

Abstract
In this study, a series of new BF2-chelated tetraarylazadipyrromethane dyes are synthesized and are shown to be suitable for the preparation of on/off photoinduced electron transfer modulated fluorescent sensors. The new indicators are noncovalently entrapped in polyurethane hydrogel D4 and feature absorption maxima in the range 660–710 nm and fluorescence emission maxima at 680–740 nm. Indicators have high molar absorption coefficients of ∼80 000 M–1 cm–1, good quantum yields (up to 20%), excellent photostability and low cross-sensitivity to the ionic strength. pKa values of indicators are determined from absorbance and fluorescence measurements and range from 7 to 11, depending on the substitution pattern of electron-donating and -withdrawing functionalities. Therefore, the new indicators are suitable for exploitation and adaptation in a diverse range of analytical applications. Apparent pKa values in sensor films derived from fluorescence data show 0.5–1 pH units lower values in comparison with those derived from the absorption data due to Förster resonance energy transfer from protonated to deprotonated form. A dual-lifetime referenced sensor is prepared, and application for monitoring of pH in corals is demonstrated.
Design, synthesis, and spectroscopic/photophysical characterization of novel fluorescent chemosensors remains a central research field in analytical chemistry.1 The measurement of pH by fluorescence-based techniques is well established for both imaging and sensing applications2,3 in various fields of experimental science, such as biomedical research,4−10 marine biology,11,12 and biotechnology.13,14 The most frequently used fluorescent pH indicators are 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS), carboxyfluorescein derivatives, seminaphthorhodafluors (SNARFs), and hydroxycoumarins.15−19 However, these indicators suffer from several drawbacks. For example, carboxyfluorescein has only moderate photostability, and the photostability of 2′,7′-dihexylfluorescein (suitable for measurements in seawater) is very poor;20 the pKa value of HPTS is highly dependent on ionic strength of solution; and most coumarins are excitable only by high-energy radiation in the range from 350 to 450 nm.
In biological applications, it is desirable to use fluorophores with absorption/emission profiles in the red or near-infrared (NIR) spectral regions because they have many advantages: significant reduction of the background signal due to the low absorption and autofluorescence of biomolecules in the NIR region, low light scattering and deep penetration of the NIR light, and the possibility to use low-cost excitation light sources. Despite the optical benefits, there is a surprising scarcity of pH indicators that have such desired absorption and emission properties. Although the water-soluble SNARF indicators and their lipophilic derivatives do absorb at ∼630 nm, they possess only moderate brightness and photostability.21 The same holds for the cyanine dyes, which are well-known NIR chromophores that have only scarcely been proposed as pH indicators.22,23
In contrast, BF2-chelated tetraarylazadipyrromethane dyes (aza-BODIPYs) represent an interesting class of NIR chromophores that are amenable to structural modification and exhibit excellent photophysical properties.24 Several fluorescent pH indicators based on aza-BODIPY dyes bearing amino- or hydroxy-functionalized substituents were reported by O’Shea and co-workers.25−27 These on/off pH indicators show photoinduced electron transfer from an amino group or a twisted phenolate to the aza-BODIPY subunit, causing fluorescence quenching in the deprotonated state. However, these probes cover only acidic and near-neutral range. A systematic study of the properties of these pH indicators and possible synthetic modifications wtih respect to tuning the pKa values has not previously been reported in the literature.
In the present study, we investigated the synthesis and characterization of aza-BODIPY fluorophores that can probe pH changes by large associated changes in their emission intensity around 700 nm and possess pKa values in the physiological and alkaline pH range. Eight 4,4-difluoro-4-bora-3a,4a-triaza-s-indacene dyes that can detect pH through a photoinduced electron transfer process were synthesized and characterized. We show that the pKa values of the new indicators can be tuned over a wide range, and simple prediction rules can be derived. This enables a variety of potential applications for sensors and imaging. As an example, an application of the new sensing materials for fiber-optic pH measurements in marine biology is demonstrated.
Experimental Section
Materials
3′-Chloro-4′-hydroxyacetophenone, 1,3-diphenyl-2-propenone, tera-tert-butyl-29H,31H-phtalocyanine, N,N-diisopropylethylamine, ammonium acetate, benzaldehyde, seminaphthorhodafluor decyl ester (SNARF-DE, chromoionophore XIII), boron trifluoride diethyl etherate, MOPS buffer salt, and anhydrous sodium sulfate were purchased from Sigma Aldrich (www.sigmaaldrich.com). 3′-Hydroxyacetophenone, 4-hydroxychalcone, 4′-hydroxychalcone, 4′-methoxychalcone, 4,4′-dimethoxychalcone, and nitromethane were obtained from ABCR (www.abcr.com). 4′-Hydroxy-3′-methylacetophenone was obtained from TCI Europe (www.tcieurope.de). Deuterated dimethyl sulfoxide was obtained from Euriso-top (www.eurisotop.com). All other solvents (synthesis grade) as well as sodium chloride and the buffer salts CHES, MES, and CAPS were supplied by Carl Roth (www.roth.de). Silica-gel (0.04–0.063 mm) was from Acros (www.fishersci.com). Polyurethane hydrogel (Hydromed D4) was purchased from AdvanSource biomaterials (www.advbiomaterials.com). Poly(ethylene glycol terephthalate) support (Mylar) was obtained from Goodfellow (www.goodfellow.com). Microcrystalline powder of phosphor chromium(III)-activated gadolinium aluminum borate (Gd3Al4.75Cr0.25O12, Cr-GAB) was prepared as described previously.28
Synthesis
The synthetic concept is exemplified by the following synthesis of 1. The other dyes were obtained in a similar way, and their preparation is described in detail in the Supporting Information.
BF2 Chelate of [5-(4-Hydroxyphenyl)-3-phenyl-1H-pyrrol-2-yl]-[5-phenyl-3-phenylpyrrol-2-ylidene]amine (1)
1-(4-Hydroxyphenyl)-4-nitro-3-phenylbutan-1-one (1a). A solution of 1-(4-hydroxyphenyl)-3-phenylpropenone (1 equiv, 2 g, 8.9 mmol), nitromethane (20 equiv, 9.63 mL, 178 mmol), and KOH (1.2 equiv, 0.6 g, 10.68 mmol) in EtOH (10 mL) was heated at 60 °C under reflux for 12 h. After cooling to room temperature, the solvent was removed in vacuo, and the oily residue obtained was acidified with 4 M HCl and partitioned between EtOAc (50 mL) and H2O (50 mL). The organic layer was separated, dried over sodium sulfate, and evaporated under reduced pressure. The obtained product was used for further synthesis without purification (2.06 g, 80%).
