

Abstract
The CYP46A1 gene codes for the cholesterol 24-hydroxylase, a cytochrome P450 specifically expressed in neurons and responsible for the majority of cholesterol turnover in the central nervous system. Previously, we have demonstrated the critical participation of Sp transcription factors in the CYP46A1 response to histone deacetylase (HDAC) inhibitors, and in this study we investigated the involvement of intracellular signaling pathways in the trichostatin A (TSA) effect. Our results show that pretreatment of neuroblastoma cells with chemical inhibitors of mitogen-activated kinase kinase (MEK)1 significantly potentiates the TSA-dependent induction of cholesterol 24-hydroxylase, whereas inhibition of protein phosphatases by okadaic acid (OA) or overexpression of MEK1 partially impairs the TSA effect without affecting histone hyperacetylation at the promoter. Immunoblotting revealed that TSA treatment decreases ERK1/2 phosphorylation concomitantly with a decrease in Sp3 binding activity, which are both reversed by pretreatment with OA. Chromatin immunoprecipitation analysis demonstrated that TSA induces the release of p-ERK1/2 from the CYP46A1 proximal promoter, whereas pretreatment with OA restores the co-occupancy of Sp3-ERK1/2 in the same promoter fragments. We demonstrate for the first time the participation of MEK-ERK1/2 signaling pathway in HDAC inhibitor-dependent induction of cytochrome P450 gene expression, underlying the importance of this regulatory signaling mechanism in the control of brain cholesterol elimination.
Keywords: gene regulation, cytochrome P450, brain cholesterol metabolism, SP transcription factors, HDAC inhibitors, MAPK signaling pathway and protein phosphatase inhibition
The brain-specific cholesterol 24-hydroxylase (CYP46A1) is a member of the cytochrome P450 superfamily of enzymes that catalyzes the hydroxylation of cholesterol into 24(S)-hydroxycholesterol (24OHC) (1). The flux of this oxysterol across the blood-brain barrier represents the major mechanism of cholesterol elimination from the brain having, therefore, a crucial role in reverse cholesterol transport and in the maintenance of brain cholesterol homeostasis (2). Cyp46a1 −/− knockout mice exhibit severe deficiencies in spatial, associative and motor learning, and hippocampal long-term potentiation (3), which are related to a 40% reduction in brain cholesterol synthesis (4). In fact, the decreased availability of geranylgeraniol, a nonsterol isoprenoid generated by the mevalonate pathway, but not cholesterol, was found to be essential for long-term potentiation, suggesting a major role of neuronal cholesterol turnover, and directly of CYP46A1, in processes such as learning and memory (4, 5). Moreover, an increasing number of studies suggest that CYP46A1 affects the pathophysiology of Alzheimer's disease (AD) (6–8). A study from Hudry and coworkers (9) highlighted the possibility that CYP46A1 overexpression can be a therapeutic approach in AD because it significantly reduced the amyloid β pathology in mouse models of the disease before and after the onset of the amyloid plaques.
Epigenetic modifiers are the only compounds known to induce CYP46A1 expression (10, 11). Characterization of the molecular mechanisms involved in the trichostatin A (TSA)-mediated derepression of CYP46A1 gene revealed that HDAC inhibition specifically induced histone hyperacetylation of CYP46A1 promoter, concomitantly with an increase in the recruitment of RNA polymerase II (11). Interestingly, the proximal promoter region, encompassing four Specificity protein-responsive elements (Sp-RE) that we have shown to be indispensable for basal CYP46A1 promoter activity (12, 13), is also essential for the TSA-mediated activation. Despite the requirement of Sp proteins binding to this proximal promoter region for the activation by HDAC inhibitors (HDACi), we have verified that a decrease in Sp3 binding at specific responsive elements is important for the shift in HDAC/histone acetyltransferase (HAT) equilibrium that leads to dynamic changes in chromatin structure (11). Moreover, pretreatment of neuroblastoma cells with the demethylating agent 5-aza-2-deoxicytidine before TSA treatment significantly potentiates the TSA-mediated CYP46A1 activation in a DNA methylation independent mechanism, inducing a decrease in Sp3/HDAC binding to the promoter of this neuronal specific gene (14). Nevertheless, the fact that histone deacetylation was evident 6 h after TSA treatment, at a time point when the HDAC/HAT ratio should still favor acetylation, led us to investigate if mechanisms besides histone hyperacetylation could participate in the TSA-mediated derepression of the CYP46A1 gene. Because Sp1/Sp3 members of the Sp-family of transcription factors are ubiquitously expressed, post-translational modifications assume a key role in the regulation of their transcriptional activity (15) and might explain the stimulatory changes induced by the HDACi in CYP46A1 transcription, as already described for other genes (16–19). In addition, Sp proteins have been described to recruit histone-modifying enzymes and chromatin remodeling complexes to specific gene promoters. Sp1 and Sp3 can recruit Sin3A HDAC1/HDAC2 complex (20) or the coactivators CPB/p300 (21) and act, respectively, as repressors or activators of transcription.
In the present study, we aimed to identify the putative participation of specific signaling pathway(s) in the TSA-mediated activation of the CYP46A1 gene transcription and further elucidate the molecular mechanisms governing the expression of this brain-specific gene and involved in the control of brain cholesterol homeostasis. We clearly demonstrate the participation of the mitogen-activated kinase kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway in the CYP46A1 derepression by TSA treatment. Modulation of Sp3 binding activity, in a ERK1/2-dependent manner, was identified as a crucial step for the TSA effect independently of histone hyperacetylation, underlying the importance of this regulatory signaling mechanism in the control of brain cholesterol elimination.
MATERIALS AND METHODS
Reagents and antibodies
All chemical inhibitors (TSA, okadaic acid [OA], H89, U0126, SP600129, PD98059, and Gö6983) were from Sigma (Sigma Aldrich Inc., St Louis, MO). The antibodies used in this work were anti-p-ERK1/2 (Santa Cruz Biotechnology Inc., Santa Cruz, CA); -ERK1/2, -p-JNK, and -JNK (Cell Signaling Technology, Danvers, MA) for Western blot; and anti-Sp3 (Santa Cruz Biotechnology Inc.), -acetyl-histone H4, and –RNA polymerase II (Millipore, Bedford, MA) for chromatin immunoprecipitation (ChIP).
Cell culture, reporter gene constructs, and transactivation assays
The SH-SY5Y human neuroblastoma cell line was maintained and transiently transfected as previously described (12). The different recombinant wild-type and mutated plasmids derived from the 5′ flanking region of the human CYP46A1 gene and used in this work have also been described previously (12). NTERA-2cl.D1 (NT2) testicular embryonal carcinoma cells were cultured and differentiated as described (13, 22).
