

Abstract
We have used systematic evolution of ligands by exponential enrichment (SELEX) to isolate RNA aptamers against aminoglycoside antibiotics. The SELEX rounds were toggled against four pairs of aminoglycosides with the goal of isolating reagents that recognize conserved structural features. The resulting aptamers bind both of their selection targets with nanomolar affinities. They also bind the less structurally related targets, although they show clear specificity for this class of antibiotics. We show that this lack of aminoglycoside specificity is a common property of aptamers previously selected against single compounds and described as “specific”. Broad target specificity aptamers would be ideal for sensors detecting the entire class of aminoglycosides. We have used ligand-induced aggregation of gold-nanoparticles coated with our aptamers as a rapid and sensitive assay for these compounds. In contrast to DNA aptamers, unmodified RNA aptamers cannot be used as the recognition ligand in this assay, whereas 2′-fluoro-pyrimidine derivatives work reliably. We discuss the possible application of these reagents as sensors for drug residues and the challenges for understanding the structural basis of aminoglycoside-aptamer recognition highlighted by the SELEX results.
Aminoglycosides (AMGs) are RNA-binding antibiotics sharing a common core structure, the streptamine ring (Figure 1 and Table S1, Supporting Information). They function by binding to bacterial ribosomes, causing frame-shifting resulting in the production of nonsense peptides, eventually leading to cell death.1 Their clinical use has been restricted due to toxic side effects, especially to the kidneys and the ear.2 However, their low cost means that they are still often widely used in animal husbandry leading to potential residues in the food chain. Nucleic acid aptamers are reagents selected in vitro from degenerate sequence libraries by their ability to bind to defined molecular targets. Their affinities and specificities rival those of antibody reagents.3−5 They have a number of properties that make them superior to antibodies for a range of applications, such as primary capture ligands in sensors.6−8 These include the ease with which they can be synthesized and modified with a variety of functionalities to facilitate the production of sensing devices. There are a large number of known aptamer sequences with affinities for targets ranging from low molecular weight organic species to whole cells (Table A1, A). Most of these aptamers are based on RNA, and such molecules specific for a number of AMGs have been reported previously, together with structural studies investigating the molecular basis of target recognition.9−13 AMGs are being investigated as leads for novel RNA binding ligands although there is no simple understanding of their binding specificity.
Figure 1.

Aminoglycoside (AMG) antibiotics and biotinylation reagents. (A) 2-Deoxystreptamine core common to many AMG antibiotics. (B) Aminoglycoside antibiotics used for selection; apramycin, gentimicin (mixture of gentamicin C1 (R1=CH3 R2=CH3), C1a (R1=H R2=CH3), and C2 (R1=CH3 R2=H)), kanamycin (mixture of kanamycin A (R1=NH2 R2=OH), B (R1=NH2 R2=NH2), and C (R1=OH R2=NH2)), tobramycin, streptomycin (R=COH) and dihydrostreptomycin (R=CH2OH) and paromomycin (R=OH) and neomycin B (R=NH2). (C) NHS-PEG12-bitoin linker used to modify gentamicin, apramycin, kanamycin, tobramycin, paromomycin, and neomycin and hydrazide-PEG4-biotin linker used for modification of streptomycin. (D) Biotinylated AMG examples of monobiotinylated apramycin and monobiotinylated streptomycin.
The use of veterinary medicines in animal husbandry is regulated to ensure that people are not exposed to harmful residues (Table A1, B,C). One element of the control system is the targeted detection/monitoring of both banned and authorized veterinary medicine residues in animal matrixes such as kidney, muscle, liver, and milk.14 In the EU, Member States are required to test many thousands (Table A1, D) of samples per annum in analytical assays to ensure that residue levels to do not exceed maximum residue limits (MRLs).15 The MRL is the maximum concentration (expressed in μg/kg) of a residue that is legally permitted or acceptable in or on a food item. These values are specified for a wide range of medicine/species/matrix combinations. For example, in the aminoglycoside (AMG) class16 of antimicrobial compounds, MRLs range from 50 μg/kg for gentamicin in bovine/porcine muscle to 20 000 μg/kg for apramycin in bovine kidney. Despite legislation, excess residues of AMG compounds have been reported, particularly in calf/cattle tissues and honey (Table A1, C).
One of the key challenges for any control system is to have efficient and cost-effective analytical methods available for use. In the case of antimicrobial agents, there are a number of commercially available rapid test kits and other laboratory-based methods that utilize microbial inhibition for the detection of, e.g., β-lactams, sulfonamides, tetracyclines, and macrolides. The majority of these, however, either do not detect AMGs or their detection limits are much higher than the MRLs.17 Alternative screening/confirmation methods based on LC-MS/MS are available,18 but these approaches are costly to operate when screening many hundreds or thousands of samples. Furthermore, LC-MS/MS methods require highly qualified personnel and cannot currently be used in remote locations with limited technical services. Consequently, there is a need for new rapid, facile, and cheap screening methods for AMGs which have the potential for use both in the laboratory and at upstream locations, such as at abattoirs or border inspection posts.
