

Abstract
We describe herein the synthesis of a late-stage intermediate en route to cortistatin A. Key transformations included a Snieckus-like cascade sequence culminating in a 6π-electrocyclization, an alkylative dearomatization, and the stereoselective functionalization of the cortistatin A-ring. While the total synthesis we sought was not accomplished, the work sets the stage for several approaches to the preparation of novel analogs via diverted total synthesis.
Keywords: total synthesis, angiogenesis inhibitor, nitrone–aryne cycloaddition, 6π-electrocyclization
Introduction
Our laboratory devotes much of its effort to the discovery and development of small molecule natural product (SMNP)–derived agents of potential therapeutic value. i Under our paradigm of diverted total synthesis (DTS),1 we identify SMNPs with compelling biological activity and challenging structural features. We first undertake the total synthesis of the natural product itself and subsequently seek to adapt the synthetic route to allow access to rationally designed analogs for further investigation and SAR analysis. Through recourse to the strategy of DTS, we have discovered a range of promising SMNP–derived lead agents – including those based on the epothilone,ii migrastatin,iii and panaxytrioliv natural product frameworks – which are under development in preclinical and clinical settings.
In our ongoing pursuit of new lead structure types, we took note of a recent report by Kobayashi and co-workers, v on the isolation of a novel class of steroidal alkaloids, the cortistatins, from the marine sponge Corticium simplex (Scheme 1). Notably, members of the cortistatin family appear to exhibit potent in vitro anti-angiogenesis activity. Angiogenesis, the formation of new blood vessels in tissues, is an essential biological process, involved in embryonic development, reproduction, and wound healing.vi The process of angiogenesis is regulated by a series of stimulators and inhibitors – including vascular endothelial growth factor (VEGF) and angiopoietin 1 (ANGPT1), respectively – and is therefore typically focal and self-limited in time in regular tissues.vii Pathological angiogenesis, however, is implicated in a variety of diseases, including atherosclerotic plaques, diabetic retinopathy, and – of particular interest to our laboratory – the growth and metastasis of malignant tumors. Selective small molecule angiogenesis inhibitors, perhaps of the cortistatin genre, seemed to represent a promising avenue of research directed toward the treatment of angiogenesis-dependent diseases. Cortistatin A, in particular, has been reported to exhibit potent growth-inhibitory activity against human umbilical vein endothelial cells (HUVECs), with an IC50 of 1.8 nM and selectivity indices of above 3000 compared to normal human dermal fibroblast (NHDF) and other tumor cell lines.viii Cortistatin A also exhibits inhibitory effects against the migration of HUVECs, and against VEGF- and bFGF- induced tubular formation at a concentration of 2 nM, with comprehensive activity against neovascularization. ix Thus, cortistatin A, which exhibits strong and selective anti-angiogenesis activity, could potentially serve as an exciting lead agent in the development of anti-angiogenic therapeutics via chemical synthesis.
Scheme 1.

Cortistatin family of natural products and their IC50 values against HUVECs.
In contemplating a total synthesis, one soon comes to focus on the unique 9(10-19)-abeo-androstane-type steroidal core, incorporating an oxabicyclo[3.2.1]octene motif, shared by the cortistatins. Among the various members of this class, three different types of C17 side chains (isoquinoline, 3-methylpyridine, and methylpiperidine) and various levels of A-ring oxidation are represented. Following the initial structure-activity relationship (SAR) study reported by the Kobayashi group,ix Nicolaou and co-workers reported the results of a binding assay screening, which indicated strong binding affinity of cortistatin A toward a series of biologically essential kinases with extended C-termini, including cyclin-dependent kinases (CDK8 and CDK11) and Rho-associated coiled-coil containing protein kinases (ROCK I and ROCK II). x Docking simulations suggest that the isoquinoline moiety of cortistatin A projects inside the kinase hinge, while the steroidal skeleton blocks the ATP-binding cleft and the polar A-ring is exposed to solvent. The extended C-termini of these kinases apparently place an aromatic side chain proximal to cortistatin A, thereby encapsulating the ATP–binding site. Thus, the cytostatic activity of the cortistatins against HUVECs is proposed to arise from inhibition of HUVEC kinase activity, resulting in diminished HUVEC proliferation and migration.
Not surprisingly, the potent biological activity and unique structures of the cortistatins have prompted many laboratories to undertake their synthesis. To date, total syntheses have been reported from the Baran,xi Nicolaou,xii Shair,xiii Myers,xiv Hirama,xv and Funk groups,xvi and formal syntheses have been achieved by the Sarpongxvii and Yang groups.xviii Numerous synthetic studies toward the steroidal core of the cortistatins have been published.xix In the aggregate, these synthetic studies represent a wide range of strategies, each of which might be adapted to provide access to different types of analogs for further investigation. Indeed, a variety of synthetic cortistatin analogs have been prepared and evaluated in preclinical settings.xx
Preliminary SAR studies on the cortistatins and synthetic analogs have revealed important information with respect to the structural features required for biological activity. As shown in Scheme 2, in order to exhibit inhibitory activity, the A-ring must incorporate at least one –OH or –NMe2 group and stereointegrity at C17 must be preserved. Moreover, there is some evidence to the effect that the A–B–C ‘skeleton angle’ and core ring planarity are important factors in mediating biological activity.xxa
Scheme 2.
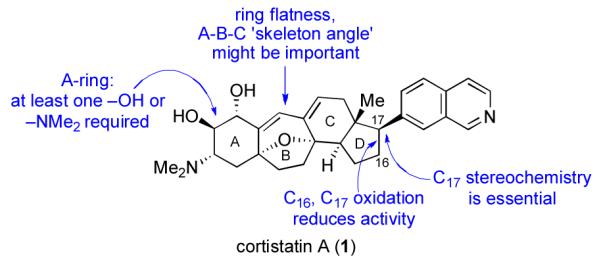
Cortistatin: Preliminary SAR Data.
Synthetic Plan
In designing our synthetic strategy toward cortistatin A, of course, we took note of its rearranged steroidal skeleton. Seeking to capitalize on the wisdom accumulated over years of research in the field of steroid synthesis,xxi we considered both B–ringxxii and C– ringxxiii disconnection strategies toward cortistatin A. In the case at hand, disconnection along the B–ring would reduce the challenge to the syntheses of the A–ring fragment, from an aromatic precursor, and the more complex C–D ring system. Pattern recognition analysis suggests that the C–D ring system could be derived from the optically enriched Hajos-Parrish ketone.xxiv,xxv As shown in Scheme 3, following some early experimentation, xxvi a convergent strategy which envisioned 1,3-dipolar cycloaddition between an aryne (2, A-ring) and an α,β-unsaturated nitrone (3, C-D ring) presented itself. The resultant benzoisoxazoline, 4, could well be converted to benzopyran intermediate 7 through a sequence involving reductive N-O bond cleavage, followed by 1,4-elimination and, finally, 6π-electrocyclization. xxvii Finally, alkylative dearomatization of 7 would deliver intermediate 8 en route to cortistatin A.xxviii
Scheme 3.