CYP46A1 expression analysis
Total cell RNA was extracted using Trizol Reagent (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Real-time quantitative qPCR analysis for CYP46A1 mRNA levels was performed as previously described (11) in an ABI 7300 sequence detection system (Applied Biosystems, Foster City, CA). Results presented are from at least three individual experiments, and each sample was assayed in triplicate. The mRNA levels were normalized to the level of β-actin and are presented as fold change from controls using the ΔΔCt method. Statistical analysis was performed using ΔCt values.
Western blot analysis
Cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% Triton-X 100) containing 1 mM DTT) in the presence of phosphatase inhibtors (10 mM NaF and 1 mM Na3VO4) and a protease inhibitor mixture (Roche Diagnostics GmbH, Penzberg, Germany). After incubation at 4°C for 30 min, samples were sonicated four times for 4 s each on ice, followed by centrifugation at 12,000 g for 15 min. Proteins were subject to SDS-PAGE gels, electroblotted onto Immobilon P (Millipore), and incubated with specific antibodies.
Electrophoretic mobility shift assay
EMSA was performed as previously prescribed (12). Briefly, 2–5 µg of nuclear extracts, prepared as described by Schreiber and coworkers (23), were incubated with γ32P-labeled oligonucleotide probe harboring sites SP-RE-B or SP-RE-D. Protein-DNA complexes were resolved on 5% native polyacrylamide gels and visualized by autoradiography.
ChIP and reChIP
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (11). For the sequential ChIP (reChIP) experiments, after the first immunoprecipitaton, the DNA/protein complexes were recovered by incubation with 100 mM DTT for 30 min at 37°C and diluted in immunoprecipitation dilution buffer (0.01% SDS, 0.5% Triton ×100, 2 mM EDTA, 16.7 mM Tris-HCl [pH 8.1], 100 mM NaCl). After dilution, a second immunoprecipitation was performed. The recovered DNA was analyzed by real-time PCR in an ABI7300 sequence detection system (Applied Biosystems) with SYBR green Master Mix (Fermentas; Thermo Fisher Scientific, Waltham, MA). Primers covering different regions of the CYP46A1 promoter were used: 5′-GCGGACCTGAGTCTGAAGAG-3′ (forward) and 5′-AATCACAACTCCGCTTCTGG-3′(reverse) for the proximal promoter (+1 region) and 5′-GGGAAGCCCTGGTCATTATT-3′ (forward) and 5′-GTTGGAGTTGGAGGGATGAA-3′ (reverse) for the distal promoter (1 kb region).
Statistical analysis
Statistical analysis was performed using the Student's t-test and the ANOVA one-way test with the Tukey HSD post hoc test or the Tukey HSD for unequal N (Spjotvoll/Stoline test). The analysis was performed using STATISTICA (data analysis software system), version 9.1 StatSoft, Inc.
RESULTS
Inhibition of MEK1 potentiates the TSA-mediated derepression of CYP46A1 gene
To investigate whether specific signaling pathway(s) were involved in the activation of CYP46A1 gene by the histone deacetylase inhibitor TSA, SH-SY5Y cells were pretreated with specific kinase/phosphatase inhibitors for 1 h followed by 16 h of treatment with 250 nM TSA (Fig. 1). To evaluate the CYP46A1 promoter activity, the 0.12pGL2 promoter reporter construct was used because we have previously described this region as being critical for the TSA response (11). The TSA-mediated increase in CYP46A1 promoter activity was significantly potentiated in the presence of PD98059, a MEK1 inhibitor, whereas the inhibition of protein kinase A (H89), protein kinase C (Gö6983), or c-Jun N-terminal kinase (SP600125) had no effect on TSA induction (ANOVA one-way test: F = 14.57, df = 3, P < 0.001; Tukey HSD for unequal N: P < 0.05) (Fig. 1A). Conversely, the inhibition of protein phosphatases (PPs) by OA partially impaired the TSA effect in a dose-dependent manner (ANOVA one-way test: F = 8.421, df = 2, P < 0.05; Tukey HSD for unequal N: P < 0.05) (Fig. 1A). In accordance, real-time PCR analysis revealed that inhibition of MEK1 by pretreatment with PD98059 or U0126, another MEK1 inhibitor, increases the CYP46A1 mRNA levels, in comparison with levels attained in cells treated with TSA alone, whereas the inhibition of PPs by OA has the opposite effect, resulting in a decrease to approximately 30% of the TSA effect (ANOVA one-way test: F = 15.2, df = 3, P < 0.001; Tukey HSD for unequal N: P < 0.001) (Fig. 1B). To assess the specificity of the transcriptional response, we examined the effect of OA treatment on the reelin gene expression. Our results showed that OA did not induce any significant changes in the reelin mRNA level of cells treated with OA or TSA plus OA (data not shown).
Fig. 1.

Effect of kinase/phosphatase chemical inhibitors on TSA mediated induction of CYP46A1 expression. A: The 0.12pGL2 plasmid, encompassing the region −236 to −64 of CYP46A1 promoter, was transfected into SH-SY5Y cells. Twenty-four hours after transfection, cells were preincubated with 5 μM H89 (PKA inhibitor), 2 μM Go6983 (PKC inhibitor), 10 μM PD98059 (MEK1 inhibitor), 10 μM SP600125 (JNK inhibitor), or with the indicated concentrations of OA for 1 h with or without 250 nM TSA for 16 h. Normalized luciferase activities were expressed as mean values ± SEM of duplicates for a minimum of three independent experiments. B: Real-time PCR analysis of CYP46A1 mRNA transcript levels in SH-SY5Y cells pretreated 1 h with 10 μM of the MEK inhibitors U0126 and PD98059 or with the indicated concentrations of OA with or without 250 nM TSA for 16 h. Values were normalized to the internal standard β-actin. Data represent mean values ± SEM of at least three independent experiments and are expressed as percentage of induction relative to TSA-treated cells (*P < 0.05; ***P < 0.001).
These results clearly demonstrate the participation of phosphorylation/dephosphorylation events in the HDACi-mediated derepression of CYP46A1 gene, particularly the involvement of the MEK signaling pathway.