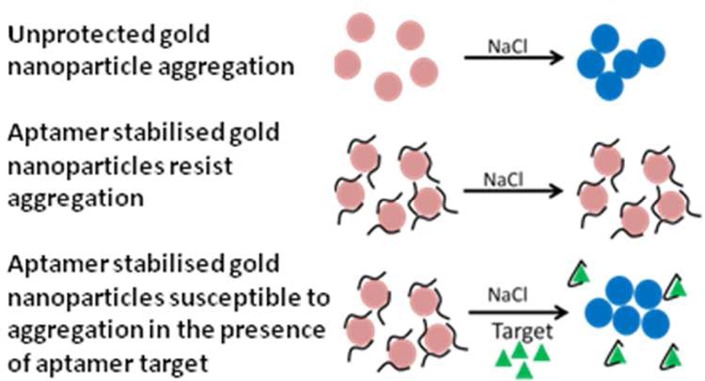
Gold nanoparticles (GNPs) have been used extensively for detection of both low molecular weight chemicals, e.g., K+,19 cocaine,20,21 adenosine,20 ATP,22 ochratoxin A,23 oxytetracycline,24 and dopamine,25 as well as larger targets, e.g., thrombin,26 in a number of formats. These include combinations with DNA oligonucleotides, including ssDNA aptamers. In their simplest format, these aptamers are physi-sorbed to the GNP surface. This has a stabilizing effect on the particles, reducing salt-induced aggregation which would lead to a color change from pink to blue, due to surface plasmon resonance coupling among neighboring particles.27 Such changes are easily observed by eye and can be quantified spectroscopically.28 To date, there are very few reports of the use of RNA oligomers with GNPs and only as 2′-O-methyl derivatives for structure probing.29,30
Here, we have combined RNA aptamer selection against AMGs, with detection in a GNP assay format. Aptamers have been selected using the systematic evolution of ligands by the exponential enrichment (SELEX) process.7 In order to obtain reagents specific for conserved functional groups/structures within the AMGs, toggle SELEX31 (Figure S1, Supporting Information) was carried out by alternating between two structurally related AMGs (referred to as toggle-pairs from here on) in successive rounds. Aptamer selection was “front-loaded”32 to incorporate 2′-fluoro-pyrimidines (2′-F), a modification that imparts stability against common environmental nucleases,33 allowing their potential use in biosensors. Our results suggest that aptamers with broad AMG binding affinity can be isolated by this route. In the GNP assays, physisorbed 2′-F RNA, but not natural RNAs, were able to stabilize GNPs against aggregation allowing detection of the AMGs in the 1–100 nM range. Surprisingly, previously reported, apparently specific anti-AMG natural RNA aptamer sequences34,35 also bind to their targets when modified to include 2′-F pyrimidines. They also showed broad, rather than specific, AMG recognition. In some cases, 2′-F modification had little or no effect on affinity, while for other AMGs there are >10-fold changes between the two forms of the aptamer. Despite these unexpected findings, our data suggest that combining GNP sensing with 2′-F equivalents of the many known RNA aptamers would provide a rapid sensing system for a wide range of analytes.
Materials and Methods
Unless indicated, general laboratory reagents including the AMGs were purchased from Sigma-Aldrich (Dorset, UK) and were the highest grade available. All assay kits were used as per the manufacturers’ instructions unless stated otherwise. AMGs were biotinylated as described in Supporting Information (Table S2). Gold nanoparticles (26 nm dia.) were prepared by standard methods (see Supporting Information)
SELEX and Cloning
The initial dsDNA library consists of approximately 1015 sequences, each containing an N30 random region, flanked by fixed primer regions; one of which carries the T7 RNA polymerase promoter. Aptamer selections were carried out on a Biomek 2000 liquid handling system, using protocols based on those described in detail elsewhere.4,36 In order to isolate aptamers which recognize the features common among the AMGs, a series of toggle selections were performed as described previously.31 The toggle pairs were as follows; gentamicin with apramycin, kanamycin with tobramycin, and paromomycin with neomycin. The toggle partner for streptomycin (dihydrostreptomycin) could not be successfully immobilized, so no toggle was carried out during that selection. PBS supplemented with 10% (v/v) methanol was used for all binding and washing steps. Further information can be found in the Supporting Information.
Analysis of SELEX Products
Biotinylated AMGs were immobilized on streptavidin coated plate wells (Roche Products Ltd., Welwyn, UK) by incubation at excess in PBS overnight at 4 °C on an orbital shaker. Unbound biotinylated AMGs were removed by copious washing with PBS. Twenty-five μL of 600 nM anti-AMG aptamers were incubated with the immobilized targets overnight, and the unbound sequences were washed away with selection buffer. Bound sequences were eluted by heat into nuclease free water, reverse transcribed, amplified and cloned using the materials outlined for SELEX and cloning. This amplified product was then diluted 1 in 5 in nuclease free water and analyzed using the 5k DNA assay chip, run on a Caliper GX II capillary electrophoresis system.
Determination of Minimum RNA Chemical Variant Concentration for GNP Stabilization
In a series of 50 mL falcon tubes, 5× master mixes were made where one reaction contained the following: 30 μL of the 26 nm GNP (see Supporting Information for preparation method) stock (0.24 nM final concentration), RNA at the appropriate concentration, and deionized water to make up the final volume to 50 μL. A series of concentrations (1–20 nM) of the 2′-F, 2′-OH, and 2′-NH2 RNA variants were made and incubated overnight at room temperature in the dark. The following day, 50 μL of the master mix was added to 50 μL of deionized water in an Iwaki polystyrene, flat bottomed, 96 well plate (the type of plate used affected the speed of the color change). To the 100 μL GNP/aptamer reaction, 6.5 μL of 1 M NaCl was added (61 mM final) and quickly mixed. The amount of salt used was the minimum concentration required to aggregate the gold nanoparticles when no RNA is present (Figure S2, Supporting Information). After 10 min, the absorbance spectrum for each well was scanned between 530 and 650 nm. The 650/530 nm ratio (A650/A530) was then plotted against RNA concentration.