[3+2] Cycloaddition–based synthetic strategy toward cortistatin A.
Results and Discussion
Nitrone-Aryne [3+2] Cycloaddition–Based Route to the Cortistatin Core
At the outset of our studies, few examples of nitrone-aryne [3+2] cycloadditions, such as 2+3→4, had been reported. Model studies, performed with simple nitrone substrates (11), revealed that high levels of regioselectivity could be achieved when the aryne dipolarophiles were equipped with ortho-substituents (Scheme 4). xxix Interestingly, ortho-TMS aryne, 10a, underwent cycloaddition to yield adduct 12a as a single regioisomer. By contrast, ortho-OMe aryne 10b generated adduct 12b, with completely inverted regioselectivity. These findings seem to reflect the interplay of competing steric and electronic factors. It was also observed that meta- and para- substituted aryne substrates (cf. 13) generally underwent cycloaddition to afford mixtures of products with poor levels of regiocontrol (Scheme 4). Similar regioselectivity trends have been reported previously.xxx
Scheme 4.

Regioselectivity of nitrone-aryne [3+2] cycloaddition
Having probed the nature of the nitrone-aryne [3+2] cycloaddition, we now sought to prepare a more sophisticated model system, through which we would explore the viability of the proposed alkylative dearomatization sequence. In order to achieve the requisite sense of regioselectivity in the cycloaddition step, we elected to install an inductively operating electron-withdrawing group, –OMe, at the ortho position of the aryne substrate. This functionality would ultimately become the C1 hydroxyl group of cortistatin A. In the event, 1,3-dipolar cycloaddition between nitrone 15 and the aryne generated from 16 afforded benzoisoxazoline 17. Treatment under mild N–O cleavage conditions followed by 1,4-elimination served to generate the intermediate o-quinomethide, which readily underwent 6π–electrocyclization to deliver adduct 18, as planned, in high yield. Deprotection and subsequent bromination afforded 19, which, upon exposure to TBAF, smoothly underwent alkylative dearomatization to provide the key model system, 20 (Scheme 5). Given this encouraging demonstration of “reduction to practice”, we hoped to build in to our scheme a sufficient functionality to enable progression, hopefully to cortistatin A itself.
Scheme 5.

[3+2] cycloaddition: Model studies toward the cortistatin core structure.
Key: a) n-BuLi, THF, −78 °C; b) Zn, HOAc, RT; then 170 °C, toluene, 55% from 15; c) Me2BBr, i-Pr2NEt, anisole, CH2Cl2, −78 °C, 84%; d) CBr4, PPh3, CH2Cl2, 87%; e) TBAF, THF, RT; then 50 °C, 20 min, 52%.
Snieckus Cascade–Based Route to the Cortistatin Core
An alternative route toward a more highly functionalized version of the quinomethide system was also pursued. Based on the elegant precedent of Snieckus, xxxi with a later adaptation by Alvarez-Manzaneda, xxxii we envisioned a cascade sequence commencing with an aryllithium substrate, 21, and a bicyclic aldehyde of the type 22xvc (Scheme 6). According to our plan, 1,2-addition of 21 to 22 would generate an alkoxide intermediate (23), which would undergo carbamate migration to afford 24. A facile 1,4-elimination would give rise to quinomethide 25, which was expected to undergo 6π-electrocyclization. Subsequent alkylative dearomatization would generate an advanced intermediate of the type 27. At the outset of our studies, there was uncertainty regarding the stereochemistry at C8 following electrocyclization (see asterisk, 26). We postulated that the isomer required for cortistatin would be the thermodynamic product, as it encompasses trans/anti stereoconnectivity between the angular methyl (C18), the C14 hydrogen, and the angular two-carbon chain.
Scheme 6.

Snieckus cascade-based route toward cortistatin A.
We first set out to synthesize the A ring precursor fragments. In order to preserve maximum synthetic flexibility in the later stages of the synthesis, we prepared both the substrate bearing a MOM protecting group at the eventual C1 (31a) and the fragment possessing a C1 methoxy group (31b). As outlined in Scheme 7, the route commenced with commercially available 28. Bromination,xxxiii followed by selective acylation, afforded carbamate 29. The remaining free hydroxyl was protected as a TBS group, and Baeyer-Villiger oxidation with subsequent saponification delivered 30. This intermediate was converted to both 31a and 31b under the conditions shown.
Scheme 7.