ERK1/2 signaling pathway mediates the increase in CYP46A1 expression by TSA
To confirm the participation of the MEK-ERK signaling pathway in the TSA-mediated derepression of CYP46A1 gene, overexpression of the wild-type and mutated forms of ERK1 and ERK2 was performed in SH-SY5Y cells (Fig. 2A). The transactivation studies revealed that the TSA induction of the 0.12pGL2 promoter construct activity was significantly diminished in the presence of the wild-type forms of ERK1 and ERK2, whereas the mutated proteins did not have any effect on the promoter activity (ANOVA one-way test: F = 11.69, df = 4, P < 0.01; Tukey HSD: ERK1 wt P < 0.01, ERK2 wt P < 0.01) (Fig. 2A). Because we detected a similar effect of the overexpression of ERK1 and ERK2 in the CYP46A1 induction by TSA, we performed the reporter activity analysis after transfection of an expression plasmid coding for a constitutively active form of MEK1 (MEK1*), which is the upstream kinase of ERK1/2. Western blot analysis confirmed the significant increase in ERK1/2 phosphorylation after MEK1* transfection (Fig. 2B). As expected, overexpression of MEK1 resulted in a significant decrease in the TSA induction of the 0.12pGL2 promoter construct activity (ANOVA one-way test: F = 22.3, df = 3, P < 0.001; Tukey HSD: 0.05 μg and 0.25 μg MEK* P <0 .01; 0.15 μg MEK* P < 0.001) (Fig. 2C). Transfection of MEK1 expression plasmid also inhibited the TSA effect on the CYP46A1 mRNA level. Indeed, by real-time PCR we observed a decrease of 30% in the TSA-mediated increase of CYP46A1 mRNA levels (Fig. 2D).
Fig. 2.

Overexpression of the MEK-ERK kinases partially impairs TSA-mediated increase in CYP46A1 expression. A: The 0.12pGL2 plasmid was transfected with ERK1 and ERK2 wild-type and mutated forms into SH-SY5Y cells. Twenty-four hours after transfection, cells were treated with or without 250 nM TSA for 16 h. Normalized luciferase activities were expressed as mean values ± SEM of duplicates for a minimum of three independent experiments. B: Western blot analysis of p-ERK1/2 and ERK1/2 protein level in SH-SY5Y cells after overexpression of a constitutively active MEK expression plasmid (MEK*). C: The 0.12pGL2 plasmid was transfected with different amounts of MEK* plasmid into SH-SY5Y cells. Twenty-four after transfection, cells were treated with or without 250 nM TSA for 16 h. Normalized luciferase activities were expressed as mean values ± SEM of duplicates for a minimum of three independent experiments. D: Real-time PCR analysis of CYP46A1 mRNA transcript levels in SH-SY5Y cells overexpressing the MEK* plasmid and treated with or without 250 nM TSA for 16 h. Values were normalized to the internal standard β-actin and are expressed as percentage of induction relative to TSA-treated cells. Data represent mean values ± SEM of at least three independent experiments (*P < 0.05; **P < 0.01; ***P < 0.001).
TSA treatment promotes ERK1/2 dephosphorylation that is prevented in the presence of okadaic acid
Because the MEK-ERK signaling pathway clearly participates in the TSA-mediated derepression of CYP46A1 gene, we investigated whether TSA treatment could affect ERK1/2 activity. Western blot analysis of total protein extracts from SH-SY5Y cells treated with 250 nM TSA for different time points revealed that TSA decreases ERK1/2 phosphorylation as soon as 30 min after treatment, with the control levels being restored 2 h after the insult (Fig. 3A). Moreover, the TSA effect on ERK1/2 activity was found to be dose dependent because treatment with increasing concentrations of TSA for 30 min induced a gradual dephosphorylation of these kinases. Western blot analysis revealed that phosphorylation levels of JNK, another member of the MAPK family, remained unchanged after TSA treatment, indicating that TSA specifically affects the MEK-ERK pathway (Fig. 3A).
Fig. 3.

Okadaic acid prevents ERK1/2 dephosphorylation induced by TSA treatment. Western blot analysis of p-ERK1/2, ERK1/2, p-JNK, and JNK protein level in SH-SY5Y cells after treatment with 250 nM TSA for the indicated time points or with different concentrations of TSA for 30 min (A) and with 10 nM OA for 1 h, with or without 250 nM TSA, for 16 h (B). The results shown are representative of those obtained in three independent experiments.
Pretreatment of SH-SY5Y cells with OA reversed the decrease in ERK1/2 phosphorylation (Fig. 3B). In fact, inhibition of PP1 and PP2A has been described to positively modulate ERK1/2 activity (24, 25). Nevertheless we did not identify the specific protein phosphatase inhibited by OA and involved in CYP46A1 induction by TSA. On one hand, the TSA effect was not affected by pretreatment with tautomycin at the concentrations that specifically inhibit PP1; on the other hand, silencing of PP2A catalytic subunit by siRNA transfection had no effect on CYP46A1 activation (data not shown). Taken together, these results suggest that early modulation of ERK1/2 activation by TSA can be critical for the end point effect on CYP46A1 transcription. Because protein phosphatase inhibition by OA prevents ERK1/2 dephosphorylation, an okadaic acid-sensitive protein phosphate that is different from PP1 and PP2A seems essential for the modulation of ERK1/2 activity by TSA.
The impairment of TSA induction of CYP46A1 expression by protein phosphatase inhibition is independent of histone hyperacetylation
Due to the drastic effect of OA pretreatment on the TSA induction of CYP46A1 mRNA levels, we evaluated if the histone hyperacetylation of the promoter induced by TSA (11) was being affected by OA administration. ChIP was performed with an antibody against tetra-acetylated histone H4 (K5, K8, K12, and K16) and chromatin isolated from SH-SY5Y cells treated with 250 nM TSA for 30 min (Fig. 4A). As expected, an increase in H4 acetylation level was observed in the −1 kb region of the CYP46A1 promoter after TSA exposure (11); however, pretreatment with OA did not induce any significant change in the level of this histone marker (ANOVA one-way test: F = 55.515, df = 7, P < 0.001; Tukey HSD: P < 0.001). Concomitantly, no differences in the recruitment of RNA pol II to the proximal promoter were observed between cells treated with 250 nM TSA for 30 min in the presence or absence of OA, suggesting that at least the initial transcription activation is not being affected (ANOVA one-way test: F = 31.184, df = 7, P < 0.001; Tukey HSD: TSA P < 0.001, OA and TSA, P < 0.05) (Fig. 4B). Indeed, a time-course analysis of CYP46A1 mRNA accumulation after TSA treatment, with or without OA, by real-time PCR corroborated this result (Fig. 5). The repressive effect of OA on CYP46A1 activation by TSA was only evident from 6 h treatment onward. The OA repression of CYP46A1 activation by TSA is therefore independent of the early and transient histone hyperacetylation at the promoter region, demonstrating that later events other than chromatin remodeling are also crucial for the HDACi effect.
Fig. 4.