Detection of Aminoglycosides with the GNP-Aptamer Sensor
Master mixes were used where possible to reduce sample–sample variation. Each assay contained: 30 μL of 26 nm gold (0.24 nM), 5 nM 2′-F aptamer, and deionized water up to 50 μL. The master mix was incubated overnight, in the dark, at room temperature to allow the aptamer to saturate the GNPs. AMG detection assays were performed on a Biomek 2000 liquid handing robot with 96 well plates (Iwaki); 2-fold serial dilutions of 1 μM AMGs were made in deionized water to 15.6 nM, and 50 μL was mixed with 50 μL of aptamer-GNP master mix. Finally, 6.5 μL of 1 M NaCl (61 mM final concentration) was then added to each well and rapidly mixed. After incubation for 20 min, the plates were moved to a POLARstar Galaxy plate reader (BMG Labtech, Germany) and scanned at 530 and 650 nm and the A650/A530 was plotted against AMG concentration.
Aptamer SPR
All SPR experiments were carried out on a BIAcore 3000 system (GE Healthcare, Buckinghamshire, UK). Aptamer sequences were prepared and purified by phenol chloroform extraction and ethanol precipitation. Samples were then resuspended in DEPC treated water and desalted using G25 Sephadex spin columns (GE Healthcare, Buckinghamshire, UK) and then dialyzed against their respective selections buffers overnight at 4 °C. Monobiotinylated AMGs were immobilized to ∼500 RU on SA-sensor (Streptavidin) chips (GE Healthcare, Buckinghamshire, UK) in PBS following preconditioning buffer injections. One flow-cell was left “blank” on each chip for background correction, and monobiotinylated tobramycin was included on each chip for control purposes. Selection buffers were used to equilibrate the chips for a minimum of 2 h prior to sample injections.
Thermal Melting of AMG-Aptamer Complexes
Aptamers were incubated with target AMGs, each 800 nM, for 30 min and then diluted to 400 nM in warmed urea solution (final concentration 6 M). Absorbance at 260 nm was recorded during a cycled gradient of 25 to 90 °C (1 °C/min gradient), and data were corrected for background and plotted as the percentage change in absorbance at 260 nm.
Results and Discussion
RNA SELEX Against AMG Targets
We used toggle SELEX to probe the recognition of conserved functional groups in pairs of AMG targets known to be likely food contaminants (Figures 1 and S1, Supporting Information). AMGs were modified with NHS-PEG12-biotin using standard amide forming chemistry, resulting in mono- and dibiotinylated AMG products (as detected by LC-TOF-MS), for all but streptomycin and dihydrostreptomycin. The PEG linker was used to minimize steric hindrance for aptamer binding. Streptomycin was successfully biotinylated using a hydrazide-PEG4-biotin which reacts with the sole aldehyde group differentiating streptomycin from dihydrostreptomycin. As it was not possible to modify and immobilize dihydrostreptomycin, aptamers were raised against streptomycin alone (not toggle-selected as for the other AMGs). The biotinylated AMGs were immobilized on streptavidin-coated magnetic beads, then washed, and resuspended in PBS. LC-TOF-MS was then used to estimate the levels of free biotinylated AMG before and after incubation with the beads and after a subsequent 100-fold molar excess of biotin to compete off bound species. As expected, biotinylated AMGs were only detected before incubation with beads and after biotin competition. These data suggest the targets were immobilized successfully.
The selection buffer consisted of PBS containing methanol, present to mimic a food extract which commonly would contain this reagent. Automated SELEX was carried out using a Biomek 2000 liquid handling system (Beckman Coulter) using previously established protocols.4,36−38 The naive (N30) library of 2′-F oligonucleotides was exposed to underivatized beads prior to positive selection in each of the first 5 rounds of SELEX in order to remove nonspecific binders. The toggle-pairs of immobilized AMGs were as follows, gentamicin with apramycin, kanamycin with tobramycin, and paromomycin with neomycin (Figure 1). The selection target was alternated at each round of selection (Figure S1, Supporting Information), e.g., round 1 was against gentamicin, round 2 was against apramycin, round 3 was against gentamicin, and so on. The resulting aptamer populations were denoted LGA#, LPN#, and LKT#, i.e., Leeds derived aptamer against Gentamicin and Apramycin, clone number, etc. Since streptomycin selection occurred without toggle rounds, its aptamers were designated LS#. AMG binding RNAs were eluted by thermal denaturation during the first 5 SELEX rounds and then by competition with the AMG selection targets. The streptomycin selection was identical except that alternating rounds were eluted by competition with streptomycin or dihydrostreptomycin. As the SELEX rounds progressed, the stringency was increased by reducing the amount of selection target, reducing incubation times with the RNA pools, and increasing the number and length of washing steps. Gel electrophoresis (Figure S3, Supporting Information) of the amplified selected products confirmed that selection had been successful.