Synthesis of aromatic A ring fragments (31a and 31b).
Key: a) Br2, CHCl3; b) ClCONEt2, pyridine, DMAP, reflux, 48% over 2 steps; c) TBSOTf, lutidine, CH2Cl2, 0 °C→RT, 97%; d) mCPBA, CH2Cl2; e) K2CO3, MeOH; f) MOMCl, i-Pr2NEt, 31a: 58% over 3 steps; g) TMSCHN2, PhH, MeOH, 31b: 65% over 3 steps.
With fragments 31 and 32xvc in hand, we were able to reduce the proposed Snieckus-type cascade sequence to practice (Scheme 8). Interestingly, isomers bearing the undesired C8 stereochemistry (epi-33a and epi-33b) were preferentially formed (10:1 dr), presumably due to unfavorable 1,3-diaxial interactions between C18 and C6-C7 in the 6π-electrocyclization event. Fortunately, however, the electrocyclization step (cf. 25→26, Scheme 6) proved to be reversible, and thermally induced epimerization of isolated epi-33a and epi-33b afforded isomeric mixtures, of which the thermodynamic products, 33a and 33b, were predominant (>2:1 dr).
Scheme 8.

Snieckus-type domino route to cortistatin core.
Key: a) t-BuLi, then 32, −78→130 °C, 50% (epi-33a), 60% (epi-33b); b) 192–198 °C.
The diastereomeric product mixtures, which were not readily separable, were subjected to I2-mediated TBS deprotection (Scheme 9).xxxiv Chromatographic separation was achieved at this stage, to afford diastereomerically pure adducts 34a and 34b. The primary alcohol functionality of 34a was subjected to tosylation conditions to provide 35a, which, upon exposure to TBAF, underwent the hoped-for alkylative dearomatization to generate pentacyclic adduct 36a in excellent overall yield. A similar sequence was employed for the conversion of 34b to adduct 36b (Scheme 9).
Scheme 9.

Alkylative dearomatization.
Key: a) I2, MeOH; chromatographic separation (34a), 54% (34b), 62%; b) TsCl, pyridine, DMAP, CH2Cl2, 95%; c) TBAF, 70 °C, 94%; d) MsCl, pyridine, CH2Cl2, 90%; e) TBAF, 130 °C, 95%.
Functionalization of the A ring en route to cortistatin A
With the cortistatin core framework in place, we now turned our attention to the functionalization of the A-ring. In the natural product, the C1, C2, and C3 functionalities (OH, OH, and NMe2, respectively) all occupy equatorial orientations, thus raising the possibility that they might be stereoselectively installed through reduction of iminium and ketone precursors, for instance, with sodium borohydride.xxxv
We first explored methods for the introduction of the C3 amine functionality. This turned out to be a surprisingly challenging task. Methods based on electrophilic amination,xxxvi Neber reaction,xxxvii silyl enol ether aziridination,xxxviii and dimethylamine displacement of a C3 bromide derived from a 3,4-dihydro version of 36b (vide infra) were all unsuccessful. Finally, following extensive experimentation, an α-azido ketone fragmentation method xxxix was found to successfully enable emplacement of the C3 amine. As shown, compound 36a was advanced to the C3-bromo intermediate 38a through exposure to L-selectride, followed by the electrophilic brominating agent, 37 (Scheme 10). Upon treatment with Bu4NN3 in THF, intermediate 38a was susceptible to displacement by azide. Subsequent fragmentation of 39a yielded enamine 40a, as shown. The stereoselective reduction of 40a was accomplished through treatment with NaBH3(CN) in CH2Cl2. The air-sensitive intermediate 41a was rapidly reduced under Luche conditions,xl to provide intermediate 42a. The latter was then protected in the form of either Boc or Fmoc carbamates. A similar sequence (unoptimized) served to advance intermediate 36b, bearing the C1 methoxy group, to compound 43b, as shown.
Scheme 10.

A-Ring functionalization: Installation of C2 and C3 stereocenters.
Key: a) L-Selectride; then 37, 62%, 1.25:1 dr; b) n-Bu4NN3, THF; c) HOAc, NaBH3(CN), CH2Cl2; d) NaBH4, CeCl3•7H2O, 42% from 38a; e) Boc2O, NaOH, THF/H2O (43a); f) Fmoc-OSu, Na2CO3, THF/H2O (44a); g) L-Selectride, −78 °C, 80%; h) LiHMDS, then 37, −78 °C, 73%, 1:1 dr; i) NaN3, DMF; then NaBH(OAc)3, HOAc, (HCHO)X, 50 °C, 69%; j) NaBH(OAc)3, HOAc, CH2Cl2; k) DIBAL-H, −78 °C, < 20% from 40b.
With the A-ring C2 and C3 stereocenters in place, the task would now involve deprotection of the C1 MOM (43a) or Me (43b) protecting group. Unfortunately, all efforts to achieve nucleophilic deprotection of the methoxy functionality of 43b (and its precursor 40b) were unsuccessful. Accordingly, we focused our efforts on the removal of the MOM protecting group. Extensive experimentation in the context of model systems revealed surprising complexities associated with this apparently straightforward proposed transformation. In summary, perhaps due to the destabilizing presence of the conjugated trienyl system, standard acid-catalyzed methods for the removal of the MOM group were not productive.
We sought to exploit the reactivity of the triene as a means to achieve the hoped-for MOM deprotection. We postulated that, if an appropriate electrophile were to attack the triene at its C12 terminus, then C1 might be rendered more susceptible to hydrolysis. In fact, in model systems, mild bromine-induced removal of the MOM enol ether could, in fact, be achieved (Scheme 11, 45→46).xli Interestingly, the ω-bromodienone adducts were unstable, and under standard work-up and purification conditions (evaporation of EtOAc solution, silica gel), these products underwent apparently rapid HBr elimination to generate trienones of the type 47. Though unexpected, we postulated that these HBr elimination products might offer a new avenue for the installation of the C1 and C2 stereocenters. We envisioned a sequence involving reduction of the C2 ketone, trienol–dienone tautomerization, and C1 ketone reduction to stereoselectively generate the C1 and C2 trans-diol system. We reasoned that protonic solvents might serve to effectively facilitate proton transfer, and Luche conditions would ensure selective 1,2 reduction.
Scheme 11.