Histone hyperacetylation induced by TSA at CYP46A1 promoter is not affected by OA pretreatment. ChIP was performed using chromatin prepared from SH-SY5Y control cells and cells pretreated 1 h with 10 nM OA and treated for 30 min with or without 250 nM TSA. After chromatin precipitation with an anti-AcH4 (A) and –RNA pol II (B) antibodies, the recovered DNA was evaluated by real-time PCR. Results represent means ± SEM of at least three independent experiments and are expressed as percentage of total input (*P < 0.05; **P < 0.01; ***P < 0.001).
Fig. 5.
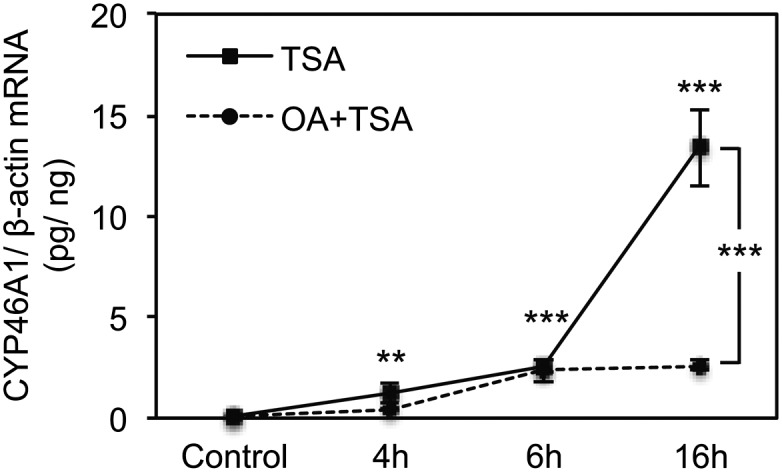
Okadaic acid inhibits the TSA-mediated activation of CYP46A1 gene in a time-dependent manner. Real-time PCR analysis of CYP46A1 mRNA transcript levels in SH-SY5Y cells treated with 250 nM TSA for different time points pretreated or not 1 h with 10 nM OA. Values were normalized to the internal standard β-actin and are expressed as pg of CYP46A1 mRNA per ng of β-actin mRNA. Data represent means ± SEM of at least three independent experiments (**P < 0.01; ***P < 0.001).
TSA treatment triggers the release of p-ERK1/2 from CYP46A1 promoter in a time-dependent manner
The presence of MAPKs on gene promoters, as part of the transcriptional complexes, has already been suggested, although ERK1/2, in contrast to ERK5, which contains a transcriptional activation domain (26), is unable to directly bind DNA. To investigate whether the effect of TSA on ERK1/2 can affect the binding and activity of transcription factors to the CYP46A1 promoter, we considered the idea that ERK1/2 dephosphorylation may induce its dissociation from chromatin-bound transcription factor complexes. To test this idea, we evaluated the recruitment of this kinase to the CYP46A1 proximal promoter by ChIP (Fig. 6). The analysis by real-time PCR of the DNA fragments recovered after immunoprecipitation with an antibody against the phosphorylated form of ERK1/2 (p-ERK1/2) revealed the presence of this kinase in the proximal promoter of CYP46A1 in untreated cells. Concomitantly with the decrease in the total levels of p-ERK1/2, 1 h after TSA treatment we could no longer detect an association of ERK1/2 to the promoter. Interestingly, ERK1/2 remained dissociated from the CYP46A1 promoter even after 6 h of TSA treatment. In contrast to what we previously observed for total protein levels, the pretreatment with OA does not impair detachment of p-ERK1/2 from the promoter in the first 6 h of treatment (data not shown).
Fig. 6.

TSA treatment promotes p-ERK1/2 detachment from the CYP46A1 promoter. ChIP was performed using chromatin prepared from SH-SY5Y control cells and cells treated with 250 nM TSA for the indicated time points. After chromatin precipitation with an anti-p-ERK1/2 antibody, the recovered DNA was evaluated by real-time PCR. Results represent means ± SEM of at least three independent experiments and are expressed as a percentage of total input (*P < 0.05).
Overall, these results suggest that the transient reduction in ERK1/2 activity 30 min after TSA treatment induces a release of p-ERK1/2 from CYP46A1 promoter that lasts at least for 6 h and is most likely required for the increase of CYP46A1 mRNA. Although OA prevents TSA-dependent ERK1/2 dephosphorylation very rapidly, as previously assessed by immunodetection in whole cell lysates (Fig. 3), the impairment of TSA-mediated activation of CYP46A1 transcription is probably due to an accumulation of p-ERK1/2 at the promoter at later time points that gradually counteracts the TSA effect.
TSA affects Sp3 activity at the CYP46A1 promoter level in a MEK-ERK1/2-dependent manner
In our previous work regarding the effect of HDAC inhibition on CYP46A1 transcription (11), we described that Sp3 binding activity to specific Sp-responsive elements of the CYP46A1 promoter was decreased in nuclear extracts from SH-SY5Y treated with TSA from 4 h onward, attaining control levels 24 h after TSA treatment. Due to the striking effect of OA on TSA activation of CYP46A1 expression 16 h after HDAC inhibition (Fig. 5), we decided to evaluate if pretreatment with OA was affecting Sp3 binding activity at this time point. We performed an EMSA with a probe encompassing one of the Sp-binding sites on the CYP46A1 proximal promoter, the Sp-RE-B probe, and nuclear extracts from SH-SY5Y cells treated with 250 nM TSA for 16 h in the presence or absence of OA (Fig. 7A). At this time point in the TSA treatment, a decrease in Sp3 binding activity was still evident, and the inhibition of PPs with OA completely reversed the TSA effect, suggesting that Sp3 binding activity is regulated by transient phosphorylation and dephosphorylation. To evaluate if OA was also affecting the binding and recruitment of Sp3 to the CYP46A1 proximal promoter, we performed ChIP assays (Fig. 7B). No significant changes in Sp3 binding were observed after 16 h treatment with TSA, OA, or both. We had previously observed a discrepancy between the results obtained with gel-shift regarding differences in Sp3 binding activity and the recruitment of this transcription factor to the promoter determined by ChIP (11). These differences are most likely due to the distance between the several Sp-RE sites in the GC-rich proximal promoter (10 putative Sp-binding sites in the region encompassed by nucleotides −417 to −64), which is smaller than average chromatin fragments produced during sonication (500–900 bp) and therefore makes it impossible to discriminate between binding of Sp proteins to the different binding sites. Because the MEK-ERK pathway mediates the TSA activation of CYP46A1 gene and PPs inhibition affects Sp3 binding activity, we hypothesized that ERK1/2 was modulating Sp3 activity. To assess if ERK1/2 affects Sp3 phosphorylation at the chromatin level, we performed reChIP analysis and evaluated the association of Sp3 and p-ERK1/2 to the same chromatin fragments of the CYP46A1 promoter (Fig. 7C). In control cells, we observed that Sp3 and p-ERK1/2 share the same promoter fragments, whereas at 16 h after TSA treatment we could no longer detect the association of these two proteins to the same regions. Pretreatment of SH-SY5Y cells with OA completely restored the cooccupancy of Sp3-p-ERK1/2 in the CYP46A1 promoter fragments.