1,3-Diphenyl-4-nitro-butan-1-on (1b). A solution of 1,3-diphenyl-2-propenone (1 equiv, 2 g, 9.6 mmol), nitromethane (20 equiv, 10.37 mL, 192 mmol) and KOH (0.2 equiv, 0.106 g, 1.9 mmol) in EtOH (10 mL) was heated at 60 °C under reflux for 12 h. After cooling to room temperature, the solvent was removed in vacuo, and the oily residue obtained was partitioned between EtOAc (50 mL) and H2O (50 mL). The organic layer was separated, dried over sodium sulfate, and evaporated under reduced pressure. The obtained product was used for further synthesis without purification (1.52 g, 80%).
[5-(4-Hydroxyphenyl)-3-phenyl-1H-pyrrol-2-yl]-[5-phenyl-3-phenylpyrrol-2-ylidene]amine (1c). Compound 1a (1 equiv, 1.0 g, 4.4 mmol), compound 1b (1 equiv, 0.93 g, 4.4 mmol), and ammonium acetate (35 equiv, 8.06 g, 245 mmol) in butanol (50 mL) were heated under reflux for 24 h. The reaction was cooled to room temperature, and the crude product was purified by column chromatography on silica-gel eluting with dichloromethane (after eluting symmetric byproduct with hexane/dichloromethane 3:1 v/v) to yield 1c as a blue-black solid. The product was recrystallized from hexane/tetrahydrofuran mixture to give green metallic crystals (0.42 g, 25%). For the calculation, the theoretical yield of the asymmetrical product is set as 100%). 1H NMR (300 MHz, DMSO-d6) δ: 8.13–8.08 (m, 6H), 7.94 (s, 2H), 7.92 (s, 1 H), 7.80 (s, 1H), 7.63–7.58 (m, 1H), 7.5–7.33 (m, 8H), 7.02 (d, J = 8.8 Hz, 2H). Electron impact direct insertion time-of-flight (EI-DI-TOF) m/z [MH+] found 465.1822, calcd 465.1841.
BF2 Chelate of 1c
Compound 1c (0.34 g, 0.73 mmol) was dissolved in dry CH2Cl2 (50 mL), treated with diisopropylethylamine (10 equiv, 0.54 mL, 3.11 mmol) and BF3 diethyletherate (15 equiv, 0.55 mL, 4.35 mmol),. and stirred under argon for 24 h. Purification by column chromatography on silica gel eluting with CH2Cl2 and recrystallization from hexane/tetrahydrofurane gave the final product 1 as a red metallic solid (0.201 g, 43%). 1H NMR (300 MHz, DMSO-d6) δ: 10.72 (s, 1H), 8.23–7.99 (m, 8H), 7.77 (s, 1H), 7.61–7.36 (m, 10H), 6.95 (d, J = 8.8 Hz, 2H). Electron impact direct insertion time-of-flight (EI-DI-TOF) m/z [MH+] found 512.186, calcd 512.186.
Preparation of Sensor Foils and Fiber-Optic Microsensors
A “cocktail” containing an indicator dye (0.25 mg), hydrogel D4 (100 mg) in 700 μL EtOH/H2O (9:1 v/v), and tetrahydrofurane (300 μL) was knife-coated on a dust-free Mylar support to obtain a ∼2.5 μm thick sensing layer after solvent evaporation.
The “cocktail” for the dual-lifetime referenced sensor was prepared similarly from 0.1 mg of indicator dye 1 with addition of 200 mg of Cr-GAB particles. Subsequently, it was coated on the tip of an optical fiber (Ø 400 μm) from Specialty Photonics (www.specialtyphotonics.com). The sensors were allowed to dry 30 min in the ambient to ensure complete evaporation of the solvent. Between measurements, the sensors were stored in darkness at room temperature.
Methods
Absorption measurements were performed on a Cary 50 UV–vis spectrophotometer from Varian (www.varianinc.com). The molar absorption coefficients were determined as an average of three independent measurements for the concentrations of the dyes 2.5–3.4 × 10–6 M. Fluorescence spectra were recorded on a Hitachi F-7000 spectrofluorometer (www.hitachi.com). Relative fluorescence quantum yields were determined according to Demas and Crosby29 using tera-tert-butyl-29H,31H-phtalocyanine (Fluka, www.sigmaaldrich.com) as a standard (quantum yield = 0.44).30 Two independent measurements were performed to obtain the average value. The concentration of the indicators was kept below 1.5 × 10–6 M to avoid dye aggregation and reabsorption of the fluorescence. NMR spectra were recorded on a 300 MHz instrument (Bruker) in DMSO-d6 or CDCl3 with TMS as a standard. Electron impact (EI, 70 eV) mass spectra were recorded on a Waters GCT Premier equipped with direct insertion (DI). The pH of the buffer solutions (CHES, MES, and CAPS) was controlled by a digital pH meter (InoLab pH/ion, WTW GmbH & Co. KG, www.wtw.com) calibrated at 25 °C with standard buffers of pH 7.0 and pH 4.0 (WTW GmbH & Co. KG, www.wtw.com). The buffers were adjusted to constant ionic strength (IS = 0.02 or 0.15 M) using sodium chloride as the background electrolyte. Dual-lifetime referenced measurements were performed with a fiber-optic Firesting fluorometer from Pyroscience (www.pyro-science.com) with a modulation frequency of 4 kHz.
Photobleaching experiments in solutions were performed by irradiating the samples with light from a 642-nm high-power 10 W LED array (www.led-tech.de) focused through a lens purchased from Edmund optics (www.edmundoptics.de). The photodegradation profiles were obtained by monitoring the absorption spectra. For the leaching test, sensor foil (D4) was placed in a flow-through cell, and the absorption of the films was monitored while aqueous buffer (IS = 0.02 M) was passed through it. A detailed description of the pH measurements in the gastric cavity of a symbiont bearing coral (Goniopora sp.) is contained in the Supporting Information.