Bromine-induced MOM deprotection
Key: a) Br2, H2O, THF, 0 °C; b) silica gel or evaporate EtOAc solution.
In the context of our target system, intermediate 43a underwent MOM deprotection followed by rapid HBr elimination to generate adduct 48, as shown. To our surprise, however, exposure of either intermediate 48 or its C1 TBS ether derivative to Luche conditions gave rise to intermediate 49, incorporating an extra hydroxyl on C12. At the time, the mechanism of this transformation was unclear; however, subsequent studies provided a plausible explanation (vide infra).
We now refocused our efforts on derivatizing the C12 brominated system arising from Br2-induced MOM deprotection (cf. 46). Based on the Hirama precedent, we were optimistic that we could achieve C12 debromination through recourse to radical methods.15a,c Surprisingly, however, in a model system, exposure of compound 50 to radical conditions yielded an isomeric mixture,xlii of which the diene 52 was the major product (Scheme 13). Moreover, when the reaction mixture was subjected to prep-TLC in air, 52 was partially converted to 53. The structural assignment of 53 was supported by 1H NMR and LR-MS analysis. The facile formation of 53 under these conditions was quite unexpected. We postulate that, considering the ease with which 52 isomerizes to its trienol form (54) under acidic conditions, it may well be that 54 is the reactive intermediate en route to 53. Similar systems are known to react with O2 in a similar manner,xliii,xliv and electron paramagnetic resonance (EPR)/ spin trapping studies support a stepwise radical pathway involving triplet oxygen. xlv Small scale attempts to prevent peroxidation through freeze-pump-thaw degassing were unsuccessful.
Scheme 13.

Radical debromination: Model studies.
Key: a) AIBN, n-Bu3SnH, PhH, 80–90 °C.
The mechanistic analysis advanced above might also serve to explain the unexpected and remarkable C12 formal oxidation observed in the context of the Luche reduction, described above (Scheme 12, 48→49). According to this reasoning, compound 48 did, perhaps, indeed undergo Luche reduction at the C2 ketone, to generate intermediate 56 (Scheme 14). Presumably, C12 peroxidation would be more rapid than enol-ketone tautomerization, and intermediate 57 would be formed. Further reduction would deliver the observed adduct, 49. It should be noted that 56 might exist in its enolate form during the reaction.xlvi Enolate peroxidation by O2 has also been suggested to proceed through a chain-reaction-based radical pathway.xlvii
Scheme 12.

MOM deprotection and attempted Luche reduction of ring A.
Key: a) Br2, H2O, THF, 0 °C; b) evaporate in EtOAc; c) CeCl3•7H2O, NaBH4, MeOH, 0 °C.
Scheme 14.

Mechanistic rationale for formation of compound 49.
Key: a) CeCl3•7H2O, NaBH4, MeOH, air, 0 °C.
Having realized a two-step sequence for the deprotection of the C1 MOM functionality in a model setting, we sought to accomplish the transformation in the context of the cortistatin core system. First, the C2 hydroxyl of compound 43b was acylated to generate 58 (Scheme 15). In the event, exposure to mild bromination conditions afforded intermediate 59, which subsequently underwent radical debromination to furnish the target compound 60, albeit in disappointingly low yield (ca. 10% from 58). In this more complex setting, the debromination reaction was quite sensitive, and signficant levels of decomposition were observed. Thorough degassing through repeated freeze-pump-thaw was required. Finally, compound 60 was advanced to 61 through Luche reduction followed by acylation of the resultant C1 hydroxyl. In principle, this compound could be advanced to 62, a late-stage intermediate in the Shair synthesis, upon Fmoc removal followed by reductive methylation. However, at this stage, we no longer had adequate levels of material on hand to pursue even a formal relay total synthesis. What was clear is that this route, which began on a very promising note, would not lead to cortistatin A in a useful way.
Scheme 15.