Analysis of SELEX Products
The aptamer pools were then cloned, and twenty clones from each selection pool were sequenced (Table S3, Supporting Information). Sequence alignment between pools using AliBee (Table A1, E) did not identify obvious conserved extended binding motifs nor was there any homology to known AMG specific aptamers described previously.34,35 The results are consistent with isolation of novel AMG binding aptamers as expected. The AMG recognition properties of all the individual sequences were then tested for target specificity in a high-throughput pull-down assay using 96-well streptavidin-coated plates. Individual, biotinylated AMGs were immobilized in each well of a plate and then incubated with ∼600 nM RNA aptamers overnight. After thorough washing, the remaining bound RNAs were eluted by thermal denaturation, amplified by RT-PCR, and the amount of binding was estimated from the intensity of signals in a capillary electrophoretogram. The GA and KT toggle pairs produced the most binders, whereas the PN selection did not apparently produce any binders. Some of the isolated aptamers also showed some binding to the linker. A couple of aptamers that only bound AMGs, one showing fairly specific binding, LGA11 (although not for its selection target), and one exhibiting broad specificity (LS13) and with the highest relative affinities, were then chosen for additional characterization (Tables 1 and S4, Supporting Information).
Table 1. Capillary Electrophoretic Analysis of the AMG Binding Profiles of the SELEX Productsa.
| aptamer | L | G | A | K | T | P | N | S |
|---|---|---|---|---|---|---|---|---|
| LGA11 | – | – | + | – | – | + | +++ | – |
| LS13 | – | ++ | ++ | ++ | – | ++ | ++ | ++ |
| TOBR12CA | + | ++ | ++ | +++ | +++ | +++ | ++ | ++ |
The amounts of DNA fragments generated from aptamers eluted from each of the targets in the 96 well plate were semiquantitatively assessed from their peak areas on a capillary electrophoresis trace: (−) no products observed (equivalent to no AMG binding); (+), (++), and (+++) correspond to increasing amounts of DNA and should therefore correlate with increased aptamer binding and affinity. Here and in all the other tables, AMGs are referred to by their initial letters. L represents the PEG12-biotin linker reagent used for modification of the AMGs.
From the results, it appears that individual aptamers exhibit large variability in their AMG recognition patterns. The selected sequences mostly have broad specificity for many AMGs rather than for individual AMGs, suggesting that the toggle selection had successfully isolated aptamers recognizing generic features, although these were not restricted to the toggle pairs. Surprisingly, an aptamer reported previously to be selective for tobramycin, TOBR12CA, showed similar promiscuous AMG recognition when produced as a 2′-F-pyrimidine derivative. The solution secondary structures of LGA11 and LS13 were then determined by enzymatic structure probing (Figure S4, Supporting Information). The results are consistent with the idea that novel anti-AMG aptamer motifs have been isolated. Aptamers LGA11 and LS13 were selected for incorporation into a GNP-based assay system.
Development of GNP Colorimetric Assays
We have previously demonstrated that the sensitivity of ssDNA aptamer-GNP assays can be improved by working with the minimum amount of DNA required to protect the GNPs from salt-induced aggregation (unpublished data). Under these conditions, the addition of sub-KD quantities of target analyte displaces enough aptamer from the GNP to permit aggregation. GNP-aptamer assays are illustrated in cartoon form in Figure S5, Supporting Information. In such assays, the detection limit for lysozyme using an aptamer with a KD ∼ 31 nM37 was ∼4 nM. This compares to a detection limit for thrombin in a nonoptimized assay21 of 83 nM with an aptamer KD of ∼0.5 nM.
Initial assay development for AMG detection was carried out using the TOBR12CA aptamer.34 Transcripts encompassing the TOBR12CA were produced as per Goertz et al.,34 using 2′-OH NTPs, and tested for their ability to stabilize the GNPs. It proved impossible to find a stabilizing RNA concentration below 20 nM, in contrast to the widespread use of 2′-deoxy-oligonucleotides for this purpose. There is evidence that the nucleotide bases are the principal feature that results in immobilization on citrate-stabilized GNPs,27 but the hydrophobicity and/or conformational preferences of the sugar residue may also play a role. Since our anti-AMG aptamers were 2′-F modified at pyrimidines, we compared GNP stabilization with TOBR12CA transcripts produced using 2′-OH, 2′-amino (2′-NH2), or 2′-F modified pyrimidines. 2′-F and 2′-NH2 modified RNAs both stabilized the GNPs at ∼5 nM, whereas 2′-OH RNA did not (Figure 2). It appears that the 2′-OH groups may prevent physi-sorption to the gold surface and that this can be overcome by substitution of the pyrimidine riboses with more hydrophobic functional groups. This observation is consistent with previous reports where 2′-O-methyl RNA transcripts on GNPs were used as probes of RNA secondary structure.29,30 In order to explore whether the secondary structures of the RNAs were important for stabilization, we assayed a range of oligonucleotides carrying 2′-F modifications at pyrimidines, ranging in length from 19 to 121 nts, and encompassing either only a stem-loop or a mixture of base-paired and single-stranded regions. The results (Figure S6A, Supporting Information) show that there is little difference between these oligos implying that it is the sugar residue modification that is the critical feature of the physi-sorption step.
Figure 2.