Results and discussion
Synthesis
Two general methods to prepare azadipyrromethene chromophore are known. In the first method, 2,4-diarylpyrroles act as precursors, and are converted into their 5-nitroso derivatives, which are then condensed with a second molecule of pyrrole to yield unsymmetric derivatives.31 In the second method, nitromethane adducts of chalcones are reacted with ammonium salts at elevated temperatures to give symmetric azadipyrromethenes.32 Conversion of the diarylnitroketones into pyrroles and 5-nitrosopyrroles and the isolation of these products is not required in this method.
Unsymmetrical derivatives of the aza-BODIPYs bearing only one pH-sensitive group were strongly preferable to the symmetrical ones for several reasons: (i) a simple acid–base equilibrium including only two forms of the indicator; (ii) significantly higher hydrophobic character of the monosubstituted derivatives compared to the symmetrical ones, which prevents the dye from leaching into an aqueous environment, particularly in case of the deprotonated form; (iii) a low charge of the deprotonated form (1−), which is expected to minimize the effect of the ionic strength on the sensing properties.
Keeping practical applications in mind (in which accessibility of the indicators is very important) we decided to employ the second method for preparation of the unsymmetrical dyes. In this approach, condensation of two different nitrochalcones results in a mixture of the aimed unsymmetrical aza-BODIPY and two symmetrical derivatives (Scheme 1), which are easily separated via chromatography on silica gel. The starting compounds for the synthesis were the diaryl α,β-unsaturated ketones (chalcones) that are either commercially available (for the synthesis of 1, 5, 7, and 8) or prepared by Claisen–Schmidt condensation (for the synthesis of 2–4, 6). These were synthesized from the corresponding aldehyde and acetophenone with KOH as a base in all cases, except for 6, for which NaH was used. The Michael addition of nitromethane to the chalcones, with KOH as base,33 yields the 1,3-diaryl-4-nitrobutan-1-ones in essentially quantitative yields after aqueous workup, which were then used without further purification. Condensation with ammonium acetate in refluxing butanol gave the azadipyrromethenes via a cascade of events (in situ formation of the pyrrole and corresponding nitrosopyrrole and subsequent condensation of those two entities). The obtained mixture was purified by chromatography with hexane/dichloromethane in the case of 1–6 or toluene/dichloromethane in case of 7 and 8. Finally, complexation of the azadipyrromethenes with boron trifluoride gave the aza-BODIPYs in good yields.
Scheme 1. Synthesis of aza-BODIPY Probes.

Photophysical Properties
The new aza-BODIPY compounds were dissolved in an EtOH/aqueous buffer mixture (1:1), and their spectroscopic properties were investigated (Figure S1 of the Supporting Information, Table 1). The absorption of the probes bearing a hydroxyl group in the p-position (R1) of Ar4 is rather similar (668–677 nm). The shortest wavelengths of the absorption maxima are observed for 5 and 6 (656 and 650 nm, respectively). These values are very close to the tetraphenyl-substituted aza-BODIPY chromophore lacking any substituents (650 nm).32 The absorption of 7 (bearing a methoxy group in the p-position of the Ar3 ring) is bathochromically shifted ∼20 nm as compared with 1. On the other hand, introduction of the second methoxy group in the p-position of the Ar2 ring (8) results in very minor changes compared with 7.
Table 1. Photophysical Properties of the aza-BODIPY Probes: Absorbance Maxima for the Acidic (λabs-acid) and the Basic Forms (λabs-base), Emission Maxima for the Acidic Form (λem-acid), Molar Absorption Coefficients (ε), and Luminescence Quantum Yields (QY).
| dye | λabs-acid/ λabs-base (EtOH/H2O-1:1) (nm) | λem-acid (EtOH/H2O-1:1) (nm) | λabs-acid/ λabs-base (hydrogel D4) (nm) | λem-acid (hydrogel D4) (nm) | ε ± 5% (M–1 cm–1)(THF)a | QY ± 20% (THF)a | QY ± 20% (EtOH/buffer-1:1)a |
|---|---|---|---|---|---|---|---|
| 1 | 670/726 | 702 | 687/742 | 718 | 84 000 | 0.15 | 0.11 |
| 2 | 668/734 | 699 | 683/754 | 730 | 80 600 | 0.16 | 0.08 |
| 3 | 675/735 | 709 | 692/750 | 720 | 86 200 | 0.18 | 0.08 |
| 4 | 677/741 | 714 | 694/752 | 722 | 89 700 | 0.16 | 0.05 |
| 5 | 656/782, 605 | 691 | 672/859, 615 | 702 | 69 500 | 0.07 | 0.03 |
| 6 | 650/656 | 676 | 660/663 | 686 | 71 100 | 0.10 | 0.02 |
| 7 | 687/743 | 722 | 707/760 | 736 | 80 100 | 0.17 | 0.14 |
| 8 | 690/750 | 721 | 708/768 | 736 | 74 000 | 0.16 | 0.11 |
For the protonated form.
These data indicate pronounced conjugation of the Ar3 and Ar4 rings with the aza-BODIPY chromophore and the absence of such conjugation for the Ar1 and Ar2 rings. This conjugation can be caused by a hydrogen bond between the fluorine atom and the hydrogen atom located in the o-positon of Ar3 and Ar4 rings. Evidently, the electron-donating substituents (−OH or O–CH3) in the p-position of the Ar3 and Ar4 rings result in the bathochromic shift of the absorption spectrum but have no effect on the spectral properties if located in the Ar1 and Ar2 rings. As expected, the hydroxyl group in the m-position of the Ar4 has virtually no effect on the spectral properties. The molar absorption coefficients for all aza-BODIPY derivatives are in the 70 000–90 000 M–1 cm–1 range.
