

Abstract
Modulation of co-inhibitory and co-stimulatory receptors of the immune system has become a promising new approach for immunotherapy of cancer. With the recent FDA approval of CTLA-4 blockade serving as an important proof of principal, many new targets are now being translated into the clinic. Preclinical research has demonstrated that targeting glucocorticoid-induced tumor necrosis factor (TNF) receptor related gene (GITR), a member of TNF receptor superfamily, by agonist antibodies or natural ligand, can serve as an effective anti-tumor therapy. In this review, we will cover this research and the rationale that has led to initiation of two phase 1 clinical trials targeting GITR as a new immunotherapeutic approach for cancer.
Introduction
Although evidence has suggested for well over 100 years that the immune system could detect and eliminate neoplastic growth, immunotherapeutic approaches have only very recently become an option for cancer therapy. Immunotherapy has now reached a new level of application since the first approved immunotherapies (adjuvant IFNα and high dose IL-2 monotherapy) with the recent FDA approval of the CTLA-4 blocking antibody ipilimumab (discussed in this issue by Allison and Sharma). While ipilimumab is the first agent to ever demonstrate improved overall survival in patients with advanced metastatic melanoma, additional options are needed to extend therapeutic benefit beyond the 20–30% of patients who have durable disease control with CTLA-4 blockade. Fortunately, the validation of checkpoint blockade as a viable cancer therapy has added new vigor to the development of additional immunotherapies. Blockade of co-inhibitory checkpoint PD-1 and its ligand (PD-L1, B7-H1) along with agonistic therapies of the co-stimulatory tumor necrosis factor (TNF) receptor family members OX40 and 4-1BB have already demonstrated promise in early phase trials. In this review, we will discuss an additional pathway of immune modulation through activation of glucocorticoid-induced TNF receptor related gene or ‘GITR’. This additional target was listed by the National Cancer Institute in 2006 as the 12th most promising immunotherapy for cancer and two phase 1 trials modulating GITR have opened in the past year. Below we will discuss the role of GITR in the immune system along with the evidence of immunotherapeutic potential, which has supported translation of GITR ligation therapy into the clinic.
GITR is a co-stimulatory receptor
GITR was originally discovered by Nocentini et al. as a gene upregulated in dexamethasone-treated murine T cell hybridomas [1]. The human ortholog was subsequently characterized in human lymphocytes and shown to share 55% identity with murine GITR. Although dexamethasone treatment played a role in the discovery of GITR, it was subsequently shown that glucocorticoid treatment has no impact on GITR expression in human cells and is unnecessary in mice [2,3]. GITR has low basal expression on naïve murine CD4+ and CD8 T+ cells, and very low expression on human T cells, similar to TNFR family members 4-1BB and OX40 [4-7]. This is in contrast to murine and human regulatory T cells (Tregs) which constitutively express GITR and to varying degrees OX40 and 4-1BB. Upon activation, naïve T cells and Tregs upregulate GITR 24–72 h after an initial stimulus, with expression lasting several days [8] (Table 1). This delayed expression pattern on effector T cells (Teff) somewhat mirrors 4-1BB and OX40 and suggests that GITR does not play a predominant role in early T cell priming, but rather exerts its effects at later time points [9]. In fact, GITR knockout mice have intact T cell development and display relatively normal priming [10]. Consistent with the ligands of OX40 and 4-1BB, GITR ligand (GITR-L) is expressed at low levels by antigen-presenting cells such as macrophages, dendritic cells (DCs), and B cells and is upregulated upon activation [7,8,11•,12]. GITR-L has also been found on endothelial cells and activated T cells; however, the role GITR-L expression plays on these cells is unclear [13]. Like most TNFR family members, human GITR-L binds GITR in a trimeric fashion while the murine GITR:GITR-L interaction is thought to be dimeric [14,15]. Currently, the significance of the differential ligand binding between human and murine GITR–GITR-L and whether it translates into differential functions of the receptor has not been described.
Table 1.
GITR is expressed on many immune cell types and is often upregulated upon activation.
| Cell type | GITR expression
|
|
|---|---|---|
| Naïve | Activated | |
| Regulatory T cells | High | Very high |
| T cells (CD4/CD8) | Intermediate | High |
| NK cells | Intermediate | High |
| Granulocytes | Intermediate | High |
| Mast cells | Intermediate | Intermediate |
| Eosinophils | Intermediate/low | |
| Basophils | Intermediate/low | |
| Monocytes/macrophages | Low | Intermediate |
Multiple studies have shown that GITR can provide a co-stimulatory signal to both CD4+ and CD8+ naïve T cells, enhancing proliferation and effector function, particularly in the setting of suboptimal T cell receptor (TCR) stimulation [5,8,16,17]. Additionally, GITR−/− T cells are more prone to activation-induced cell death (AICD), suggesting that GITR stimulus may protect T cells from AICD. Conversely, in the setting of very strong TCR stimulus, GITR ligation can actually enhance AICD of CD4+ effector T cells [8,16,18].