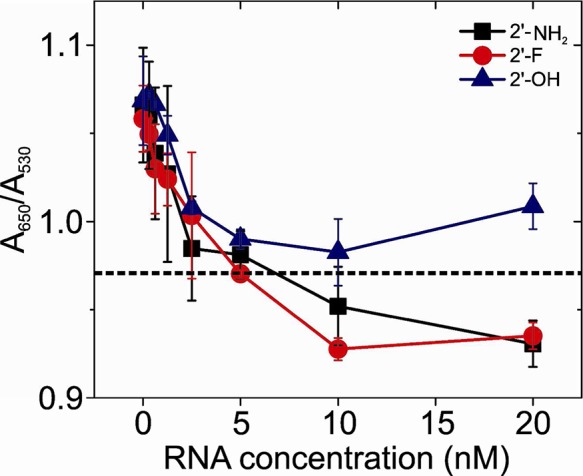
Stabilization of gold nanoparticles with variant RNAs.
GNP stabilization experiments using 2′-OH, 2′-NH2, and 2′-F modified RNA were analyzed spectroscopically. Different concentrations of each aptamer were mixed with 26 nm GNPs at 0.24 nM and incubated overnight (room temperature, in the dark). 61 mM NaCl (final concentration) was then added, and the samples were equilibrated for a further 20 min; the absorbance at 530 and 650 nm was measured. The ratio of A650/A530 is a useful diagnostic of GNP aggregation. The dashed line in the graph highlights the ratio at which the visible color change occurs: above the line the solutions are blue, and below it they are red.
As a further control, we also prepared transcripts encompassing the streptomycin binding aptamer, SB84.35 Again, this control aptamer was initially selected using 2′-OH nucleotides, but we produced it with 2′-F modified pyrimidines, in the hope that some AMG-binding activity would remain. The control aptamer and LGA11 and LS13 transcripts stabilized the GNPs at ∼5 nM, and this was confirmed by transmission electron microscopy (Figure S6B,C, Supporting Information). Although RNA-GNP incubation times as long as 18 h were often used for convenience, we have shown that stabilization is very rapid and is complete after 5 min. Furthermore, once stabilized, the GNPs were stable at room temperature in the dark and showed identical ligand binding properties for at least 6 months.
Figure 3A shows the result of GNP-LGA11 assays for apramycin (red) and gentamicin (blue). The buffer control in the absence of analyte is shown as black symbols. LGA11 clearly binds to both of its selection targets but with differing apparent affinities. Apramycin binding was detectable, using spectroscopy at low nM concentrations and was visibly red by ∼62.5 nM. In contrast, gentamicin binding is not apparent until ≥100 nM and is not detectable by the eye until ∼250 nM. The reason for these different affinities is not obvious from the structures or physical properties of either AMG (Table S1, Supporting Information). Both would be expected to carry 5 positive charges under our assay conditions and have similar numbers of hydrogen bond donor groups (11 for apramycin vs 8 for gentamicin). It appears therefore that the toggle SELEX was successful. However, given the lack of specificity suggested in the pull down assays (Table 1), we assayed LGA11’s ability to detect the other AMGs (Figure 3 B, C). All AMGs were detectable at concentrations below 500 nM, except streptomycin and dihydrostreptomycin. Strikingly, the aptamer showed higher sensitivity for neomycin than for either of its selection targets, displaying a color change at ∼50 nM (Figure 3C), consistent with the pull down assay. Tobramycin and paromomycin had binding profiles similar to apramycin, and the cognate gentamicin was the least well recognized with the LGA11 aptamer. These results do not seem to follow any obvious trend based on the chemical structures and properties of the AMGs (Table S1, Supporting Information). They are, however, due to specific recognition of the AMGs by the aptamer, since GNPs stabilized with the naïve starting pool RNAs show no binding below 125 nM AMG concentrations (Figure S7, Supporting Information).
Figure 3.
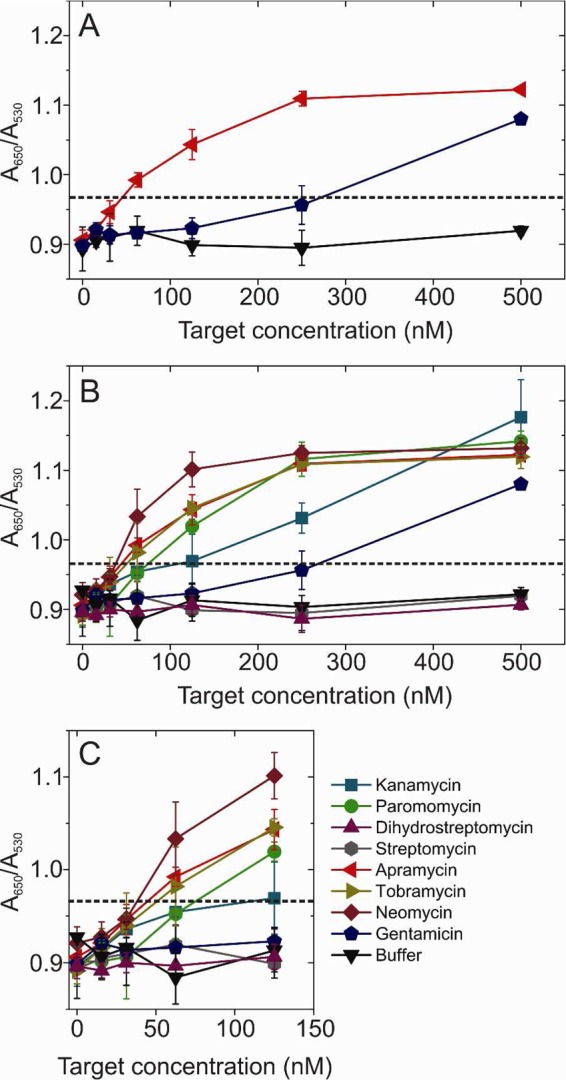
GNP-aptamer sensor detection of aminoglycosides. Shows the results of the GNP-LGA11 aptamer assay against the selection targets gentamicin and apramycin (A) and against 8 aminoglycosides (B). (C) shows chart B, but it is expanded for clarity.