It should be mentioned that precision of determination of molar absorptivities as well as fluorescence quantum yields can be affected by inpurities of the probes. Although all the indicators were obtained as single crystals, several indicators show solvent impurities in the 1H NMR spectrum. As revealed by crystallographic study of 8 (Figure S2 of the Supporting Information), this is explained by incorporation of solvent molecules (tetrahydrofurane and hexane) into the crystal structure. Evidently, this slightly affects the calculated molar absorption coefficients. For instance, the crystals of 8 contain one molecule of tetrahydrofurane per each molecule of the dye, and this results in the reduction of molar absorption coefficients by about 8.8%.
The trend observed in absorption maxima was the same for fluorescence emission maxima (Table 1). The emission maxima of the probes bearing a hydroxyl group in the p-position (R1) of Ar4 were located between 699 and 714 nm. The compounds 5 and 6 clearly showed a hypsochromic shift (676 and 691 nm, respectively), and the emission of 7 and 8 was shifted bathochromically (721 and 722 nm, respectively) as compared with the parent compound 1. The trends in the absorption and fluorescence maxima of indicators in hydrogel D4 mirrored those in solution with an additional bathochromic shift of ∼15 nm (Table 1).
The fluorescence quantum yields (QYs) of most indicators were very similar (0.14–0.17 in tetrahydrofurane, Table 1). Again, the dyes 5 and 6 represent a notable exception as they exhibited significantly lower quantum yields. As can be seen, all the values are lower in EtOH/aqueous buffer. Although very low concentrations of the dyes were used (<1.5 × 10–6 M), some aggregation of the dyes in EtOH/water mixture cannot be excluded. On the other hand, lower quantum yields in this media can be explained by more efficient radiationless deactivation involving O–H vibrations. It should be mentioned that the QYs of most reported NIR-emitting dyes are generally lower than for those emitting in the visible part of the spectrum. In summary, the photophysical properties of the new aza-BODIPY derivatives retain most of the advantages of the aza-BODIPY fluorophores (except for 5 and 6), including narrow bandwidth, high molar absorption coefficient, and acceptable fluorescence quantum yields.
pH-Sensing Properties
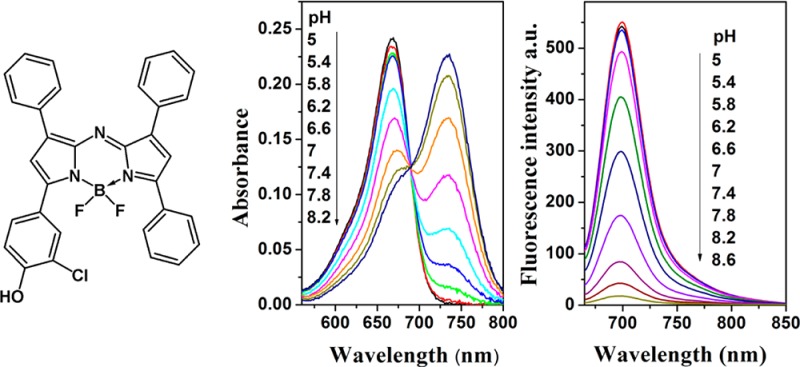
pKa values of the new probes were determined both in ethanol/aqueous buffered solution (1:1) and in a hydrogel D4 film from the absorption measurements. As can be seen, the absorption spectra shift bathochromically upon deprotonation of the hydroxyl group in all cases except for 6 as a result of the absence of conjugation with the aza-BODIPY core (Figure 1, Table 1). Notably, the absorption spectrum of deprotonated 5 is rather unusual and shows two peaks (Figure 1b). The pKa values determined at two ionic strengths of the solution (0.02 and 0.15 M) were very similar (Table 2). Such low cross-sensitivity to the ionic strength is explained by the low charge of the indicator molecule (0 and −1 for the protonated and deprotonated forms, respectively).
Figure 1.

pH dependence of absorbance for 2 (a), 5 (b), and 6 (c) in ethanol/aqueous buffer solution (1:1, IS 0.02 M).
Table 2. pKa Values of Fluorescent aza-BODIPY Probes.
| dye no. | pKabs (IS = 0.02M) (EtOH/H2O-1:1) | pKem (IS = 0.02M) (EtOH/H2O-1:1) | pKabs (IS = 0.15M) (EtOH/H2O-1:1) | pK′em (IS = 0.15M) (EtOH/H2O-1:1) | pKabs (IS = 0.02M) (hydrogel D4) | pK′em (IS = 0.02M) (hydrogel D4) |
|---|---|---|---|---|---|---|
| 1 | 8.35 | 8.38 | 8.36 | 8.41 | 8.47 | 8.09 |
| 2 | 7.00 | 7.01 | 6.92 | 6.98 | 6.73 | 6.08 |
| 3 | 8.36 | 8.47 | 8.42 | 8.45 | 8.49 | 7.82 |
| 4 | 8.06 | 8.05 | 8.11 | 8.15 | 7.94 | 7.19 |
| 5 | 9.69 | 9.68 | 9.69 | 10.88 | 9.84 | 9.67 |
| 6 | 11.05 | 10.88 | 11.02 | 9.69 | 12.35 | 11.4 |
| 7 | 8.71 | 8.65 | 8.76 | 8.74 | 8.81 | 8.23 |
| 8 | 8.92 | 8.89 | 8.90 | 8.94 | 9.11 | 8.56 |
The pKa of the aza-BODIPY derivatives can be tuned over a wide range by introducing electron-withdrawing/donating (remote) neighboring functionalities or changing the position of the hydroxyl group. If probe 1 (with pKa of 8.38) is substituted at the m-position with a chlorine atom, the pKa drops to 7.01 (2). When (inductively) electron-donating methyl groups are introduced in the m-positions, the pKa is 8.47 for one methyl group (3) and 8.05 for two methyl groups (4). The increased acidity of a dimethyl-substituted derivative is surprising, but can be explained by the difficulties of formation of the solvent adduct due to steric hindrances. If a hydroxyl group is introduced in the p-position of the Ar1 aryl ring instead of the Ar4 aryl ring, the pKa increases to 9.68 (5). This is very close to the pKa value of phenol (pKa = 10),34 which indicates the absence of the conjugation with the aza-BODIPY core. This observation is in good agreement with the trends obtained from the absorption spectra. A drastically high pKa of 6 (10.88) compared with the pKa of parent compound 1 (8.38) was attributed to the formation of an intramolecular hydrogen bond between the hydroxyl group and the neighboring fluorine atom. Introduction of a remote electron-donating substituent (methoxy group) in the p-position of the Ar3 ring (7) slightly increases the pKa value (by about 0.3 units), relative to the parent compound 1. Evidently, the second methoxy group in the p-position on the Ar2 aryl ring (8) has less effect on the pKa value. Effects observed in this study are in line with the electron-withdrawing (or -donating) power of the neighboring (remote) substituents. In summary, pH indicators with tailored pKa values can be realized, and the indicators can be easily adapted to particular applications.