Signaling downstream of GITR results in the activation of NF-κB and along with members of the MAPK pathway including p38, JNK and ERK [1,5,19]. The downstream events in turn are believed to enhance T cell survival by upregulating IL-2Rα, IL-2 and IFNγ [5]. Although the three cysteine-rich extracellular domains and cytoplasmic tail of GITR share significant homology with 4-1BB and OX40, recent research has demonstrated that GITR signals through different TNF receptor associate factor (TRAF) proteins [20,21••]. Meticulously reviewed by Snell et al., GITR signals through a trimer consisting of a single TRAF5 and a two TRAF2 proteins [11•]. This was found to contrast with 4-1BB which combines a TRAF1 with two TRAF2s [11•]. How the differential use of TRAF1 and TRAF5 results in the non-redundant roles of GITR and 4-1BB has yet to be determined.
Role in immune activation
The lack of a profound phenotype in GITR−/− mice had for some time confounded the understanding of the physiological role of GITR in the immune system. Recently, research has begun to elucidate its role comparing the immune response to various pathogens. Studies of murine responses to influenza found that GITR−/− mice were able to resist a mild challenge of influenza X31, but succumbed to infection with the more virulent influenza A/PR8 [21••]. Even though GITR−/− mice could reject the X31 infection, GITR−/− CD8+ cells displayed reduced expansion and survival compared to wild type CD8+ T cells. Interestingly, lack of GITR did not appear to change the percentage of CD8+ T cells that became IFNγ+ [21••]. This finding was similar to a previous one by the same group demonstrating reduced survival of 4-1BB−/− mice challenged with the PR8 strain [22•]. Thus, although GITR and 4-1BB may be similar, they seem to serve non-redundant roles. Both appear to be equally important for enhancing T cell persistence necessary to reject complex immune challenges. Different from CD8-mediated influenza responses, GITR does not play a major role in CD4+ Th2 responses to Helminth infection. While the GITR agonist antibody DTA-1 was able to enhance typical Th2 responses, blockade of the GITR:GITR-L interactions in vivo did not appear to alter Th2 priming [23]. Furthermore, the ability of GITR to enhance Th2 responses was short-lived, whereas Th1 responses remained elevated 60 days after treatment [23]. Studies of several inducible inflammatory disease models have provided additional clues to how GITR stimulus may normally be intercalated during activation of the immune response. GITR−/− mice have less severe phenotypes in models of splanchnic artery occlusion shock, collagen-induced arthritis, bleomycin-induced and carrageenan-induced lung injury, TNBS-induced colitis, and cerulein-induced pancreatitis [24-30]. In these extreme models of inflammatory diseases, GITR−/− mice exhibited decreased levels of proinflammatory cytokines in the affected tissues along with reduced expression of adhesion molecules such as ICAM-1, P-selectin, and E-selectin on the vasculature [25,26,29]. This in turn appears to alter the recruitment of innate immune cells to sites of tissue damage. Taken together, current evidence suggests that GITR may play a role skewing immune responses toward Th1 instead of Th2 (Figure 1).
Figure 1.
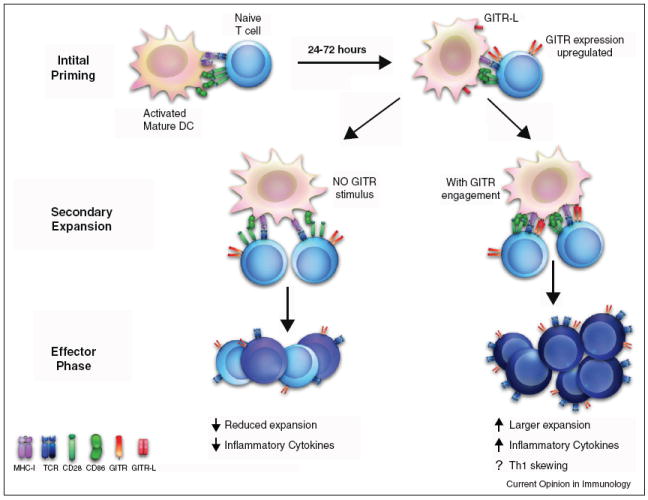
Model of GITR function during an immune response. During the initial phase of priming, naïve T cells are activated by interactions between the TCR and MHC molecules, receiving co-stimulation through CD28 binding to either CD80 or CD86. After passing this checkpoint, activated T cells enter secondary rounds of priming and expansion, upregulating GITR 24–72 h after this initial activation. If GITR-L is expressed by DCs, it alters both the quality and the quantity of the ensuing immune response. The net result of which is enhanced inflammatory response, with increased persistence of the antigen-specific T cells.
In addition to T cells, NK cells express high levels of GITR. Studies investigating the roles of GITR, OX40, and 4-1BB on NK cells have shown that engagement of these TNFR members leads to increased cytotoxicity and IFNγ production [31,7,32]. However, a consensus for the role of GITR in NK cells has not been reached. In one study, human NK cells exhibited increased cytotoxicity and cytokine production when cultured with target cells transfected with GITR-L [33]. Conversely another group showed that an anti-human GITR agonist antibody inhibited proliferation and cytokine production in human NK cells [34]. Subsequent research using the same anti-human GITR antibody has confounded the story further, showing that NK cell-mediated lysis of GITR-L expressing human tumor cells was increased [35]. Though the later work was attributed to blocking of GITR–GITR-L ligand interaction by the antibody, it is unclear if this was due to blockade or stimulation of GITR. Further studies are needed to fully determine the role GITR in the NK cell-mediated immune response. It is entirely possible that the GITR–GITR-L interaction has opposite effects on mouse and human NK cells, as has been shown for 4-1BB [36].