Given the apparent promiscuity of our AMG aptamers and the poor recognition of the streptomycin targets by LGA11, we then screened LS13 and both control aptamers against all AMG SELEX targets (Figure 4). In order to increase throughput and reproducibility, the assay was adapted to a 96 well plate format and carried out on a Biomek 2000 liquid handling robot. Remarkably, 2′-F pyrimidine modified TOBR12CA aptamer retained its affinity for tobramycin but was also able to detect all the other AMGs, except the streptomycin compounds (Figure 4 A). The greatest sensitivity was shown for kanamycin with the poorest binding for gentamicin and dihydrostreptomycin. Binding to streptomycin only becomes apparent at ∼500 nM. This cross-reactivity in an aptamer reported to have been selected against a single AMG target suggests that our result with LGA11 is likely to be common for anti-AMG aptamers. Indeed, this seemed to be true for the GNP-SB84 assay, which failed to detect either of its original cognate targets at concentrations <500 nM but bound all the other targets with roughly similar affinities in the 50 nM range (Figure 4B). The behavior of the GNP-LS13 version of the sensor was consistent with these results (Figure 4C) showing binding to all SELEX targets, except the streptomycins, and with the noncognate kanamycin being the best recognized. Binding appears specific for this class of compound since no binding was seen with the unrelated compounds, tetracycline, ampicillin, ATP, BSA, and the PEG12 biotin linker. (The data for LGA11 binding to these reagents is representative and is shown in Figure S8, Supporting Information.)
Figure 4.

Promiscuity of aminoglycoside binding by GNP-aptamer sensors. 2′-F modified TOBR12CA (A), SB84 (B), and LS13 (C) against a panel of 8 AMGs at concentrations ranging from 0 to 500 nM (see key). Expansions of the 0–125 nM binding curves are shown on the right. The vertical red line shows the MRL value for gentamicin which is the lowest of all the AMGs tested.
It is surprising that the streptomycins were so poorly detected in these assays especially by aptamer sequences selected against the parent compound. SB84 is reported to have a KD for its cognate target of 1 μM.35 The difference with the promiscuous binding to the other AMGs is suggestive of a different type of interaction. This is consistent with the reported structure of the streptomycin complex with an RNA based on a sequence selected by Wallace and Schroeder35 which reveals that the AMG is completely encased within the RNA.39 That aptamer was also selected in the presence of magnesium ions, which were not present in our assays. We therefore repeated the GNP-aptamer assays with LS13, TOBR12CA, and SB84 across a wider concentration range (1–6 μM) and in the presence or absence of magnesium ions (Figure S9, Supporting Information). The results show that both streptomycin and dihydrostreptomycin can be detected readily in this format.
The highest affinity for the streptomycins is with TOBR12CA, which was visibly blue at ≥500 nM, but all aptamers were saturated by 1.5 μM. TOBR12CA binding was largely unaffected by the presence of magnesium ions while LGA11 and, remarkably, SB84 were less sensitive in the presence of the divalent metal ion than in its absence.
Validating the GNP Assay
In order to be able to compare the results of the GNP assays with other well established binding assays, surface plasmon resonance (SPR)40,41 was used to determine the KDs of each aptamer in their selection buffer for the various AMGs. Prior to immobilization on streptavidin sensor chips, the biotinylated AMGs used for selection were further purified using reverse phase HPLC, to isolate monobiotinylated AMGs (final purity >95%) in order to reduce the heterogeneity of the interactions being observed. The amount of immobilized ligand was kept constant (±5%) for all AMGs. The data were analyzed by fitting the association and dissociation phases separately using the BIAevaluation 3.0 software (Table 2). KD values for LGA11 and LS13 were in the range of ∼10–100 nM for all of the AMGs, including streptomycin and dihydrostreptomycin which were poorly detected in the GNP assays. TOBR12CA showed some binding to all AMGs with affinities ranging from ∼2–600 nM for either the 2′-F or 2′-OH forms. SB84 bound some AMGs with affinities in the low nM range and others in the μM range, i.e., it is much more specific for neomycin and paromomycin, with some recognition of streptomycin and tobramycin under these conditions (Table 2). The aptamers selected here recognize all AMGs in the low nM range; i.e., they appear to be recognizing a generic feature.