The pKa values determined in this study differ significantly from the reported pKa of 6.9 of a phenolic derivative with a hydroxyl group in the p-position of the Ar4 ring (similar to the compound 1 in our study),26 possibly because of different media used by Murtagh et al. for the calibration of the pH indicator (nonionic surfactant) and higher concentration of the dye in these micelles. As will be demonstrated in the following, the concentration of the indicators is rather critical for determination of apparent pKa values via fluorescence measurements.
The fluorescence of the indicators is quenched upon deprotonation (Figure 2), which is attributed to efficient photoinduced electron transfer.35−37 In fact, no emission is detected for the deprotonated form of the dyes. The apparent pKa values (pK `a) obtained from the emission measurements in solution are very close to those determined from the absorption spectra (Table 2).
Figure 2.
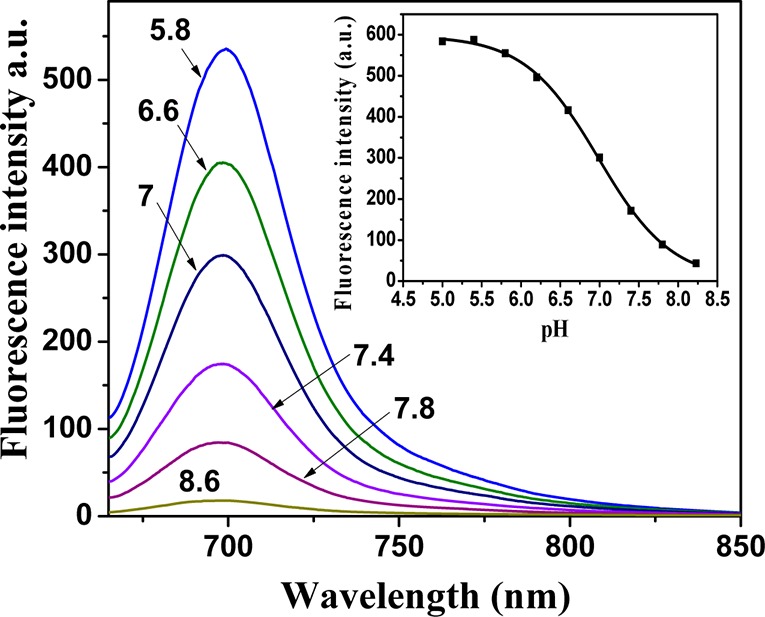
pH dependence of fluorescence for 2 (1.41 × 10–6 M) in ethanol/aqueous buffer solution (1:1) and the corresponding calibration curve (insert).
Optical pH sensors were prepared by noncovalent entrapment of the indicators in the hydrogel D4 matrix, which is a highly proton-permeable, uncharged polyurethane derivative with a water uptake capacity of about 50%. The pKa values obtained for the sensing materials from the absorption measurements were very close to those obtained for the solutions of the indicators (Table 2). Unexpectedly, the apparent pKa values in D4 as derived from fluorescence data were 0.5–1 pH units lower than those obtained from absorption data (Table 2). This discrepancy can be explained by the fact that the concentration of the indicator is much higher in the hydrogel than in the solution. Therefore, the dye molecules are close enough to enable Förster resonance energy transfer (FRET) from the protonated to the deprotonated form (Figure S3 of the Supporting Information). Considering that FRET is concentration-dependent, this offers another possibility of tuning the dynamic range of the materials. In fact, pḰa values derived from the fluorescence data are lower for higher concentration of the indicators in hydrogel D4 (e.g., pḰa of 6.65, 6.14, 6.03, and 5.89 for the sensor films containing 0.1, 0.25, 0.5 and 1% of the indicator 2), Supporting Information Figure S3c. The pḰa values obtained from the absorption measurements (Supporting Information Figure S10b) are 6.92, 6.83, 7.04, and 7.32 for the same concentrations. This increase may be due to different localization patterns of the dye in hydrogel D4, which is known to be composed of the hydrophilic and hydrophobic regions.
We compared the pH-sensing properties of the complexes and the respective ligands. The ligands were virtually nonfluorescent, possibly due to their nonplanar structure. However, distinct changes in the absorption spectra were observed (Figure S4a of the Supporting Information). The pKa values were ∼1 unit higher than for the corresponding complexes (Supporting Information Figure S4b). For example, the pKa values were 8.35 and 7.00 for 1 and 2, but 9.39 and 7.97 for the corresponding ligands. This effect is likely due in part to the lower degree of the π-conjugation between the phenolic substituents and the chromophore core (which is reflected by the smaller shift between the λmax of both forms of the dye compared with the corresponding complexes). The electron-withdrawing effect of the BF2 group can also contribute here. Despite the absence of fluorescence, the nonchelated dyes can be promising as absorption-based indicators.
Dye leaching out of the sensing matrix may be a problem in the case of physically entrapped indicators. It was tested in the case of 1 by monitoring the absorption of the sensing foils. No evident leaching into the aqueous solution was detectable for the protonated form of the dye. In the case of the charged deprotonated form, the decrease in the absorption was very low (0.8% per 24 h, Figure S5 of the Supporting Information), which is within the experimental error. Consequently, leaching is not critical for the investigated sensors due to the pronounced hydrophobicity of the indicator systems.