Radical debromination and synthesis of compound 61 en route to cortistatin A.
Key: a) Ac2O, pyridine, DMAP, CH2Cl2 (70% from 42); b) Br2, H2O, THF, 0 °C; c) AIBN, n-Bu3SnH, C6D6, 70 °C, ~10% from 58; d) CeCl3•7H2O, NaBH4, MeOH, 0 °C; e) Ac2O, pyridine, DMAP, CH2Cl2.
Conclusion
In summary, the synthesis of a late-stage intermediate en route to cortistatin A has been accomplished. Key transformations include a Snieckus-like cascade sequence and the development of methods for the functionalization of the challenging cortistatin A-ring. In this context, our attempts to manipulate A-ring functionalities led to surprising findings, which served to deepen our understanding of the reactivities of the complex cortistatin core system. While even a formal total synthesis of the natural product was not achieved, the ability to generate serious amounts of look-alike structures (cf. inter alia: 38, 40, 42) might well be exploitable to what is now the important problem of producing an accessible and therapeutically useful agent based on cortistatin A.
Experimental Section
General Methods
Unless mentioned otherwise, all non-aqueous reactions were carried out in vacuum-flame-dried glassware under a balloon-pressure of argon; commercially available reagents were used as received; anhydrous solvents were purchased as the highest grade from Sigma-Aldrich, or passed through a solvent-purification system, or purified as follows: THF was distilled from Na-benzophenone, CH2Cl2 was distilled from CaH2. Reactions were monitored by thin-layer chromatography on Merck silica gel 60-F254 coated 0.25 mm plates. Flash column chromatography was performed using RediSep® pre-packed disposable silica gel columns (normal phase, 230-400 mesh) from Teledyne Isco. Prep-TLC was performed on Merck silica gel 60-F254 coated 0.50 or 1 mm plates. Yields are reported for isolated, spectroscopically pure compounds. NMR spectra were obtained on Bruker DRX 300 or 400 MHz, or Bruker DMX 500 MHz spectrometers. CDCl3, dried by standing over K2CO3, was used for NMR samples and chemical shifts were referenced on residual solvent peaks (δ = 7.26 for 1H NMR and 77.0 for 13C NMR). Abbreviations for 1H NMR: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or br = broad. IR spectra were obtained on a Perkin-Elmer Paragon 1000 FTIR spectrometer. High resolution mass spectra were acquired at the Columbia University Mass Spectral Core facility on a JEOL HX110 spectrometer. Optical rotations were measured on a JASCO DIP-1000 spectrometer.
Synthesis of compound 29
To a mixture of 2-bromo-3,6-dihydroxybenzaldehydexlviii (9.25 g, 42.6 mmol) and DMAP (98.6 mg, 0.81 mmol) in 35 mL pyridine was added diethyl carbamyl chloride (5.40 mL, 44 mmol) at room temperature. The reaction mixture was stirred at reflux for 13 h under argon atmosphere. Then the reaction mixture was poured into ice water and extracted with ether 3 times. The combined organic phases were washed with 1N HCl, brine and dried with anhydrous MgSO4 and then filtrated. The residue was purified by flash chromatography (hexane / EtOAc: 92 / 8) to give 6.42 g desired product 29 in 48% yield (the synthesis of 2-bromo-3,6-dihydroxybenzaldehyde from 28 was quantitative).
1H NMR (400 MHz, CDCl3): δ 11.91 (s, 1H), 10.33 (s, 1H), 7.32 (d, J = 9.2 Hz, 1H), 6.94 (d, J = 9.2 Hz, 1H), 3.50 (q, J = 7.2 Hz, 2H), 3.38 (q, J = 7.2 Hz, 2H), 1.30 (t, J = 7.2 Hz, 3H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 196.9, 160.6, 152.4, 141.1, 131.9, 120.4, 117.3, 116.8, 41.9, 41.5, 13.7, 12.7. IR (NaCl, cm−1): 2976, 2935, 1725, 1652, 1607, 1580, 1462, 1420, 1380, 1294, 1265, 1240, 1220, 1176, 1152. HR-MS (FAB+) calcd for C12H15O4N79Br (M + 1): 316.0184, found 316.0176.
Synthesis of 2-bromo-4-((tert-butyldimethylsilyl)oxy)-3-formylphenyl diethylcarbamate
To a solution of 29 (1.37 g, 4.34 mmol) and 2,6-lutidine (1.15 mL, 13.0 mmol) in 20 mL of CH2Cl2 was added TBSOTf (1.19 mL, 5.16 mmol) slowly at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched with saturated aqueous NH4Cl at 0 °C and extracted with ether 3 times. The combined organic layers were washed with 1N HCl, saturated aqueous sodium bicarbonate, water and brine, sequentially. The combined organic layer was dried over anhydrous MgSO4, concentrated and then purified by flash chromatography (hexane / EtOAc: 92 / 8) to give 1.88 g desired product in 97% yield.