Modulation of GITR for tumor immunotherapy
Interest in GITR as a target for cancer immunotherapy developed from reports showing that agonist anti-GITR antibodies (the rat monoclonal DTA-1 and goat polyclonals) could overcome self-tolerance and reverse in vitro Treg suppression [37,38]. Originally, both groups attributed this to direct effects on Tregs; however, it was subsequently shown with the use of GITR−/− T cells that GITR ligation worked primarily by making Teffs resistant to Treg suppression [39•].
In collaboration with Shimon Sakaguchi, our group was the first to demonstrate that DTA-1 could be used for immunotherapy in a model of concomitant immunity to B16 melanoma. Unlike other models, secondary B16 tumor challenge tumors cannot be rejected during the early stages of primary tumor growth, unless CD4+CD25+ Tregs are depleted [40,41•]. Consistent with the ability of DTA-1 to interfere with Treg suppression in vitro, 100% of mice treated with DTA-1 (0 and 7 days after primary B16 tumor challenge) rejected the secondary tumor challenge [41•]. We recently expanded on this observation, demonstrating that a single dose of DTA-1 could cure small established B16 tumors when administered 4 days after challenge [42••]. Additionally, the Sakaguchi group independently reported that a single treatment with DTA-1 could cure 8-day established Meth-A sarcomas [43]. The effect of DTA-1 on tumor immunity was long-lived, as mice were resistant to a secondary challenge 70 days later [43]. Therapeutic effects of DTA-1 have also been shown in multiple tumor models including CT26 colon tumors, A20 lymphoma and various vaccine models discussed below [44,45] (Cohen A, unpublished).
GITR can also be modulated through the therapeutic use of GITR-L; however, most evidence in murine models suggests that this approach works best when combined with tumor antigen vaccination and not as monotherapy. Boczkowsk et al. observed that vaccination with DCs expressing melanocyte differentiation antigen TYRP-2 provided only a short delay in tumor growth after B16 tumor challenge. When combined with DCs engineered to secrete either a GITR-L-Fc fusion protein or DTA-1, tumor survival was enhanced to 50% [46••]. Likewise, combining DNA vaccines encoding tumor antigens from the CMS5 sarcoma with DNA encoding GITR-L dramatically delayed tumor growth [47•]. In a similar fashion, DTA-1 improved protection of xenogeneic DNA vaccines against B16 melanoma from 20–40% to 80–100% [48]. DTA-1 can also extend the effectiveness of prophylactic vaccines into therapeutic settings. Treatment with an adenovirus HPV vaccine alone only causes temporary regression of TC-1 tumors, with 90% of tumors reappearing [49]. The addition of DTA-1 8 days following vaccination prevented tumor recurrence [49]. These additive effects have also been demonstrated when DTA-1 was combined with adoptive T cell therapy or CTLA-4 blockade, synergistically enhancing tumor regression of CMS5 fibrosarcoma and CT26 tumors, respectively [43,50•,51]. Pruitt et al. has recently shown that addition of DCs expressing anti-CTLA-4 to their original GITR-L and TYRP-2 expressing DC vaccine further enhanced the efficacy of this approach [52•].
Mechanisms of GITR tumor immunotherapy
The extensive studies on GITR immunotherapy in tumor models have helped develop a model of how GITR modulation enhances anti-tumor immune responses. As might be expected, depletion of CD8+ and CD4+ T cells abrogates or reduces the effects of both GITR-L and DTA-1 on tumor growth in CMS5, CT26, A20 and melanoma tumor models, with NK or NKT depletion having an effect in some models as well [44,47•,53]. Although a role for the humoral anti-tumor response has not been demonstrated, B cells, possibly as APCs, appear critical for the DTA-1-mediated anti-CT26 response [54]. In most cases, GITR ligation results in enhanced effector responses as measured by IFNγ and activation phenotypes [42••,47•]. Additionally, DTA-1 appears to enhance the polyclonality of the tumor antigen-specific response [53,55•]. It is interesting to note that in the setting of vaccination, immunization with concurrent DTA-1 results in reduced responses compared to use of GITR-L [47•]. Our group has observed a detrimental effect on tumor protection if DTA-1 is given together with the first of three xenogeneic melanoma vaccines, instead of being delayed until the second vaccination [48]. Likewise, when administered on the day of tumor challenge, DTA-1 provides only ~30% tumor-free survival with B16 melanoma, compared to 80% when a single dose was administered at day 4 following challenge [42••]. This shows that the method, timing and strength of GITR ligation can dramatically influence the outcome.