Table 2. Aptamer AMG Affinities Determined by SPRa.
| KD (nM) ±
SE |
||||||||
|---|---|---|---|---|---|---|---|---|
| aptamer | mod. | A | G | K | N | P | S | T |
| LGA11 | 2′-F | 28.4 ± 0.20 | 22.3 ± 0.13 | 52.8 ± 0.09 | 40.3 ± 0.38 | 21.4 ± 0.16 | 49.9 ± 0.11 | 19.2 ± 0.13 |
| LS13 | 2′-F | 35.9 ± 0.27 | 26.7 ± 0.18 | 69.9 ± 0.07 | 37.0 ± 0.55 | 19.8 ± 0.19 | 69.6 ± 0.09 | 24.8 ± 0.13 |
| TOBR12CA | 2′-OH | 570 ± 0.94 | 53.3 ± 0.56 | 117 ± 1.16 | 17.1 ± 0.36 | 2.54 ± 10.24 | 9.68 ± 0.48 | 25.8 ± 0.31 |
| TOBR12CA | 2′-F | 58.4 ± 0.53 | 34.6 ± 0.29 | 47.8 ± 1.55 | 19.9 ± 0.32 | 30.3 ± 0.36 | 74.1 ± 0.31 | 22.4 ± 0.29 |
| SB84 | 2′-OH | no binding | no binding | no binding | 8.41 ± 0.13 | 40.5 ± 0.17 | 133 ± 0.36 | 112 ± 0.23 |
| SB84 | 2′-F | 3320 ± 1.27 | 694 ± 1.57 | 27200 ± 4.80 | 15.1 ± 0.19 | 42.3 ± 0.21 | 212 ± 1.58 | 132 ± 0.47 |
Analysis of anti-AMG aptamer-ligand affinities. The apparent kinetic parameters for aptamers in their selection buffers binding to immobilized monobiotinylated AMGs were determined using the BIAevaluation software, assuming a 1:1 interaction model. Aptamers SB84 and TOBR12CA were analyzed as both 2′-OH and 2′-F RNA. The apparent equilibrium dissociation constants are listed from the values of kass and kdiss. The standard errors (SE) were calculated from the SEs of kass and kdiss.
The SPR experiments confirm promiscuous AMG binding by the toggle aptamers, even though their binding profiles differ slightly. In both the pull down and SPR assays, recognition is occurring with an immobilized partner. In order to confirm that such promiscuous binding also occurs in solution, we used melting curve profiles (Figure S10, Supporting Information) of 2′-F modified TOBR12CA and LGA11 in the presence of different AMGs. The transcripts for these experiments are 80 nt long, 2′-F modified RNAs which would be expected to have high Tms and be capable of forming several differing conformations in solution, leading to complex unfolding curves. In order to ensure that the RNA secondary structures would be completely unfolded at high temperature, we conducted these experiments in urea which reduces the Tm of melting but does not alter the unfolding/folding pathway.42 The aptamers still showed complex unfolding curves in the absence of AMGs, but in their presence, the differing AMGs resulted in unfolding curves with differing Tms depending on the AMG present. With the exception of the effects on the melting of LGA11 by streptomycin, the stabilization effects mirror expectations based on the GNP assays (Figures 3A and 4). The streptomycins are poorly recognized in the GNP assays but bind much tighter in SPR (Table 2). The solution behavior is therefore closer to the result obtained with the GNP-RNA aptamer assays.
These data demonstrate the potential of GNP-RNA aptamer sensors for rapid and cheap detection of low molecular weight analytes, such as the AMGs. Although a number of apparently specific anti-AMG aptamers have been reported previously, no other aptamers selected for broad specificity are currently known. The lack of sequence and/or secondary structure motif matches in the final selection pools obtained here (Table S3 and Figure S4, Supporting Information) suggests that the structure–function space for AMG recognition is far from exhausted. This is perhaps to be expected from the wide range of known natural RNA targets for these compounds.40 The molecular basis of AMG recognition by RNA is also complex.43 This may account for the lack of consistency in some of the binding assays reported here. The GNP assay is perhaps the most complex of these requiring adsorbed aptamers to dissociate, fold into the ligand binding conformation, and form a complex. Various equilibria are involved in the end result, but for a sensor the critical issue is sensitive ligand detection; here, we have demonstrated facile detection of the AMGs down to the minimum MRL value. The ease of coupling the aptamers with the GNPs suggests that there will be many other applications of this approach.
Acknowledgments
We thank Dr. Jack Kay of the Veterinary Medicines Directorate of the UK Department of Food, Environment and Rural Affairs for supporting this project [project code VM02162 to Dr. Jack Kay], and it was also supported in part by The Wellcome Trust (grant 062164) and by the University of Leeds. We also thank Amy Barker for help with thermal melting measurements and Martin Huscroft for help with purification of monobiotinylated AMGs. N.D. thanks the UK BBSRC and Dionex UK Ltd for studentship support. L.S. thanks the University of Leeds for FFIRS scholarship support.