1H NMR (300 MHz, CDCl3): δ 10.32 (s, 1H), 7.22 (d, J = 9.0 Hz, 1H), 6.81 (d, J = 9.0 Hz, 1H), 3.47 (q, J = 7.2 Hz, 2H), 3.35 (q, J = 7.2 Hz, 2H), 1.27 (t, J = 7.2 Hz, 3H), 1.15 (t, J = 7.2 Hz, 3H), 0.96 (s, 9H), 0.21 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 189.6, 155.7, 153.0, 143.4, 128.6, 126.2, 119.6, 118.0, 42.3, 41.9, 25.5, 18.1, 14.1, 13.1, −4.5. IR (NaCl, cm−1): 2932, 2859, 1727, 1702, 1591, 1562, 1462, 1420, 1393, 1243, 1221, 1153. HR-MS (FAB+) calcd for C18H27O4N79BrSi (M − 1): 428.0893, found 428.0899.
Synthesis of compounds 31a and 31b
To a stirred solution of 2-bromo-4-((tert-butyldimethylsilyl)oxy)-3-formylphenyl diethylcarbamate (1.89g, 4.40 mmol) in 50 mL dichloromethane was added mCPBA (3.79g, 77% max.) at 0 °C, and the solution was slowly warmed to room temperature and stirred for 1 day. The reaction was quenched by NH4Cl (sat.) solution at 0 °C, and after separation, the aqueous phase was re-extracted by EtOAc three times, the combined EtOAc solution was to added hexanes, and then washed with NaHCO3 (sat.) and brine sequentially and dried over MgSO4. The filtrate was evaporated and the desired crude 2-bromo-6-((tert-butyldimethylsilyl)oxy)-3-((diethylcarbamoyl)oxy)phenyl formate was directly used in the next step.
The crude 2-bromo-6-((tert-butyldimethylsilyl)oxy)-3-((diethylcarbamoyl)oxy)phenyl formate from the last step was dissolved in 50 mL methanol and K2CO3 (1.50 g) was added at 0 °C. The reaction mixture was warmed to room temperature and stirred for 0.5 hr. The reaction was quenched by NH4Cl (sat.) at 0 °C, and the aqueous phase was extracted with Et2O three times. The organic phase was evaporated and then re-dissolved in Et2O, and hexanes were added, then washed with NH4Cl (sat.) and brine. After drying over MgSO4, the filtrate was evaporated and the crude 30 was directly used in the next step.
The crude 30 was dissolved in 50 mL dichloromethane and cooled to 0 °C, then diisopropylamine (6.17 mL, 35.2 mmol) was added, followed by MOMCl (1.34 mL, 17.6 mmol). The reaction mixture was evaporated 1 h later and the residue was chromatographed (hexanes:EtOAc = 9:1 to 7:1) to give the desired product 31a (1.17 g, 2.53 mmol, 58% yield for 3 steps).
In another batch, the crude phenol 30 (prepared from 1.11 g 2-bromo-4-((tert-butyldimethylsilyl)oxy)-3-formylphenyl diethylcarbamate) in 10 mL of MeOH and 10 mL of PhH was added TMSCHN2 (1.94 mL, 1.5 equiv.) dropwise. The reaction mixture was stirred at room temperature for 2 h. After removal of the solvents, the residue was purified by flash chromatography (hexane / EtOAc : 90 / 10) to give 0.75 g desired product 31b in 65% yield over three operations.
31a
1H NMR (400 MHz, CDCl3): δ 6.87 (d, J = 8.8 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 5.16 (s, 2H), 3.63 (s, 3H), 3.48 (q, J = 7.2 Hz, 2H), 3.38 (q, J = 7.2 Hz, 2H), 1.29 (t, J = 7.2 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H), 0.98 (s, 9H), 0.19 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 153.3, 146.4, 146.3, 143.6, 119.4, 118.7, 113.3, 98.7, 58.2 42.3, 41.9, 25.6, 18.1, 14.2, 13.3, −4.5. IR (NaCl, cm−1): 2955, 2931, 2857, 1727, 1483, 1469, 1416, 1388, 1241, 1221, 1154. HR-MS (FAB+) calcd for C19H31O4N79BrSi (M − 1): 460.1155, found 460.1161.
31b
1H NMR (400 MHz, CDCl3): δ 6.87 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 3.82 (s, 3H), 3.49 (q, J = 6.8 Hz, 2H), 3.39 (q, J = 6.8 Hz, 2H), 1.30 (t, J = 7.2 Hz, 3H), 1.21 (t, J = 6.8 Hz, 3H), 1.01 (s, 9H), 0.18 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 153.2, 149.2, 146.9, 143.4, 119.5, 118.6, 112.8, 60.0, 42.2, 41.8, 25.4, 18.0, 14.0, 13.1, −4.9. HR-MS (FAB+) calcd for C18H31O4N79 BrSi (M + 1): 432.1206, found 432.1185.
Synthesis of epi-33a
(Caution: the sealed tube must be thick and strong enough, and a blast shield is needed) In a flame-dried sealed tube was charged bromide 31a (37 mg, 0.080 mmol) in 1 mL Et2O, and the mixture was cooled to −78 °C. Next, t-BuLi (1.7 M in pentane, 0.160 mmol) was added dropwise and stirring continued for 10 min. Then aldehyde 32xv (32.8 mg, 0.073 mmol) in 2 mL Et2O was added dropwise, and the stirring continued for another 10 min. The reaction mixture was warmed to room temperature and then heated at 130 °C in an oil bath for 4 hrs before being quenched by brine at room temperature. The two phases were separated and the aqueous phase was re-extracted by Et2O twice. The combined organic phase was dried over MgSO4, and after filtration and evaporation, the residue was chromatographed by prep-TLC (hexanes:EtOAc = 50:1) and gave desired product epi-33a (10:1 d.r. at C8, 31.1 mg, 60% yield).