The question of whether GITR ligation directly affects Tregs has remained a source of contention. We and others have demonstrated that Tregs isolated from the periphery of mice treated with DTA-1 or GITR-L remain suppressive ex vivo [42••,47•]. Instead, antigen-specific CD8+ T cells treated in vivo with GITR-L were resistant to Treg suppression during subsequent co-culture, without additional ex vivo GITR stimulation [47•]. A recent study also showed that DTA-1-mediated concomitant immunity was maintained in Teff and Treg reconstituted Rag−/− mice even when donor Tregs came from GITR−/− mice, suggesting that DTA-1 exerts its protective effects primarily through modulation of Teff cells [55•]. However, it is evident that GITR, like OX40, can have different effects on intra-tumor and peripheral Tregs [42••,56,57]. Agonist OX40 therapy had been shown to cause loss of intra-tumor Tregs in combination with chemotherapy, and also to directly alter intra-tumor Treg suppressive function [57,58]. While GITR has not been directly shown to alter in vivo Treg suppression in a similar fashion, we have shown that treatment with DTA-1 can cause intra-tumor Tregs to become unstable and lose expression of Foxp3, which is required for their suppressive ability [42••]. This results in an enhanced intra-tumor Teff:Treg ratio, correlating with therapeutic benefit, similar to what has been reported with OX40 and CTLA-4, [42••,57,59]. A recent report by Coe et al. has suggested that DTA-1 directly depletes Tregs as part of its mechanism. We believe that the phenotype described may be related to Treg stability, as we have not seen evidence of antibody-mediated depletion in our experiments [42••,60] (Schaer, unpublished). Still, the effect of DTA-1 on Tregs appears to be important. RAG−/− reconstitution experiments in the DTA-1 monotherapy setting, in contrast to the concomitant immunity model, show that optimal treatment of melanoma requires both Teffs and Tregs to express GITR [42••]. Thus, while it is clear in some models that GITR ligation primarily modulates Teffs, there is developing evidence that effects on Tregs, which express GITR constitutively, may also play an important role (Figure 2).
Figure 2.
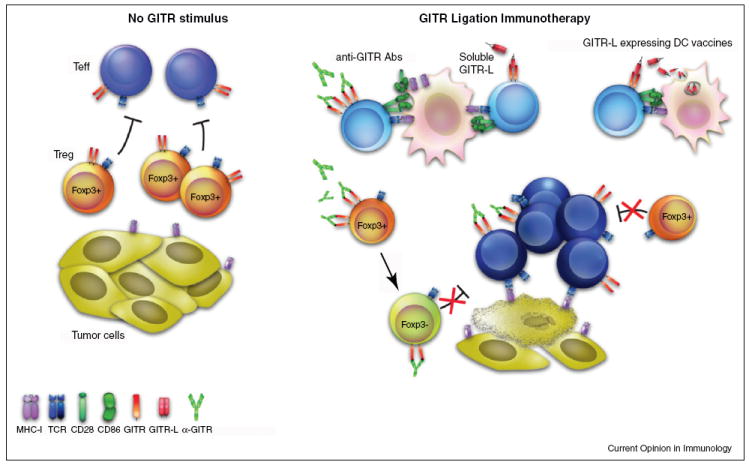
Modulating GITR to enhance immunotherapy. Under steady-state conditions, this immune response is insufficient to mediate tumor regression. Teff generated are unable to overcome an inhibitory tumor environment and remain vulnerable to regulatory T cell (Treg)-mediated suppression. Provision of additional GITR stimulus during the secondary stages of priming and expansion, through agonist anti-GITR antibody, soluble GITR ligand or DC vaccines expressing soluble GITR modulates the outcome of both Teff and Treg tumor responses. Teffs demonstrate greater proliferation, effector function, and resistance to suppression, while Tregs show decreased tumor infiltration and stability, with loss of foxp3 expression. This results in augmented intra-tumor Teff:Treg ratio which favors tumor immunity and regression.
The road ahead; translating GITR immunomodulation to the clinic
In collaboration with Tolerx Inc, we opened a phase I dose escalation trial in melanoma testing the safety of the humanized anti-human GITR mAb, TRX518. TRX518 has been shown to have similar agonist activity on human T cells compared to DTA-1 on murine T cells in vitro [61,62]. Duke University has also opened a phase I trial in melanoma patients testing DC vaccines alone or in combination with DCs expressing GITR-L, anti-CTLA-4 or both. Pruitt et al. have recently demonstrated the ability of this approach to enhance the generation of anti-tumor CTLs from human PBMCs in vitro to melanoma or breast cancer antigens [52•]. Safety is a primary concern in consideration of experiences with OX-40 and 4-1BB antibodies which have already entered the clinic. The phase I trial of mouse anti-human OX40 completed in 2009 was reported to have low toxicity; however trials of 4-1BB agonist antibodies were put on hold after the occurrence of fatal hepatic adverse events [63,64•]. Although GITR therapy can aggravate mice predisposed to autoimmune diseases including colitis, diabetes and experiential autoimmune encephalitis, when DTA-1 has been used in mice that are not predisposed to autoimmunity, the same effect is not generally seen [17,42••,43,46••,65-67].
Conclusion
Preclinical evidence has demonstrated that signaling through GITR can enhance activation of Teff and modulate the activity of intra-tumor Tregs during immunotherapy. The preservation of peripheral Treg function during GITR agonist therapy may make widespread loss of tolerance and immune related adverse events resulting from in vivo GITR ligation less likely. However, differences in both the physical structure (trimer vs. dimer) and the cellular expression pattern between human and murine GITR make it difficult to predict the exact in vivo results. In consideration of this and the valuable clinical lessons of 4-1BB agonist antibodies and co-inhibitory blockade of CTLA-4, we have entered the phase 1 GITR trials with heightened vigilance regarding immune-related adverse events. Only through the current clinical trials together with careful immune monitoring will we learn whether targeting GITR will emerge as a therapeutic option for treating human cancer.