Fig. 7.

Sp3 binding activity is affected by okadaic acid treatment. EMSA (A), ChIP (B), and reChIP (C) were performed using nuclear extracts and chromatin, respectively, prepared from SH-SY5Y cells pretreated 1 h with 10 nM OA or treated for 16 h with or without 250 nM TSA. For the EMSA, a double-stranded oligonucleotide corresponding to the Sp-RE-B site was used as a probe. In the ChIP and reChIP assay, after chromatin precipitation with an anti-Sp3 and -p-ERK1/2 antibodies, the recovered DNA was evaluated by real-time PCR. Results represent means ± SEM of at least three independent experiments and are expressed as percentage of total input (*P < 0.05; **P < 0.01; ***P < 0.001).
Overall, these results demonstrate that TSA affects Sp3 activity in a MEK-ERK dependent manner, which is an essential event for the HDACi-mediated derepression of CYP46A1 gene. In the presence of OA, an accumulation of p-ERK1/2 in the Sp3-containg fragments of CYP46A1 promoter occurs, leading to a phosphorylation-dependent modulation of Sp3 activity and consequently to a decrease in the TSA induction of CYP46A1 expression.
Okadaic acid inhibits CYP46A1 basal expression in NT2 human post-mitotic neurons
Because changes in the activity of protein phosphatases (27, 28) and kinases (29–31) and in the levels of 24(S)-hydroxycholesterol (32–34) have been described in patients with AD, we were interested in investigating whether these pathways could act directly as modulators of CYP46A1 transcription. We started by evaluating CYP46A1 promoter activity after treatment with kinase and phosphatase inhibitors (Fig. 8A). The 0.12pGL2 promoter reporter construct was transfected in SH-SY5Y cells and 24 h after cells were treated for 16 h with the kinase inhibitors H89, PD98059, SP600125 or with the protein phosphatase inhibitor OA. As previously observed for the TSA effect, specific inhibition of the MEK-ERK pathway significantly increased CYP46A1 promoter activity (ANOVA one-way test: F = 4.255, df = 3, P < 0.05; Tukey HSD: P < 0.05), whereas OA treatment had the opposite effect, resulting in a decrease in the activity to approximately 50% of the control levels (ANOVA one-way test: F = 6.647, df = 3, P < 0.01; Tukey HSD: P < 0.01). Accordingly, real-time PCR analysis revealed that MEK-ERK inhibition, by treatment with U0126 and PD98059 for 16 h, induces a 2.5-fold increase in SH-SY5Y CYP46A1 mRNA level (ANOVA one-way test: F = 10.403, df = 2, P < 0.001; Tukey HSD: U0126 P < 0.01, PD98059 P< 0.05) (Fig. 8B). Because SH-SY5Y cells express very low levels of CYP46A1 mRNA, making it impossible to determine differences in gene expression that result in transcription inhibition, we treated human NT2 post-mitotic neurons (NT2N) with different concentrations of OA for 16 h to evaluate if protein phosphatase inhibition also affects the basal transcription of this gene (Fig. 8B). Indeed, we have previously shown that NT2N cells are a suitable model to study CYP46A1 transcription and neuronal cholesterol metabolism because these cells express high levels of CYP46A1 mRNA and protein and increased production of 24OHC (13, 22). Real-time PCR analysis revealed that OA (10 nM) is able to inhibit the basal expression of CYP46A1 gene, resulting in a decrease of approximately 50% in the mRNA transcripts level (ANOVA one-way test: F = 18.974, df = 2, P < 0.001; Tukey HSD: P < 0.001). These results indicate that both serine-threonine protein phosphatases and the MEK-ERK signaling pathway are involved in the regulation of CYP46A1 basal transcription, apart from their pivotal role in the derepression by histone deacetylase inhibition.
Fig. 8.

Effect of kinase/phosphatase chemical inhibitors on CYP46A1 basal expression. A: The 0.12pGL2 plasmid was transfected into SH-SY5Y cells. Twenty-four hours after transfection, cells were incubated with 5 μM H89, 10 μM PD98059, 10 μM SP600125, or with the indicated concentrations of OA for 16 h. Normalized luciferase activities were expressed as mean values ± SEM of duplicates for a minimum of three independent experiments. B: Real-time PCR analysis of CYP46A1 steady-state mRNA transcripts level in SH-SY5Y and NT2N cells treated, respectively, with 10 μM U0126 or 10 μM PD98059 and with the indicated doses of OA for 16 h. Values were normalized to the internal standard β-actin. Data represent means ± SEM of at least three independent experiments and are expressed as percentage of induction relative to vehicle-treated cells (NT2N) or as pg of CYP46A1 per ng of β-actin (SH-SY5Y) (*P < 0.05; **P < 0.01; ***P < 0.001).
DISCUSSION
Understanding CYP46A1 transcriptional regulation required a new perspective because of the beneficial effects arising from overexpressing this enzyme in a mouse model of AD (9). We and others have shown that HDACi are one of the few classes of compounds known to induce CYP46A1 expression (10, 11). In our previous study, we showed that the histone deacetylase inhibitor TSA induced a transient histone hyperacetylation of CYP46A1 promoter, which significantly increased CYP46A1 transcription in SH-SY5Y neuroblastoma cells (11). In the same work, a specific region in the proximal promoter that encompasses four Sp-binding sites was identified as essential for the TSA activation of CYP46A1 gene. Binding of Sp1 and Sp4 transcription factors was found to be indispensable for the TSA effect. On the other hand, a decrease in Sp3 binding activity was observed after TSA treatment.
The fact that histone deacetylation was observed at a time point in TSA treatment where the HDAC/HAT equilibrium should still be favoring acetylation led us to investigate which mechanism(s) other than histone hyperacetylation were involved in the TSA-mediated increase in CYP46A1 gene expression. The studies presented here clearly identify the MEK-ERK signaling pathway as critical for the modulation of Sp3 transcriptional activity and the activation of CYP46A1 by TSA. The SP family of transcription factors, including Sp3, is highly regulated at the post-translational level. Although Sp3 is able to up- (35, 36) and down-regulate gene transcription (37–39), phosphorylation is generally associated with increased DNA binding activity and, consequently, increased transcriptional activation (35, 40). Modifications such as acetylation have also been described to activate (21) and repress (41) Sp3 transcriptional activity, whereas sumoylated Sp3 significantly represses transcription (42–44).