In a large scale reaction, 11.27 g bromide 31a and 9.98 g aldehyde 32 (both divided into two identical batches for the reaction and then combined during work-up) gave 8.14 g (51% yield) of the desired product epi-33a, and only 20 min was needed for heating at 130 °C.
Synthesis of 34a and epi-34a
(Caution: the sealed tube must be thick and strong enough, and a blast shield is needed) In a sealed tube was charged epi-33a (446 mg, 0.623 mmol) in 10 mL THF, and the vessel was placed in a 192 °C oil bath and heated for 11 hrs. The sealed tube was cooled to room temperature and 10 mL MeOH and I2 (14.7 mg, 0.058 mmol) were added. 3 hrs later, the reaction was quenched by 10% Na2S2O3 solution until the color faded. The mixture was evaporated and then partitioned by Et2O and brine and separated. The aqueous phase was re-extracted by Et2O twice and the combined organic phase was dried over MgSO4, and the filtrate was evaporated and chromatographed, providing 34a (204.4 mg, 0.339 mmol, 54% yield) and epi-34a (81.4 mg, 0.135 mmol, 22% yield).
Synthesis of epi-33a from epi-34a
epi-34a (20.4 mg, 0.034 mmol) was dissolved in 1 mL dichloromethane, and imidazole (31.8 mg, 0.467 mmol) and TBSCl (43.9 mg, 0.291 mmol) were added sequentially. The reaction mixture was stirred overnight before being quenched by NH4Cl (sat.). Et2O and brine were added, and the separated aqueous phase was re-extracted with Et2O twice. The combined organic phase was dried over MgSO4 and filtered, and the filtrate was evaporated and prep-TLC (hexanes:EtOAc = 10:1) gave the desired product epi-33a (22.0 mg, 0.031 mmol, 91% yield). This product can be used for the thermal C8 epimerization reaction.
epi-33a
1H NMR (400 MHz, CDCl3): δ 6.62 (d, J = 8.8 Hz, 1H), 6.55 (s, 1H), 6.51 (d, J = 8.8 Hz, 1H), 6.25 (d, J = 9.6 Hz, 1H), 6.17 (d, J = 9.2 Hz, 1H), 5.16 (d, J = 6.0 Hz, 1H), 5.10 (d, J = 6.0 Hz, 1H), 3.83 (t, J = 8.4 Hz, 1H), 3.53 (s, 3H), 3.65 – 3.50 (m, 2H), 2.15 – 1.91 (m, 4H), 1.86 – 1.78 (m, 1H), 1.75 – 1.67 (m, 1H), 1.61 – 1.51 (m, 1H), 1.00 (s, 9H), 0.903 (s, 9H), 0.896 (s, 3H), 0.83 (s, 9H), 0.18 (s, 3H), 0.17 (s, 3H), 0.07 (s, 3H), 0.05 (s, 3H), −0.05 (s, 3H), −0.07 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.0, 144.1, 141.5, 139.1, 134.7, 126.7, 120.2, 119.5, 114.4, 111.9, 98.8, 76.8, 76.8, 59.1, 57.6, 47.8, 46.3, 39.5, 30.7, 25.8, 25.8, 19.2, 18.2, 18.0, 12.8, −4.4, −4.4, −4.5, −4.8. [α]D20: +41.44 (c = 0.93, CH2Cl2). IR (NaCl, cm−1): 2955, 2929, 2893, 2857, 1570, 1471, 1387, 1251, 1176, 1162, 1094, 1000. HR-MS (FAB+) calcd for C39H68O6Si3 (M+): 716.4324, found 716.4312.
33a
1H NMR (400 MHz, CDCl3): δ 6.60 (d, J = 8.4 Hz, 1H), 6.53 (s, 1H), 6.46 (d, J = 8.8 Hz, 1H), 6.01 (d, J = 9.6 Hz, 1H), 5.92 (d, J = 9.6 Hz, 1H), 5.13 (d, J = 6.0 Hz, 1H), 5.10 (d, J = 6.0 Hz, 1H), 3.82 – 3.63 (m, 3H), 3.52 (s, 3H), 2.25 (dd, J = 13.2, 6.4 Hz, 1H), 2.21 – 1.87 (m, 4H), 1.85 – 1.72 (m, 1H), 1.55 – 1.45 (m, 1H), 1.07 (s, 3H), 0.98 (s, 9H), 0.91 (s, 9H), 0.80 (s, 9H), 0.16 (s, 3H), 0.15 (s, 3H), 0.05 (s, 3H), 0.03 (s, 3H), −0.08 (s, 3H), −0.10 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.4, 144.0, 141.7, 137.0, 136.5, 125.4, 120.3, 118.6, 115.0, 111.4, 98.9, 78.8, 78.0, 59.4, 57.7, 50.6, 47.1, 38.4, 30.6, 25.9, 25.8, 25.8, 20.9, 18.3, 18.2, 18.1, 14.0, −4.4, −4.5, −4.5, −4.9, −5.3, −5.3. [α]D19: −1.19 (c = 2.20, CH2Cl2). IR (NaCl, cm−1): 2953, 2928, 2887, 2857, 1571, 1471, 1384, 1295, 1252, 1161, 1121, 1088. HR-MS (FAB+) calcd for C39H68O6Si3 (M+): 716.4324, found 716.4319.
epi-34a
1H NMR (400 MHz, CDCl3): δ 6.63 (d, J = 8.8 Hz, 1H), 6.58 (s, 1H), 6.51 (d, J = 8.8 Hz, 1H), 6.24 (d, J = 9.6 Hz, 1H), 6.19 (d, J = 9.2 Hz, 1H), 5.16 (d, J = 6.0 Hz, 1H), 5.09 (d, J = 6.0 Hz, 1H), 3.86 (t, J = 8.4 Hz, 1H), 3.75 – 3.66 (m, 1H), 3.64 – 3.57 (m, 1H), 3.53 (s, 3H), 2.21 – 2.01 (m, 3H), 1.91 (dd, J = 12.4, 7.2 Hz, 1H), 1.78 – 1.53 (m, 4H), 1.00 (s, 9H), 0.91 (s, 3H), 0.90 (s, 9H), 0.19 (s, 3H), 0.18 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 146.5, 144.1, 141.8, 139.4, 134.6, 126.4, 120.4, 119.4, 114.6, 111.8, 98.8, 77.1, 76.7, 59.3, 57.6, 48.3, 46.6, 39.6, 30.7, 25.8, 25.7, 19.6, 18.1, 18.0, 12.8, −4.3, −4.4, −4.8. [α]D21: +31.43 (c = 0.81, CH2Cl2). IR (NaCl, cm−1): 3417, 2954, 2929, 2886, 2857, 1471, 1361, 1295, 1250, 1159, 1116, 1046, 998. HR-MS (FAB+) calcd for C33H54O6Si2 (M+): 602.3459, found 602.3461.