Acknowledgments
This work was supported by NIH grants R01CA56821 and P01CA59350, Swim Across America, Melanoma Research Alliance, Cancer Research Institute, Commonwealth Cancer Research Foundation/MSKCC Experimental Therapeutics Center (J.D.W.); D.A.S.: NIH Clinical Training for Scholar Grant K12 CA120121-01, T32 CA09149-30, John D. Proctor Foundation: Margaret A. Cunningham Immune Mechanisms in Cancer Research Fellowship Award.
Footnotes
Conflict of interest statement
All authors declare no conflict of interest related to this work.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
- 1.Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, Migliorati G, Riccardi C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:6216–6221. doi: 10.1073/pnas.94.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwon B, Yu KY, Ni J, Yu GL, Jang IK, Kim YJ, Xing L, Liu D, Wang SX, Kwon BS. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J Biol Chem. 1999;274:6056–6061. doi: 10.1074/jbc.274.10.6056. [DOI] [PubMed] [Google Scholar]
- 3.Gurney AL, Marsters SA, Huang RM, Pitti RM, Mark DT, Baldwin DT, Gray AM, Dowd AD, Brush AD, Heldens AD, et al. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Curr Biol. 1999;9:215–218. doi: 10.1016/s0960-9822(99)80093-1. [DOI] [PubMed] [Google Scholar]
- 4.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 5.Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, Ayroldi E, Riccardi C. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol. 2004;34:613–622. doi: 10.1002/eji.200324804. [DOI] [PubMed] [Google Scholar]
- 6.Pollok K, Kim Y, Zhou Z, Hurtado J, Kim K, Pickard R, Kwon B. Inducible T cell antigen 4-1BB. Analysis of expression and function J Immunol. 1993;150:771–781. [PubMed] [Google Scholar]
- 7.Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annu Rev Immunol. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tone M, Tone Y, Adams E, Yates SF, Frewin MR, Cobbold SP, Waldmann H. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci U S A. 2003;100:15059–15064. doi: 10.1073/pnas.2334901100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawicki W, Watts TH. Expression and function of 4-1BB during CD4 versus CD8 T cell responses in vivo. Eur J Immunol. 2004;34:743–751. doi: 10.1002/eji.200324278. [DOI] [PubMed] [Google Scholar]
- 10.Ronchetti S, Nocentini G, Riccardi C, Pandolfi PP. Role of GITR in activation response of T lymphocytes. Blood. 2002;100:350–352. doi: 10.1182/blood-2001-12-0276. [DOI] [PubMed] [Google Scholar]
- 11•.Snell LM, Lin GHY, McPherson AJ, Moraes TJ, Watts TH. T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy. Immunol Rev. 2011;244:197–217. doi: 10.1111/j.1600-065X.2011.01063.x. This very comprehensive review covers in depth the basic molecular functions of both 4-1BB and GITR, along with comparing and contrasting the roles of these closely related TNFR family members during immune responses. [DOI] [PubMed] [Google Scholar]
- 12.Suvas S, Kim B, Sarangi PP, Tone M, Waldmann H, Rouse BT. In vivo kinetics of GITR and GITR ligand expression and their functional significance in regulating viral immunopathology. J Virol. 2005;79:11935–11942. doi: 10.1128/JVI.79.18.11935-11942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nocentini G, Riccardi C. In: GITR: A Modulator of Immune Response and Inflammation Therapeutic Targets of the TNF Superfamily. Grewal IS, editor. New York: Springer; 2009. pp. 156–173. [DOI] [PubMed] [Google Scholar]
- 14.Chattopadhyay K, Ramagopal UA, Brenowitz M, Nathenson SG, Almo SC. Evolution of GITRL immune function: murine GITRL exhibits unique structural and biochemical properties within the TNF superfamily. Proc Natl Acad Sci U S A. 2008;105:635–640. doi: 10.1073/pnas.0710529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, Tone Y, Song X, Furuuchi K, Lear JD, Waldmann H, Tone M, Greene MI, Murali R. Structural basis for ligand-mediated mouse GITR activation. Proc Natl Acad Sci U S A. 2008;105:641–645. doi: 10.1073/pnas.0711206105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanamaru F, Youngnak P, Hashiguchi M, Nishioka T, Takahashi T, Sakaguchi S, Ishikawa I, Azuma M. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol. 2004;172:7306–7314. doi: 10.4049/jimmunol.172.12.7306. [DOI] [PubMed] [Google Scholar]
- 17.Kohm AP, Williams JS, Miller SD. Cutting edge: ligation of the glucocorticoid-induced TNF receptor enhances autoreactive CD4+ T cell activation and experimental autoimmune encephalomyelitis. J Immunol. 2004;172:4686–4690. doi: 10.4049/jimmunol.172.8.4686. [DOI] [PubMed] [Google Scholar]