The participation of the MEK-ERK signaling pathway in the derepression of CYP46A1 gene by TSA was confirmed by pretreatment of neuroblastoma cells with specific inhibitors of this pathway (PD98059 and U0126) and overexpression of the wild-type and catalytic inactive forms of ERK1 and ERK2 and a constitutively active form of MEK1. From these experiments, we confirmed that inhibition of the MEK-ERK pathway resulted in the potentiation of the TSA effect. In contrast, inhibition of protein phosphatase activity impaired the TSA-mediated activation of CYP46A1 gene, pointing to a dephosphorylation event as indispensable for the HDACi effect. TSA and other HDACi have been associated with increased kinase activity; namely, sodium butyrate induces erythroid differentiation of K562 cells by inhibition of ERK and activation of p38 signal transduction pathways (45) and stimulates PKC activation in the same cellular model (46). However, TSA was also shown to significantly decrease phosphorylation of Akt and ERK1/2 in a glioblastoma cell line (19). In our cellular model, the SH-SY5Y neuroblastoma cells, TSA induced an early marked but transient decrease in ERK1/2 phosphorylation as soon as 30 min after TSA treatment, resulting in the dissociation of these kinases from the CYP46A1 promoter for at least 6 h. Because this effect perfectly correlated with the decreased binding activity of Sp3 protein in the presence of TSA and due to the established importance of the SP family of transcription factors in the HDACi derepression of CYP46A1 gene, we hypothesized that TSA was modulating Sp3 transcriptional activity in a ERK1/2-dependent manner. In fact, Sp3 has been described as a target of the ERK1/2 MAPK family (35, 40).
OA pretreatment reversed the decrease in Sp3 binding activity induced by TSA, strengthening our hypothesis. Although PP1 has been demonstrated to dephosphorylate Sp3 (47), it seems that the protein phosphatase involved in the process is not directly modulating Sp3 phosphorylation but instead modulates ERK1/2 activity because treatment with OA efficiently reverted the ERK1/2 dephosphorylation observed after TSA treatment. The concentration of the protein phosphatase inhibitor used in this study (10 nM OA) should specifically inhibit PP2A and not PP1. However, silencing of PP2A catalytic subunit α (PP2Ac) had no effect on CYP46A1 activation by TSA, confirming that another OA-sensitive phosphatase is involved in the TSA effect. In fact, at this concentration, OA can inhibit other protein phosphatases, such as PP4 or PP5, which can only be discriminated by specific siRNA silencing. We have verified that most studies using OA implicate PP1 or PP2A in biological processes and neglect the possibility that some of the biological effects of OA are due to the inhibition of PP4 or PP5. Our results highlight the importance of the identification of the protein phosphatase involved in the control of ERK1/2 activity in neuronal cells.
The fact that alterations in the phosphorylation level of ERK1/2 correlated with CYP46A1 mRNA level and Sp3 binding activity point to a relationship between Sp3 phosphorylation/dephosphorylation and the TSA effect. Importantly, impairment of TSA-mediated activation of CYP46A1 gene by inhibition of protein phosphatases activity resulted in the blockage of ERK1/2 dephosphorylation without affecting histone hyperacetylation of the promoter or the initial recruitment of RNA polymerase II. The repressive effect of protein phosphatase inhibition by pretreatment with OA led to an increase in the association of Sp3 with activated ERK1/2 at the CYP46A1 proximal promoter, which most likely induces Sp3 phosphorylation and anchoring of co-repressors complexes to the promoter at later time points. The Sp3 role in this context is distinct from what we have previously observed after treatment of neuroblastoma cells with the demethylating agent 5-aza-2-deoxicytidine (14). In that work, a decrease in Sp3 protein level was observed after drug treatment concomitantly with a detachment of Sp3 and HDACs from CYP46A1 proximal promoter, demonstrating that this transcription factor modulates transcription by recruitment and detachment of chromatin remodeling agents. An interesting hypothesis is that Sp3 phosphorylation by ERK1/2 favors other post-translational modifications, such as acetylation or sumoylation, which can also be responsible for Sp3-mediated repression. Indeed, it has been reported that sumoylation and ubiquitination of Sp1 transcription factor can be mediated by phosphorylation (48), supporting a novel mechanism of Sp-dependent gene regulation. This is also a feature of the ERK1/2 MAPK family to control transcriptional regulation as players between phosphorylation-dependent post-translational modifications that can affect protein's transcriptional activity from ubiquitination to sumoylation and acetylation (49).
We have recently described the differentiation of NT2 cells into post mitotic neurons (NT2N) as a suitable cellular model to study CYP46A1 transcriptional regulation (13, 22). Treatment of NT2N cells with 10 nM OA for 16 h significantly reduced CYP46A1 mRNA level. This effect is particularly relevant in the context of AD. This neurological condition has been widely linked to compromised protein phosphatase activity, which is responsible for one of the hallmarks of the disease, Tau protein hyperphosphorylation. Indeed, our results suggest that the decreased PP2A and PP5 activity described in the brains of patients with AD (27, 50, 51) can also have an adverse consequence in brain cholesterol metabolism. Interestingly, changes in the flux of 24OHC from the brain into the circulation have been reported in patients with AD, with reduced circulating levels of this oxysterol in the more advanced cases of neurodenegeration (52). Concomitantly, inhibition of the MEK-ERK signaling pathway increased CYP46A1 basal expression in neuroblastoma cells, suggesting that specific modulation of this intracellular pathway can also represent a pharmacological target to ameliorate amyloid β pathology in AD.
Taken together, these results demonstrate the critical participation of the MEK-ERK transduction pathway in the HDACi-mediated activation of CYP46A1 gene in a mechanism that is independent of histone hyperacetylation of the promoter. TSA treatment induces the dissociation of p-ERK1/2 from the promoter in a PP1/PP2A independent manner and specifically from the Sp3-containing DNA fragments, which leads to a decrease in Sp3 repressor transcriptional activity and probably the recruitment of other co-repressor proteins to the CYP46A1 promoter.
Acknowledgments
The authors thank Dr. Melanie Cobb (University of Texas, Southwestern Medical Center, Dallas, TX) for providing the expression vectors for the wild type and catalytic inactive forms of ERK1 and 2 (pCEP4-ERK1-wt and K71R; 3xflag-pCMV7-ERK2-wt and KR) and Dr. Roger J. Davis (Howard Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MA) for the plasmid encoding a constitutively active form of MEK1 (pCMV-deltaN3-MEK1*).