Appendix 1
Table A1.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Contributions
¶ The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
Supplementary Material
References
- Wirmer J.; Westhof E. Methods Enzymol. 2006, 415, 180–202. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P.; Tulkens M. P. Antimicrob. Agents Chemother. 1999, 43, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunka D. H.; Stockley P. G. Nat. Rev. Microbiol. 2006, 4, 588–596. [DOI] [PubMed] [Google Scholar]
- Bunka D. H.; Mantle B. J.; Morten I. J.; Tennent G. A.; Radford S. E.; Stockley P. G. J. Biol. Chem. 2007, 282, 34500–34509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann T.; Patel D. J. Science 2000, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Fang X.; Tan W. Acc. Chem. Res. 2010, 43, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan J. A.; Ohyama T.; Wang D.; Natarajan K. Nucleic Acids Res. 2000, 28, 2935–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tereshko V.; Skripkin E.; Patel D. J. Chem. Biol. 2003, 10, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; Patel D. J. Nat. Struct. Mol. Biol. 1998, 5, 769–774. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Suri A. K.; Fiala R.; Patel D. J. Chem. Biol. 1997, 4, 35–50. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Majumdar A.; Hu W.; Jaishree T. J.; Xu W.; Patel D. J. Structure 1999, 7, 817–S817. [DOI] [PubMed] [Google Scholar]
- Council Directive 96/23/EC of 29 April 1996 on measures to monitor certain substances and residues thereof in live animals and animal products and repealing Directives 85/358/EEC and 86/469/EEC and Decisions 89/187/EEC and 91/664/EEC. Official Journal of the European Union. 1996, L125: 39, 10–32. [Google Scholar]
- REGULATION (EC) No 470/2009 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 6 May 2009 laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin, repealing Council Regulation (EEC) No 2377/90 and amending Directive 2001/82/EC of the European Parliament and of the Council and Regulation (EC) No 726/2004 of the European Parliament and of the Council. Official Journal of the European Union. 2009, L152: 52, 11–22. [Google Scholar]
- McGlinchey T. A.; Rafter P. A.; Regan F.; McMahon G. P. Anal. Chim. Acta 2008, 621, 1–15. [DOI] [PubMed] [Google Scholar]
- Gaudin V.; Hedou C.; Rault A.; Verdon E. Food Addit. Contam., Part A 2010, 27, 935–952. [DOI] [PubMed] [Google Scholar]
- van Holthoon F. L.; Essers M. L.; Mulder P. J.; Stead S. L.; Caldow M.; Ashwin H. M.; Sharman M. Anal. Chim. Acta 2009, 637, 135–143. [DOI] [PubMed] [Google Scholar]
- Wang L.; Liu X.; Hu X.; Song S.; Fan C. Chem. Commun. 2006, 36, 3780–3782. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wang L.; Pan D.; Song S.; Boey F. Y. C.; Zhang H.; Fan C. Small 2008, 4, 1196–1200. [DOI] [PubMed] [Google Scholar]
- Xia F.; Zuo X.; Yang R.; White R. J.; Xiao Y.; Kang D.; Gong X.; Lubin A. A.; Vallée-Bélisle A.; Yuen J. D.; et al. J. Am. Chem. Soc. 2010, 132, 8557–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang L.; Liu X.; Liang Z.; Song S.; Li W.; Li G.; Fan C. Adv. Mater. 2007, 19, 3943–3946. [Google Scholar]
- Yang C.; Wang Y.; Marty J.-L.; Yang X. Biosens. Bioelectron. 2011, 26, 2724–2727. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Kim J. H.; Kim I. A.; Lee S. J.; Jurng J.; Gu M. B. Biosens. Bioelectron. 2010, 26, 1644–1649. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Wang Y.; Yang X. Sens. Actuators, B: Chem. 2011, 156, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Li B.; Li J.; Wang E.; Dong S. Chem. Commun. 2007, 36, 3735–3737. [DOI] [PubMed] [Google Scholar]
- Nelson E. M.; Rothberg L. J. Langmuir 2011, 27, 1770–1777. [DOI] [PubMed] [Google Scholar]
- Li H.; Rothberg L. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14036–14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Rothberg L. Anal. Chem. 2005, 77, 6229–6233. [DOI] [PubMed] [Google Scholar]
- Li H.; Liang R.; Turner D. H.; Rothberg L. J.; Duan S. RNA 2007, 13, 2034–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.; Rusconi C.; Scardino E.; Wolberg A.; Lawson J.; Hoffman M.; Sullenger B. Mol. Ther. 2001, 4, 567–574. [DOI] [PubMed] [Google Scholar]
- Gold L.; Brown D.; He Y.-Y.; Shtatland T.; Single B. S.; Wu Y. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigelman L.; McSwiggen J. A.; Draper K. G.; Gonzalez C.; Jensen K.; Karpeisky A. M.; Modak A. S.; Matulic-Adamic J.; DiRenzo A. B.; Haeberli P.; et al. J. Biol. Chem. 1995, 270, 25702–25708. [DOI] [PubMed] [Google Scholar]
- Goertz P. W.; Cox J. C.; Ellington A. D. J. Assoc. Lab. Autom. 2004, 9, 150–154. [Google Scholar]
- Wallace S. T.; Schroeder R. RNA 1998, 4, 112–123. [PMC free article] [PubMed] [Google Scholar]
- Barton J. L.; Bunka D. H.; Knowling S. E.; Lefevre P.; Warren A. J.; Bonifer C.; Stockley P. G. Nucleic Acids Res. 2009, 37, 6818–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. C.; Ellington A. D. Bioorg. Med. Chem. 2011, 9, 2525–2531. [DOI] [PubMed] [Google Scholar]
- Moore M. D.; Bunka D. H.; Forzan M.; Spear P. G.; Stockley P. G.; McGowan I.; James W. J. Gen. Virol. 2011, 92, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tereshko V.; Skripkin E.; Patel D. J. Chem. Biol. 2003, 10, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ironmonger A.; Whittaker B.; Baron A. J.; Clique B.; Adams C. J.; Ashcroft A. E.; Stockley P. G.; Nelson A. Org. Biomol. Chem. 2007, 5, 1081–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons I. D.; Persson B.; Mekhalfia A.; Blackburn G. M.; Stockley P. G. Nucleic Acids Res. 1995, 23, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gell C.; Sabir T.; Westwood J.; Rashid A.; Smith D. A.; Harris S. A.; Stockley P. G. J. Mol. Biol. 2008, 384, 264–278. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Suri A. K. J. Biotechnol. 2000, 74, 39–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.