- 18.Muriglan SJ, Ramirez-Montagut T, Alpdogan O, Van Huystee TW, Eng JM, Hubbard VM, Kochman AA, Tjoe KH, Riccardi C, Pandolfi PP, et al. GITR activation induces an opposite effect on alloreactive CD4(+) and CD8(+) T cells in graft-versus-host disease. J Exp Med. 2004;200:149–157. doi: 10.1084/jem.20040116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esparza EM, Arch RH. Glucocorticoid-induced TNF receptor functions as a costimulatory receptor that promotes survival in early phases of T cell activation. J Immunol. 2005;174:7869–7874. doi: 10.4049/jimmunol.174.12.7869. [DOI] [PubMed] [Google Scholar]
- 20.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 21••.Snell LM, McPherson AJ, Lin GHY, Sakaguchi S, Pandolfi PP, Riccardi C, Watts TH. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol. 2010;185:7223–7234. doi: 10.4049/jimmunol.1001912. Using GITR+/+ and GITR−/− T cells, Snell et al. elucidated the natural role GITR plays during anti-viral immune responses and demonstrate the specific TRAF family members responsible for downstream GITR signaling. [DOI] [PubMed] [Google Scholar]
- 22•.Lin GHY, Sedgmen BJ, Moraes TJ, Snell LM, Topham DJ, Watts TH. Endogenous 4-1BB ligand plays a critical role in protection from influenza-induced disease. J Immunol. 2009;182:934–947. doi: 10.4049/jimmunol.182.2.934. Demonstrating a phenotype similar to GITR−/− anti-viral responses, Watts’ group shows that GITR and 4-1BB have important non-redundant roles. [DOI] [PubMed] [Google Scholar]
- 23.van der Werf N, Redpath SA, Phythian-Adams AT, Azuma M, Allen JE, Maizels RM, MacDonald AS, Taylor MD. Th2 responses to helminth parasites can be therapeutically enhanced by, but are not dependent upon, GITR–GITR ligand costimulation in vivo. J Immunol. 2011;187:1411–1420. doi: 10.4049/jimmunol.1100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuzzocrea S, Ayroldi E, Di Paola R, Agostini M, Mazzon E, Bruscoli S, Genovese T, Ronchetti S, Caputi AP, Riccardi C. Role of glucocorticoid-induced TNF receptor family gene (GITR) in collagen-induced arthritis. FASEB J. 2005;19:1253–1265. doi: 10.1096/fj.04-3556com. [DOI] [PubMed] [Google Scholar]
- 25.Cuzzocrea S, Nocentini G, Di Paola R, Agostini M, Mazzon E, Ronchetti S, Crisafulli C, Esposito E, Caputi AP, Riccardi C. Proinflammatory role of glucocorticoid-induced TNF receptor-related gene in acute lung inflammation. J Immunol. 2006;177:631–641. doi: 10.4049/jimmunol.177.1.631. [DOI] [PubMed] [Google Scholar]
- 26.Cuzzocrea S, Nocentini G, Di Paola R, Mazzon E, Ronchetti S, Genovese T, Muia C, Caputi AP, Riccardi C. Glucocorticoid-induced TNF receptor family gene (GITR) knockout mice exhibit a resistance to splanchnic artery occlusion (SAO) shock. J Leukoc Biol. 2004;76:933–940. doi: 10.1189/jlb.0204110. [DOI] [PubMed] [Google Scholar]
- 27.Cuzzocrea S, Ronchetti S, Genovese T, Mazzon E, Agostini M, Di Paola R, Esposito E, Muia C, Nocentini G, Riccardi C. Genetic and pharmacological inhibition of GITR–GITRL interaction reduces chronic lung injury induced by bleomycin instillation. FASEB J. 2007;21:117–129. doi: 10.1096/fj.06-6611com. [DOI] [PubMed] [Google Scholar]
- 28.Galuppo M, Nocentini G, Mazzon E, Ronchetti S, Esposito E, Riccardi L, Di Paola R, Bruscoli S, Riccardi C, Cuzzocrea S. GITR gene deletion and GITR-FC soluble protein administration inhibit multiple organ failure induced by zymosan. Shock. 2011;36:263–271. doi: 10.1097/SHK.0b013e3182262c48. [DOI] [PubMed] [Google Scholar]
- 29.Galuppo M, Nocentini G, Mazzon E, Ronchetti S, Esposito E, Riccardi L, Sportoletti P, Di Paola R, Bruscoli S, Riccardi C, et al. The glucocorticoid-induced TNF receptor family-related protein (GITR) is critical to the development of acute pancreatitis in mice. Br J Pharmacol. 2011;162:1186–1201. doi: 10.1111/j.1476-5381.2010.01123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santucci L, Agostini M, Bruscoli S, Mencarelli A, Ronchetti S, Ayroldi E, Morelli A, Baldoni M, Riccardi C. GITR modulates innate and adaptive mucosal immunity during the development of experimental colitis in mice. Gut. 2007;56:52–60. doi: 10.1136/gut.2006.091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Placke T, Kopp HG, Salih HR. Glucocorticoid-induced TNFR-related (GITR) protein and its ligand in antitumor immunity: functional role and therapeutic modulation. Clin Dev Immunol. 2010;2010:p239083. doi: 10.1155/2010/239083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, Muller A, Pachynski R, Czerwinski D, Coutre S, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117:2423–2432. doi: 10.1182/blood-2010-08-301945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Hanabuchi S, Watanabe N, Wang YH, Ito T, Shaw J, Cao W, Qin FX, Liu YJ. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL) Blood. 2006;107:3617–3623. doi: 10.1182/blood-2005-08-3419. [DOI] [PubMed] [Google Scholar]
