., Kielar D., Kaminski W. E., Dittrich-Wengenroth E., et al. 2002. Identification of sterol-independent regulatory elements in the human ATP-binding cassette transporter A1 promoter: role of Sp1/3, E-box binding factors, and an oncostatin M-responsive element. J. Biol. Chem. 277: 14443–14450. [DOI] [PubMed] [Google Scholar]
- 44.Santamarina-Fojo S., Peterson K., Knapper C., Qiu Y., Freeman L., Cheng J. F., Osorio J., Remaley A., Yang X. P., Haudenschild C., et al. 2000. Complete genomic sequence of the human ABCA1 gene: analysis of the human and mouse ATP-binding cassette A promoter. Proc. Natl. Acad. Sci. USA. 97: 7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y., Chen X., Yang H., Zhou L., Okoro E. U., Guo Z. 2011. A novel function of apolipoprotein E: upregulation of ATP-binding cassette transporter A1 expression. PLoS ONE. 6: e21453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X., Zhao Y., Guo Z., Zhou L., Okoro E. U., Yang H. 2011. Transcriptional regulation of ATP-binding cassette transporter A1 expression by a novel signaling pathway. J. Biol. Chem. 286: 8917–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sevov M., Elfineh L., Cavelier L. B. 2006. Resveratrol regulates the expression of LXR-alpha in human macrophages. Biochem. Biophys. Res. Commun. 348: 1047–1054. [DOI] [PubMed] [Google Scholar]
- 48.D'Addario M., Arora P. D., McCulloch C. A. 2006. Role of p38 in stress activation of Sp1. Gene. 379: 51–61. [DOI] [PubMed] [Google Scholar]
- 49.Trasino S. E., Kim Y. S., Wang T. T. 2009. Ligand, receptor, and cell type-dependent regulation of ABCA1 and ABCG1 mRNA in prostate cancer epithelial cells. Mol. Cancer Ther. 8: 1934–1945. [DOI] [PubMed] [Google Scholar]