Photostability
Photostability is a very important parameter for all optical chemosensors. We investigated solutions of the new pH dyes in dimethylformamide under continuous illumination with an ultrabright 642-nm LED array. Figure 3 demonstrates the photodegradation profiles for the new indicators and for seminaphthorhodafluor decyl ester (SNARF-DE), which is used for comparison. The aza-BODIPY probes were significantly more photostable (about 30-fold) than SNARF-DE under identical conditions. In fact, after 2.5 h of illumination, only 1–8% of the dyes was decomposed compared with SNARF-DE, which degrades almost completely after 60 min of irradiation. The photostability trend for aza-BODIPY probes was 6 > 3 > 4 > 1–2 ∼ 7 > 5–8 (Figure S6 of the Supporting Information). The electron-donating substituents in the aryl rings Ar1 and Ar2 seem to be responsible for decreased photostability; nevertheless, new pH dyes retain excellent photostability of aza-BODIPY dyes,38 which makes them particularly suitable for long-duration measurements.
Figure 3.
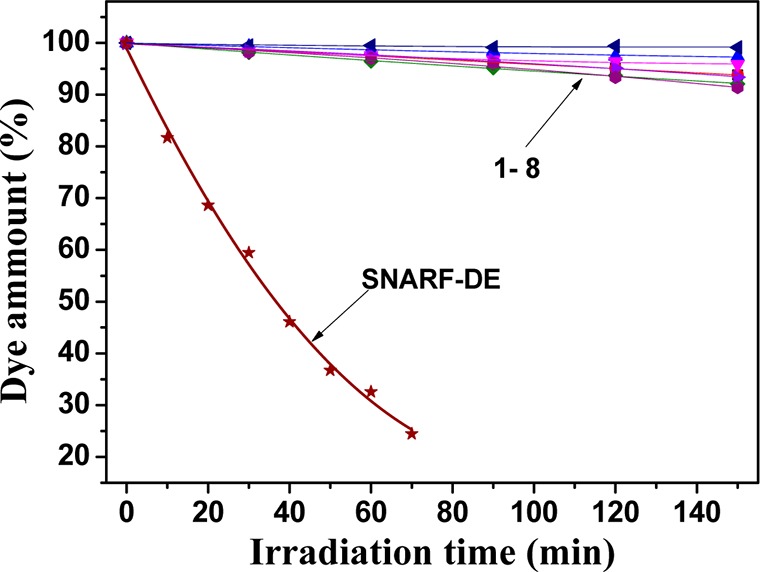
Comparison of photobleaching profiles for aza-BODIPY fluorescent pH probes and SNARF-DE in dimethylformamide determined from the absorption measurements.
Dual-Lifetime Referenced pH Sensor
Fluorescence intensity is a parameter that is easy to measure, but it is influenced by a number of factors, such as the intensity and light field of the excitation source, the sensitivity of the photodetector, and the coloration and turbidity of the measuring media. In contrast, measurements of fluorescence decay time, which is a self-referenced parameter, normally require complicated and expensive equipment. Ratiometric sensing and dual lifetime referencing are popular alternatives. In the latter approach, a referenced material having a long luminescence decay time (typically in the microseconds time domain) is added, and the overall phase shifts are measured in the frequency domain.39 Since good spectral compatibility (both for excitation and for emission) is essential, a recently reported inorganic phosphor, chromium(III)-doped gadolinium aluminum borate (Cr-GAB)28 is an excellent candidate for this purpose; its luminescence decay times of about 100 μs, and high chemical and photochemical inertness make it particularly attractive. The spectral compatibility of the indicator (in its protonated form) and the referenced phosphor is very good (Figure 4) because both are efficiently excited by red LEDs and emit in the same spectral window.
Figure 4.

Spectral properties of the luminescent materials used in the pH sensor: (a) excitation spectrum of the Cr-GAB and pH indicator 1 in protonated form; (b) emission spectra of Cr-GAB and pH indicator 1; (c) calibration plot for the DLR pH sensor containing pH indicator 1 and Cr-GAB in hydrogel D4 at 25 °C (IS = 0.02 M).
The resulting pH-sensing material is compatible with a commercially available fiber-optic phase oxygen meter (Firesting from Pyroscience), which was used for measurements (λexc ∼ 620 nm). The “sensing chemistry” applied on the tip of a glass optical fiber included 0.1% w/w of indicator 1 and 66% w/w Cr-GAB particles and 33% w/w of hydrogel D4. Different concentrations of indicator and Cr-GAB particles were tested to ensure optimal phase shift dynamics (data not shown). Figure 4c shows a calibration plot of the cotangents of the phase angle, cot Φ vs pH for the DLR pH-sensing material. The calibration showed an inflection point at pH 8.12, which is very close to the apparent pKa value obtained from the spectroscopic investigations. Photostability of the indicator at the fiber optic tip was monitored by applying 5 times stronger light intensity and 10 times longer integration times than in the standard settings. For the acidic form, no evident photobleaching was observed after 2000 measurement points, which corresponds to 100 000 measurement points in the standard settings (Figure S7 of ESI). A very small drift of the phase angle (0.2°) was observed in the basic conditions for the same measurement time. Nevertheless, it can be concluded that the fiber-optic sensors possess excellent photostability and can be used for prolonged measurements without recalibration.
Application in Marine Biology
The new pH sensing materials operate in significantly different dynamic ranges and are, therefore, suitable for a variety of important applications. For example, the apparent pK′a values of the indicators 2 and 4 make them particularly suitable for biotechnological and biological applications, respectively. On the other side, the apparent pK′a of about 8 found for indicator 1 almost ideally matches the pH of the seawater. This new sensing material thus provides a promising alternative to the state-of-the-art optical sensors for seawater pH measurements, which rely on fluorescein derivatives and possess poor photostability.40,41 We demonstrated this applicability in marine biology by measuring the pH in corals with a fiber-optic pH optode based on the DLR material described above (Figure 5). The pH in corals is affected by both the photosynthetic activity of the algal symbionts in the host tissue, which tend to increase the pH due to their phototsynthetic fixation of inorganic carbon, and the respiratory activity of the coral host cells and hetrotrophic bacteria associated with the coral, the activity of which tends to decrease pH. Although most pH measurements in corals have focused on the exposed tissues,42 the pH conditions in the gastric cavity (i.e., the coral stomach) are largely unknown; yet, such measurements are highly relevant to understand the microbial processes going on inside corals. Our pH measurements showed a pH gradient of almost 1 pH unit from the coral mouth and into the deepest part of the coral just above the calcium carbonate skeleton (Figure 5b). Evidently, the pH in these parts of the coral was not affected by photosynthetic activity, which was also seen by the absence of any accumulation of O2 (data not shown).