- 34.Liu B, Li Z, Mahesh SP, Pantanelli S, Hwang FS, Siu WO, Nussenblatt RB. Glucocorticoid-induced tumor necrosis factor receptor negatively regulates activation of human primary natural killer (NK) cells by blocking proliferative signals and increasing NK cell apoptosis. J Biol Chem. 2008;283:8202–8210. doi: 10.1074/jbc.M708944200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baltz KM, Krusch M, Bringmann A, Brossart P, Mayer F, Kloss M, Baessler T, Kumbier I, Peterfi A, Kupka S, et al. Cancer immunoediting by GITR (glucocorticoid-induced TNF-related protein) ligand in humans: NK cell/tumor cell interactions. FASEB J. 2007;21:2442–2454. doi: 10.1096/fj.06-7724com. [DOI] [PubMed] [Google Scholar]
- 36.Baessler T, Charton JE, Schmiedel BJ, Grunebach F, Krusch M, Wacker A, Rammensee HG, Salih HR. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood. 2010;115:3058–3069. doi: 10.1182/blood-2009-06-227934. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 38.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 39•.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173:5008–5020. doi: 10.4049/jimmunol.173.8.5008. The use of GITR−/− T cells showed that the DTA-1 modulated Teff cells directly and not Tregs, allowing Teff to resist in vitro Treg suppression. [DOI] [PubMed] [Google Scholar]
- 40.North RJ. Down-regulation of the antitumor immune response. Adv Cancer Res. 1985;45:1–43. doi: 10.1016/s0065-230x(08)60265-1. [DOI] [PubMed] [Google Scholar]
- 41•.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782. doi: 10.1084/jem.20041130. Turk and colleagues were the first to demonstrate GITR’s potential for tumor immunotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, Diab A, Rizzuto G, Duan F, Perales MA, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One. 2010;5:pe10436. doi: 10.1371/journal.pone.0010436. In this manuscript, we show the first evidence that GITR ligation can have direct effects on Tregs during tumor therapy. Optimal therapy required GITR expression on both Tregs and Teff and caused intra-tumor Tregs to lose Foxp3 expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J, Nomura T, Chiba T, Sakaguchi S. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–891. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Houot R, Levy R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood. 2009;113:3546–3552. doi: 10.1182/blood-2008-07-170274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou P, L’Italien L, Hodges D, Schebye XM. Pivotal roles of CD4+ effector T cells in mediating agonistic anti-GITR mAb-induced-immune activation and tumor immunity in CT26 tumors. J Immunol. 2007;179:7365–7375. doi: 10.4049/jimmunol.179.11.7365. [DOI] [PubMed] [Google Scholar]
- 46••.Boczkowski D, Lee J, Pruitt S, Nair S. Dendritic cells engineered to secrete anti-GITR antibodies are effective adjuvants to dendritic cell-based immunotherapy. Cancer Gene Ther. 2009;16:900–911. doi: 10.1038/cgt.2009.39. Here Boczkowski et al. combine DC engineered to express a melanoma vaccine with DCs engineered to express DTA-1 or GITR-L and demonstrate treatment effects similar to injected monoclonal antibody. This technique is the basis of an open trial combining DC vaccines with GITR modulation. [DOI] [PubMed] [Google Scholar]
- 47•.Nishikawa H, Kato T, Hirayama M, Orito Y, Sato E, Harada N, Gnjatic S, Old LJ, Shiku H. Regulatory T cell-resistant CD8+ T cells induced by glucocorticoid-induced tumor necrosis factor receptor signaling. Cancer Res. 2008;68:5948–5954. doi: 10.1158/0008-5472.CAN-07-5839. First evidence that in vivo GITR modulated Teff resistance to Treg suppression may be an important mechanism of GITR ligation tumor immunotherapy. [DOI] [PubMed] [Google Scholar]
- 48.Cohen AD, Diab A, Perales MA, Wolchok JD, Rizzuto G, Merghoub T, Huggins D, Liu C, Turk MJ, Restifo NP, et al. Agonist anti-GITR antibody enhances vaccine-induced CD8(+) T-cell responses and tumor immunity. Cancer Res. 2006;66:4904–4912. doi: 10.1158/0008-5472.CAN-05-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann C, Stanke J, Kaufmann AM, Loddenkemper C, Schneider A, Cichon G. Combining T-cell vaccination and application of agonistic anti-GITR mAb (DTA-1) induces complete eradication of HPV oncogene expressing tumors in mice. J Immunother. 2010;33:136–145. doi: 10.1097/CJI.0b013e3181badc46. [DOI] [PubMed] [Google Scholar]
- 50•.Mitsui J, Nishikawa H, Muraoka D, Wang L, Noguchi T, Sato E, Kondo S, Allison JP, Sakaguchi S, Old LJ, et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin Cancer Res. 2010;16:2781–2791. doi: 10.1158/1078-0432.CCR-09-3243. Here they show that in conditions where neither agent alone is effective, the combination of CTLA-4 and DTA-1 can affect tumor clearance. [DOI] [PubMed] [Google Scholar]