Footnotes
Abbreviations:
- AD
- Alzheimer's disease
- ChIP
- chromatin immunoprecipitation
- CYP46A1
- cholesterol 24-hydroxylase
- ERK
- extracellular signal-regulated kinase
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- HDACi
- histone deacetylase inhibitors
- MEK
- mitogen-activated kinase kinase
- NT2
- NTERA-2cl.D1
- NT2N
- NT2 post-mitotic neurons
- OA
- okadaic acid
- 24OHC
- 24(S)-hydroxycholesterol
- p-ERK1/2
- phosphorylated form of ERK1/2
- PP
- protein phosphatase
- Sp-RE
- Specificity protein-responsive elements
- TSA
- trichostatin A
This work was supported by Fundação para a Ciência e Tecnologia Projects PTDC/SAU-GMG/64176/2006 and PEst-OE/SAU/UI4013/2011 and by PhD grants SFRH/BD/41848/2007 (M.J.N.) and SFRH/BD/27660/2006 (I.M.).
REFERENCES
- 1.Lund E. G., Guileyardo J. M., Russell D. W. 1999. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. USA. 96: 7238–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjorkhem I., Lutjohann D., Diczfalusy U., Stahle L., Ahlborg G., Wahren J. 1998. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39: 1594–1600. [PubMed] [Google Scholar]
- 3.Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., Russell D. W. 2003. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278: 22980–22988. [DOI] [PubMed] [Google Scholar]
- 4.Kotti T. J., Ramirez D. M., Pfeiffer B. E., Huber K. M., Russell D. W. 2006. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. USA. 103: 3869–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kotti T., Head D. D., McKenna C. E., Russell D. W. 2008. Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc. Natl. Acad. Sci. USA. 105: 11394–11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown J., 3rd, Theisler C., Silberman S., Magnuson D., Gottardi-Littell N., Lee J. M., Yager D., Crowley J., Sambamurti K., Rahman M. M., et al. 2004. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J. Biol. Chem. 279: 34674–34681. [DOI] [PubMed] [Google Scholar]
- 7.Famer D., Meaney S., Mousavi M., Nordberg A., Bjorkhem I., Crisby M. 2007. Regulation of alpha- and beta-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha-secretase pathway. Biochem. Biophys. Res. Commun. 359: 46–50. [DOI] [PubMed] [Google Scholar]
- 8.Prasanthi J. R., Huls A., Thomasson S., Thompson A., Schommer E., Ghribi O. 2009. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on beta-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol. Neurodegener. 4: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudry E., Van Dam D., Kulik W., De Deyn P. P., Stet F. S., Ahouansou O., Benraiss A., Delacourte A., Bougneres P., Aubourg P., et al. 2010. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol. Ther. 18: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shafaati M., O'Driscoll R., Bjorkhem I., Meaney S. 2009. Transcriptional regulation of cholesterol 24-hydroxylase by histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 378: 689–694. [DOI] [PubMed] [Google Scholar]
- 11.Nunes M. J., Milagre I., Schnekenburger M., Gama M. J., Diederich M., Rodrigues E. 2010. Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of CYP46A1 gene transcription. J. Neurochem. 113: 418–431. [DOI] [PubMed] [Google Scholar]
- 12.Milagre I., Nunes M. J., Gama M. J., Silva R. F., Pascussi J. M., Lechner M. C., Rodrigues E. 2008. Transcriptional regulation of the human CYP46A1 brain-specific expression by Sp transcription factors. J. Neurochem. 106: 835–849. [DOI] [PubMed] [Google Scholar]
- 13.Milagre I., Nunes M. J., Castro-Caldas M., Moutinho M., Gama M. J., Rodrigues E. 2012. Neuronal differentiation alters the ratio of Sp transcription factors recruited to the CYP46A1 promoter. J. Neurochem. 120: 220–229. [DOI] [PubMed] [Google Scholar]
- 14.Milagre I., Nunes M. J., Moutinho M., Rivera I., Fuso A., Scarpa S., Gama M. J., Rodrigues E. 2010. Chromatin-modifying agents increase transcription of CYP46A1, a key player in brain cholesterol elimination. J. Alzheimers Dis. 22: 1209–1221. [DOI] [PubMed] [Google Scholar]
- 15.Waby J. S., Bingle C. D., Corfe B. M. 2008. Post-translational control of sp-family transcription factors. Curr. Genomics. 9: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Espinos E., Le Van Thai A., Pomies C., Weber M. J. 1999. Cooperation between phosphorylation and acetylation processes in transcriptional control. Mol. Cell. Biol. 19: 3474–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camarero N., Nadal A., Barrero M. J., Haro D., Marrero P. F. 2003. Histone deacetylase inhibitors stimulate mitochondrial HMG-CoA synthase gene expression via a promoter proximal Sp1 site. Nucleic Acids Res. 31: 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y., Liao M., Dufau M. L. 2006. Phosphatidylinositol 3-kinase/protein kinase Czeta-induced phosphorylation of Sp1 and p107 repressor release have a critical role in histone deacetylase inhibitor-mediated derepression [corrected] of transcription of the luteinizing hormone receptor gene. Mol. Cell. Biol. 26: 6748–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C. S., Weng S. C., Tseng P. H., Lin H. P. 2005. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 280: 38879–38887. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y., Dufau M. L. 2003. Dual mechanisms of regulation of transcription of luteinizing hormone receptor gene by nuclear orphan receptors and histone deacetylase complexes. J. Steroid Biochem. Mol. Biol. 85: 401–414. [DOI] [PubMed] [Google Scholar]
- 21.Ammanamanchi S., Freeman J. W., Brattain M. G. 2003. Acetylated sp3 is a transcriptional activator. J. Biol. Chem. 278: 35775–35780. [DOI] [PubMed] [Google Scholar]
- 22.Milagre I., Olin M., Nunes M. J., Moutinho M., Lovgren-Sandblom A., Gama M. J., Bjorkhem I., Rodrigues E. 2012. Marked change in the balance between CYP27A1 and CYP46A1 mediated elimination of cholesterol during differentiation of human neuronal cells. Neurochem. Int. 60: 192–198. [DOI] [PubMed] [Google Scholar]
- 23.Schreiber E., Matthias P., Muller M. M., Schaffner W. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 17: 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao L., Yang L., Arora A., Choe E. S., Zhang G., Liu Z., Fibuch E. E., Wang J. Q. 2005. Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. J. Biol. Chem. 280: 12602–12610. [DOI] [PubMed] [Google Scholar]