Figure 5.
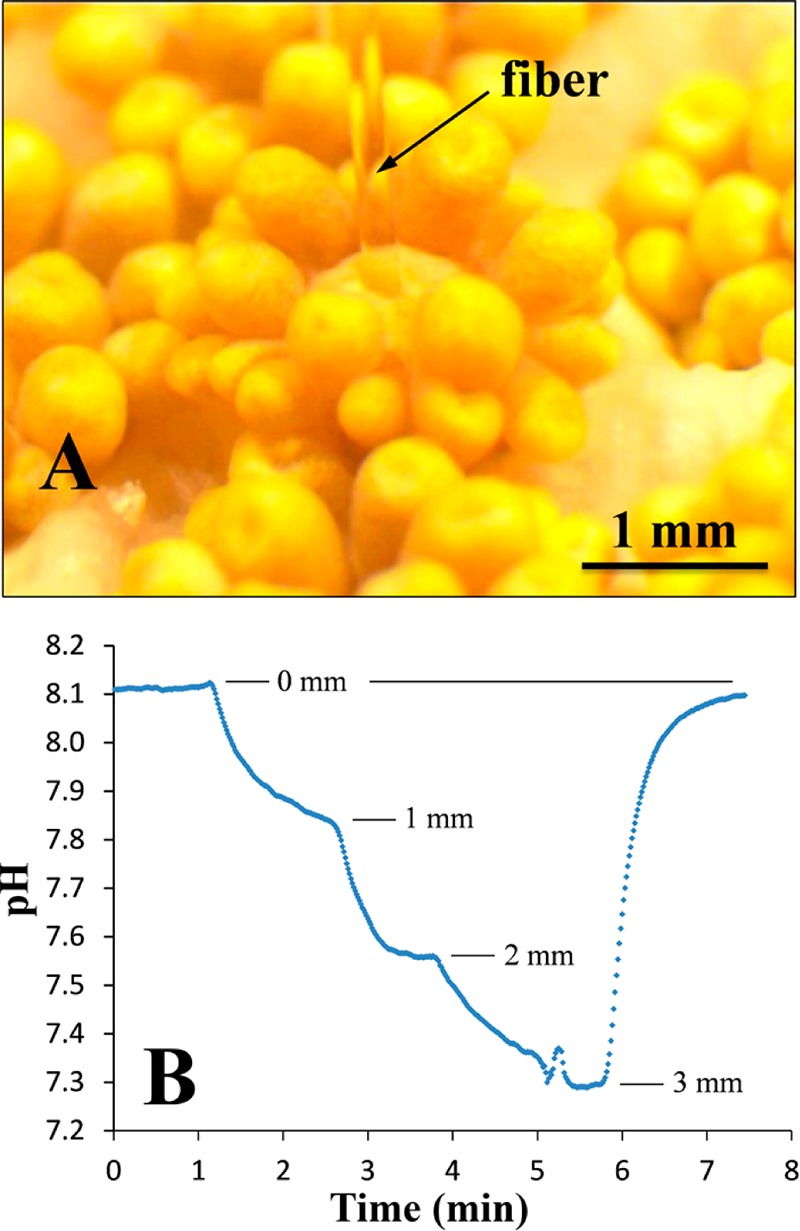
(A) Photographic image of a fiber-optic pH optode based on 1 and Cr-GAB inserted into the mouth opening of a single polyp in the coral (Goniopora sp.) under an irradiance of ∼150 μmol photons m–2 s–1. (B) pH profile in the gastric cavity of the coral showing a pH decrease at increasing distance from the mouth opening. After measurement at 3 mm depth, the pH optode was retracted into the overlaying seawater.
Conclusion
In conclusion, we prepared and characterized a series of NIR fluorescent pH indicators with potential to suit a diverse range of analytical applications. pH-sensitive functional aza-BODIPY derivatives were obtained via a simple reaction route starting from commercially available compounds. Except for probes 5 and 6, the new indicators retain the advantages of aza-BODIPY fluorescent probes, such as good brightness and excellent photostability. Variations in both the substitution pattern and the position of hydroxyl functionality allowed manipulation of pKa values over a wide range, providing valuable information that can be used for future rational design of the indicator systems. The new sensors have high potential for a variety of biotechnological, biological, environmental, etc. applications. As an example, monitoring of pH inside coral polyps was demonstrated. Ongoing work concerns the covalent immobilization of the indicators into the polymeric network, which will completely suppress their leaching, migration, and aggregation and can lead to even better shelf life stability and operating time of the optical sensors.
Acknowledgments
This study was supported by the CHEBANA Marie Curie Initial Training Network funded by the European Commission under the 7th Framework Programme (Grant Agreement No. 264772) and Austrian Science Fund FWF (Project No. P21192-N17). Additional support was due to the Danish Council for Independent Research Natural Sciences (M.K.). We thank Dr. Roland Fischer (Institute of Inorganic Chemistry, Graz University of Technology) for crystallographic study of compound 8.