- 51.Imai N, Ikeda H, Tawara I, Wang L, Nishikawa H, Kato T, Shiku H. Glucocorticoid-induced tumor necrosis factor receptor stimulation enhances the multifunctionality of adoptively transferred tumor antigen-specific CD8+ T cells with tumor regression. Cancer Sci. 2009;100:1317–1325. doi: 10.1111/j.1349-7006.2009.01179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52•.Pruitt SK, Boczkowski D, de Rosa N, Haley NR, Morse MA, Tyler DS, Dannull J, Nair S. Enhancement of anti-tumor immunity through local modulation of CTLA-4 and GITR by dendritic cells. Eur J Immunol. 2011 doi: 10.1002/eji.201141383. (Epub online September 9, 2011). In an extension of their original work on GITR ligand expressing DC vaccines, Pruitt et al. improve the effectivens of GITR-L combining it with CTLA-4 blocking antibody expressing DCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramirez-Montagut T, Chow A, Hirschhorn-Cymerman D, Terwey TH, Kochman AA, Lu S, Miles RC, Sakaguchi S, Houghton AN, van den Brink MR. Glucocorticoid-induced TNF receptor family related gene activation overcomes tolerance/ignorance to melanoma differentiation antigens and enhances antitumor immunity. J Immunol. 2006;176:6434–6442. doi: 10.4049/jimmunol.176.11.6434. [DOI] [PubMed] [Google Scholar]
- 54.Zhou P, Qiu J, L’Italien L, Gu D, Hodges D, Chao CC, Schebye XM. Mature B cells are critical to T-cell-mediated tumor immunity induced by an agonist anti-GITR monoclonal antibody. J Immunother. 2010;33:789–797. doi: 10.1097/CJI.0b013e3181ee6ba9. [DOI] [PubMed] [Google Scholar]
- 55•.Côté AL, Zhang P, O’Sullivan JA, Jacobs VL, Clemis CR, Sakaguchi S, Guevara-Patiño JA, Turk MJ. Stimulation of the glucocorticoid-induced TNF receptor family-related receptor on CD8 T cells induces protective and high-avidity T cell responses to tumor-specific antigens. J Immunol. 2011;186:275–283. doi: 10.4049/jimmunol.1001308. In a closer look at DTA-1 enhanced concomitant immunity, Turk’s group shows that GITR expression is only necessary on the Teff cells for rejection of secondary tumor challenge and expands the polyclonality of the response generated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One. 2011;6:pe19499. doi: 10.1371/journal.pone.0019499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, Weinberg AD, Wolchok JD, Houghton AN. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med. 2009;206:1103–1116. doi: 10.1084/jem.20082205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coe D, Begom S, Addey C, White M, Dyson J, Chai J-G. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol Immunother. 2010;59:1367–1377. doi: 10.1007/s00262-010-0866-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenzwieg M, Ponte J, Apostolou I, Doty D, Guild J, Slavonic M, Ponath P, Vaikus L. Development of TRX518, an aglycosyl humanized monoclonal antibody (Mab) agonist of huGITR. J Clin Oncol. 2010;28(Suppl):pe13028. [Google Scholar]
- 62.Tolerx Inc. Agonistic antibodies to human glucocorticoid-induced tumor necrosis factor receptor as potential stimulators of T cell immunity for the treatment of cancer and viral infections. Expert Opin Ther Patents. 2007;17:567–575. [Google Scholar]
- 63.Ascierto PA, Simeone E, Sznol M, Fu Y-X, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010;37:508–516. doi: 10.1053/j.seminoncol.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 64•.Weinberg AD, Morris NP, Kovacsovics-Bankowski M, Urba WJ, Curti BD. Science gone translational: the OX40 agonist story. Immunol Rev. 2011;244:218–231. doi: 10.1111/j.1600-065X.2011.01069.x. Weinberg et al. give an inside look at the development of OX-40 immunotherapy and discuss their experience running the first-in-human trial of OX40 agonist antibody in melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.You S, Poulton L, Cobbold S, Liu CP, Rosenzweig M, Ringler D, Lee WH, Segovia B, Bach JF, Waldmann H, et al. Key role of the GITR/GITR ligand pathway in the development of murine autoimmune diabetes: a potential therapeutic target. PLoS One. 2009;4:pe7848. doi: 10.1371/journal.pone.0007848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uraushihara K, Kanai T, Ko K, Totsuka T, Makita S, Iiyama R, Nakamura T, Watanabe M. Regulation of murine inflammatory bowel disease by CD25+ and CD25− CD4+ glucocorticoid-induced TNF receptor family-related gene+ regulatory T cells. J Immunol. 2003;171:708–716. doi: 10.4049/jimmunol.171.2.708. [DOI] [PubMed] [Google Scholar]
- 67.Suri A, Shimizu J, Katz JD, Sakaguchi S, Unanue ER, Kanagawa O. Regulation of autoimmune diabetes by non-islet-specific T cells — a role for the glucocorticoid-induced TNF receptor. Eur J Immunol. 2004;34:447–454. doi: 10.1002/eji.200324599. [DOI] [PubMed] [Google Scholar]