- 25.Hedou G. F., Koshibu K., Farinelli M., Kilic E., Gee C. E., Kilic U., Baumgartel K., Hermann D. M., Mansuy I. M. 2008. Protein phosphatase 1-dependent bidirectional synaptic plasticity controls ischemic recovery in the adult brain. J. Neurosci. 28: 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasler H. G., Victoria J., Duramad O., Winoto A. 2000. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 20: 8382–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong C. X., Shaikh S., Wang J. Z., Zaidi T., Grundke-Iqbal I., Iqbal K. 1995. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J. Neurochem. 65: 732–738. [DOI] [PubMed] [Google Scholar]
- 28.Qian W., Yin X., Hu W., Shi J., Gu J., Grundke-Iqbal I., Iqbal K., Gong C. X., Liu F. 2100. Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer's disease. J. Alzheimers Dis. 23: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu X., Rottkamp C. A., Boux H., Takeda A., Perry G., Smith M. A. 2000. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 59: 880–888. [DOI] [PubMed] [Google Scholar]
- 30.Zhu X., Raina A. K., Rottkamp C. A., Aliev G., Perry G., Boux H., Smith M. A. 2001. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J. Neurochem. 76: 435–441. [DOI] [PubMed] [Google Scholar]
- 31.Pei J. J., Braak H., An W. L., Winblad B., Cowburn R. F., Iqbal K., Grundke-Iqbal I. 2002. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer's disease. Brain Res. Mol. Brain Res. 109: 45–55. [DOI] [PubMed] [Google Scholar]
- 32.Lutjohann D., Papassotiropoulos A., Bjorkhem I., Locatelli S., Bagli M., Oehring R. D., Schlegel U., Jessen F., Rao M. L., von Bergmann K., et al. 2000. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 41: 195–198. [PubMed] [Google Scholar]
- 33.Papassotiropoulos A., Lutjohann D., Bagli M., Locatelli S., Jessen F., Buschfort R., Ptok U., Bjorkhem I., von Bergmann K., Heun R. 2002. 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J. Psychiatr. Res. 36: 27–32. [DOI] [PubMed] [Google Scholar]
- 34.Zuliani G., Donnorso M. P., Bosi C., Passaro A., Dalla Nora E., Zurlo A., Bonetti F., Mozzi A. F., Cortese C. 2011. Plasma 24S-hydroxycholesterol levels in elderly subjects with late onset Alzheimer's disease or vascular dementia: a case-control study. BMC Neurol. 11: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pages G. 2007. Sp3-mediated VEGF regulation is dependent on phosphorylation by extra-cellular signals regulated kinases (Erk). J. Cell. Physiol. 213: 454–463. [DOI] [PubMed] [Google Scholar]
- 36.Bergeron F., Bagu E. T., Tremblay J. J. 2011. Transcription of platelet-derived growth factor receptor alpha in Leydig cells involves specificity protein 1 and 3. J. Mol. Endocrinol. 46: 125–138. [DOI] [PubMed] [Google Scholar]
- 37.Petrovic I., Kovacevic-Grujicic N., Stevanovic M. 2009. ZBP-89 and Sp3 down-regulate while NF-Y up-regulates SOX18 promoter activity in HeLa cells. Mol. Biol. Rep. 36: 993–1000. [DOI] [PubMed] [Google Scholar]
- 38.Tsika G., Ji J., Tsika R. 2004. Sp3 proteins negatively regulate beta myosin heavy chain gene expression during skeletal muscle inactivity. Mol. Cell. Biol. 24: 10777–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seyed M., Dimario J. X. 2008. Fibroblast growth factor receptor 1 gene expression is required for cardiomyocyte proliferation and is repressed by Sp3. J. Mol. Cell. Cardiol. 44: 510–519. [DOI] [PubMed] [Google Scholar]
- 40.Bakovic M., Waite K., Vance D. E. 2003. Oncogenic Ha-Ras transformation modulates the transcription of the CTP:phosphocholine cytidylyltransferase alpha gene via p42/44MAPK and transcription factor Sp3. J. Biol. Chem. 278: 14753–14761. [DOI] [PubMed] [Google Scholar]
- 41.Braun H., Koop R., Ertmer A., Nacht S., Suske G. 2001. Transcription factor Sp3 is regulated by acetylation. Nucleic Acids Res. 29: 4994–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sapetschnig A., Rischitor G., Braun H., Doll A., Schergaut M., Melchior F., Suske G. 2002. Transcription factor Sp3 is silenced through SUMO modification by PIAS1. EMBO J. 21: 5206–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ross S., Best J. L., Zon L. I., Gill G. 2002. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol. Cell. 10: 831–842. [DOI] [PubMed] [Google Scholar]
- 44.Stielow B., Sapetschnig A., Wink C., Kruger I., Suske G. 2008. SUMO-modified Sp3 represses transcription by provoking local heterochromatic gene silencing. EMBO Rep. 9: 899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Witt O., Sand K., Pekrun A. 2000. Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood. 95: 2391–2396. [PubMed] [Google Scholar]
- 46.Rivero J. A., Adunyah S. E. 1998. Sodium butyrate stimulates PKC activation and induces differential expression of certain PKC isoforms during erythroid differentiation. Biochem. Biophys. Res. Commun. 248: 664–668. [DOI] [PubMed] [Google Scholar]
- 47.Chu S., Cockrell C. A., Ferro T. J. 2003. Expression of alpha-ENaC2 is dependent on an upstream Sp1 binding motif and is modulated by protein phosphatase 1 in lung epithelial cells. Biochem. Biophys. Res. Commun. 303: 1159–1168. [DOI] [PubMed] [Google Scholar]
- 48.Spengler M. L., Guo L. W., Brattain M. G. 2008. Phosphorylation mediates Sp1 coupled activities of proteolytic processing, desumoylation and degradation. Cell Cycle. 7: 623–630. [DOI] [PubMed] [Google Scholar]
- 49.Whitmarsh A. J. 2007. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim. Biophys. Acta. 1773: 1285–1298. [DOI] [PubMed] [Google Scholar]
- 50.Liu F., Iqbal K., Grundke-Iqbal I., Rossie S., Gong C. X. 2005. Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer's disease. J. Biol. Chem. 280: 1790–1796. [DOI] [PubMed] [Google Scholar]
- 51.Liu R., Wang J. Z. 2009. Protein phosphatase 2A in Alzheimer's disease. Pathophysiology. 16: 273–277. [DOI] [PubMed] [Google Scholar]
- 52.Papassotiropoulos A., Lutjohann D., Bagli M., Locatelli S., Jessen F., Rao M. L., Maier W., Bjorkhem I., von Bergmann K., Heun R. 2000. Plasma 24S-hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer's disease. Neuroreport. 11: 1959–1962. [DOI] [PubMed] [Google Scholar]