Supporting Information Available
Details on coral experiments, synthetic procedures, 1H NMR and MS spectra, absorption and emission spectra, calibration curves. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Supplementary Material
References
- Valeur B.Molecular Fluorescence. Principles and Applications; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Haugland R. P.The Handbook. A Guide to Fluorescent Probes and Labeling Technologies, 10th ed.; Molecular Probes: Eugene, Oregon, 2005. [Google Scholar]
- Han J.; Burgess K. Chem. Rev. 2010, 110, 2709. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Tang B.; Liu X.; Liu Y.; Xu K.; Ma J.; Tong L.; Yang G. Analyst 2009, 134, 367. [DOI] [PubMed] [Google Scholar]
- Tang B.; Yu F.; Li P.; Tong L.; Duan X.; Xie T.; Wang X. J. Am. Chem. Soc. 2009, 131, 3016. [DOI] [PubMed] [Google Scholar]
- Almutairi A.; Guillaudeu S. J.; Berezin M. Y.; Achilefu S.; Frechet J. M. J. J. Am. Chem. Soc. 2008, 130, 444. [DOI] [PubMed] [Google Scholar]
- Lee H.; Berezin M. Y.; Guo K.; Kao J.; Achilefu S. Org. Lett. 2009, 11, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Achilefu S. Chem. Commun. 2005, 5887. [DOI] [PubMed] [Google Scholar]
- Deniz E.; Isbasar G. C.; Bozdemir O. A.; Yildirim L. T.; Siemiarczuk A.; Akkaya E. U. Org. Lett. 2008, 10, 3401. [DOI] [PubMed] [Google Scholar]
- Hilderbrand S. A.; Kelly K. A.; Nieder M.; Weissleder R. Bioconjugate Chem. 2008, 19, 1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl M. Methods Enzymol. 2005, 397, 166. [DOI] [PubMed] [Google Scholar]
- Larsen M.; Borisov S. M.; Grunwald B.; Klimant I.; Glud R. N. Limnol. Oceanogr. Methods 2011, 9, 348. [Google Scholar]
- Jeevarajan A. S.; Vani S.; Taylor T. D.; Anderson M. M. Biotechnol. Bioeng. 2002, 78, 467. [DOI] [PubMed] [Google Scholar]
- John G. T.; Goelling D.; Klimant I.; Schneider H.; Heinzle E. J. Dairy Res. 2003, 70, 327. [DOI] [PubMed] [Google Scholar]
- Wolfbeis O. S.; Fürlinger E.; Kroneis H.; Marsoner H. Fresenius' J. Anal. Chem. 1983, 314, 119. [Google Scholar]
- Whitaker J. E.; Haugland R. P.; Prendergas F. G. Anal. Biochem. 1991, 194, 330. [DOI] [PubMed] [Google Scholar]
- Offenbacher H.; Wolfbeis O. S.; Fürlinger E. Sens. Actuators, B 1986, 9, 73. [Google Scholar]
- Zhujun Z.; Seitz W. R. Anal. Chim. Acta 1984, 160, 47. [Google Scholar]
- Xu Z.; Rollins A.; Alcala R.; Marchant E. J. Biomed. Mater. Res. 1998, 39, 9. [DOI] [PubMed] [Google Scholar]
- Weidgans B. M.; Krause C.; Klimant I.; Wolfbeis O. S. Analyst 2004, 129, 645. [DOI] [PubMed] [Google Scholar]
- Borisov S. M.; Gatterer K.; Klimant I. Analyst 2010, 135, 1711. [DOI] [PubMed] [Google Scholar]
- Cooper M. E.; Gregory S.; Adie E.; Kalinka S. J. Fluoresc. 2002, 12, 425. [Google Scholar]
- Briggs M. S.; Burns D. D.; Cooper M. E.; Gregory S. J. Chem. Commun. 2000, 23, 2323. [Google Scholar]
- Killoran J.; Allen L.; Gallagher J. F.; Gallagher W. M.; O’Shea D. F.. Chem. Commun., 2002, 1862. [DOI] [PubMed] [Google Scholar]
- Killoran J.; McDonnell S. O.; Gallagher J. F.; O’Shea D. F. New J. Chem. 2008, 32, 483. [Google Scholar]
- Murtagh J.; Frimannsson D. O.; O’Shea D. F. Org. Lett. 2009, 11, 5386. [DOI] [PubMed] [Google Scholar]
- McDonnell S. O.; O’Shea D. F. Org. Lett. 2006, 8, 3493. [DOI] [PubMed] [Google Scholar]
- Borisov S. M.; Gaterrer K.; Bitschnau B.; Klimant I. J. Phys. Chem. C 2010, 114, 9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas J. N.; Crosby G. A. J. Phys. Chem. 1971, 75, 991. [Google Scholar]
- Freyer W.; Mueller S.; Teuchner K. J. Photochem. Photobiol. A 2004, 163, 231. [Google Scholar]
- Hall M. J.; McDonnell S. O.; Killoran J.; O’Shea D. F. J. Org. Chem. 2005, 70, 5571. [DOI] [PubMed] [Google Scholar]
- Gorman A.; Killoran J.; O’Shea C.; Kenna T.; Gallagher W. M.; O’Shea D. F. J. Am. Chem. Soc. 2004, 126, 10619. [DOI] [PubMed] [Google Scholar]
- Loudet A.; Rakeshwar B.; Wu L.; Burgess K. Tetrahedron 2008, 64, 3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs N. S.Physical Organic Chemistry, 2nd ed.; Longman Scientific & Technical: Harlow, England, 1995, p 188. [Google Scholar]
- Daffy L. M.; de Silva A. P.; Gunaratne H. Q. M.; Huber C.; Lynch P. L. M.; Werner T.; Wolfbeis O. S. Chem.—Eur. J. 1998, 4, 1810. [Google Scholar]
- Werner T.; Huber C.; Heinl S.; Kollmannsberger M.; Daub J.; Wolfbeis O. S. Fresenius' J. Anal. Chem. 1997, 359, 150. [Google Scholar]
- de Silva A. P.; Vance T. P.; West M. E. S.; Wright G. D. Org. Biomol. Chem. 2008, 6, 2468. [DOI] [PubMed] [Google Scholar]
- Batat P.; Cantuel M.; Jonusauskas G.; Scarpantonio L.; Palma A.; O’Shea D. F.; McClenaghan N. D. J. Phys. Chem. A 2011, 115, 14034. [DOI] [PubMed] [Google Scholar]
- Huber C.; Klimant I.; Krause C.; Wolfbeis O. S. Anal. Chem. 2001, 73, 2097. [DOI] [PubMed] [Google Scholar]
- Schröder C. R.; Weidgans B. M.; Klimant I. Analyst 2005, 130, 907. [DOI] [PubMed] [Google Scholar]
- Schröder C. R.; Polerecky L.; Klimant I. Anal. Chem. 2007, 79, 60. [DOI] [PubMed] [Google Scholar]
- Kühl M.; Cohen Y.; Dalsgaard T.; Jørgensen B. B.; Revsbech N. P. Mar. Ecol.: Prog. Ser. 1995, 117, 159. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.