34a
1H NMR (400 MHz, CDCl3): δ 6.60 (d, J = 8.8 Hz, 1H), 6.55 (s, 1H), 6.46 (d, J = 8.4 Hz, 1H), 6.02 (d, J = 9.6 Hz, 1H), 5.93 (d, J = 9.2 Hz, 1H), 5.14 (d, J = 6.0 Hz, 1H), 5.07 (d, J = 6.0 Hz, 1H), 3.82 – 3.65 (m, 3H), 3.52 (s, 3H), 2.30 (dd, J = 13.6, 6.4 Hz, 1H), 2.27 – 2.18 (m, 1H), 2.11 – 1.91 (m, 3H), 1.90 – 1.78 (m, 1H), 1.73 (s, br, 1H), 1.57 – 1.47 (m, 1H), 1.06 (s, 3H), 0.98 (s, 9H), 0.91 (s, 9H), 0.17 (s, 3H), 0.16 (s, 3H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.2, 144.0, 141.9, 137.2, 136.3, 125.4, 120.4, 118.5, 115.0, 111.2, 98.8, 79.3, 78.0, 59.4, 57.6, 50.7, 47.1, 37.5, 30.6, 25.8, 25.7, 20.6, 18.1, 18.0, 14.0, −4.5, −4.9. [α]D20: +4.75 (c = 2.04, CH2Cl2). IR (NaCl, cm−1): 3413, 2955, 2929, 2892, 2857, 1472, 1386, 1296, 1252, 1159, 1120. HR-MS (FAB+) calcd for C33H54O6Si2 (M+): 602.3459, found 602.3476.
Synthesis of epi-33b
(Caution: the sealed tube must be thick and strong enough, and a blast shield is needed) To a solution of bromide 31b (72 mg, 0.166 mmol) in Et2O (3 mL) was added t-BuLi (0.215 mL, 0.366 mmol, 1.7 M in pentane) at −78 °C. The reaction mixture was stirred for 30 min, then a solution of aldehyde 32 (68 mg, 0.151 mmol) in Et2O (3 mL) was added slowly. After 30 min at −78 °C, the reaction was warmed to room temperature for 1 h, then heated at 130 °C for 5 h. After cooling to 0 °C, it was quenched with aqueous saturated NH4Cl. The mixture was extracted with ether 3 times and the combined organic layers were washed with water and brine, dried over anhydrous MgSO4 and concentrated. The residue was purified by flash chromatography (hexane / EtOAc : 98 / 2 to 97 / 3) to give 52 mg of 33b in 50% yield (10:1 dr).
1H NMR (400 MHz, CDCl3): δ 6.61 (d, J = 8.4 Hz, 1H), 6.49 (d, J = 9.2 Hz, 1H), 6.47 (s, 1H), 6.24 (d, J = 9.2 Hz, 1H), 6.17 (d, J = 9.2 Hz, 1H), 3.83 (t, J = 8.4 Hz, 1H), 3.80 (s, 3H), 3.53-3.62 (m, 2H), 1.92-2.12 (m, 4H), 1.78-1.85 (m, 1H), 1.67-1.71 (m, 1H), 1.54-1.59 (m, 1H), 1.01 (s, 9H), 0.90 (s, 9H), 0.89 (s, 3H), 0.82 (s, 9H), 0.17 (s, 3H), 0.16 (s, 3H), 0.07 (s, 3H), 0.05 (s, 3H), −0.06 (s, 3H), −0.08 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.0, 146.9, 142.1, 139.1, 134.9, 126.7, 120.5, 118.9, 113.9, 111.7, one overlaps with CDCl3, 76.9, 60.8, 59.2, 48.0, 46.4, 39.8, 30.7, 25.84, 25.76, 19.3, 18.2, 18.1, 12.8, −4.3, −4.58, −4.63, −4.8, −5.37, −5.43. HR-MS (FAB+) calcd for C38H66O5Si3: 686.4218. found 686.4204.
Synthesis of 34b and epi-34b
(Caution: the sealed tube must be thick and strong enough, and a blast shield is needed) The solution of epi-33b (420 mg, 0.611 mmol) in 20 mL THF in a sealed tube was heated at 198 °C for 36 h. After the reaction mixture was cooled to room temperature, 20 mL of MeOH and 7.8 mg of I2 (0.05 equiv., 0.031 mmol) were added. After 8 h at room temperature, the reaction was quenched with saturated aqueous NaHCO3 and 20% aqueous Na2S2O3. The mixture was extracted twice with ether. The combined organic layer was washed with saturated aqueous NaHCO3, 20% aqueous Na2S2O3, water and brine, dried over MgSO4. Concentration and flash column chromatography (hexane / EtOAc : 96 / 4) gave 34b (215 mg) in 62% yield as well as epi-34b (87 mg) in 25% yield.
34b
1H NMR (400 MHz, CDCl3): δ 6.60 (d, J = 8.4 Hz, 1H), 6.46 (s, 1H), 6.45 (d, J = 8.8 Hz, 1H), 6.02 (d, J = 10.0 Hz, 1H), 5.94 (d, J = 9.2 Hz, 1H), 3.76 (s, 3H), 3.71-3.81 (m, 3H), 2.10-2.33 (m, 2H), 1.78-2.09 (m, 4H), 1.48-1.56 (m, 1H), 1.07 (s, 3H), 1.01 (s, 9H), 0.92 (s, 9H), 0.18 (s, 3H), 0.16 (s, 3H), 0.06 (s, 3H), 0.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.1, 146.8, 142.6, 137.1, 136.5, 125.3, 120.6, 117.8, 114.5, 111.1, 79.3, 78.0, 60.8, 59.4, 50.7, 47.1, 37.5, 30.6, 25.8, 25.7, 20.7, 18.2, 18.0, 14.0, −4.5, −4.6, −4.7, −4.9. HR-MS (FAB+) calcd for C32H52O5Si2: 572.3353, found: compound is not stable under various mass conditions.
Synthesis of 35a
To a stirred solution of 34a (338.4 mg, 0.56 mmol) in 2 mL dichloromethane was added pyridine (0.20 mL), 4-dimethylaminopyridine (10.5 mg), and p-toluenesulfonyl chloride (300.2 mg, 1.57 mmol). The reaction mixture was stirred overnight before being quenched by NH4Cl (sat.), then Et2O and brine were added, and the separated aqueous phase was re-extracted with Et2O twice. The combined organic phase was dried over MgSO4 and filtered, and the filtrate was evaporated and flash column chromatography (hexanes:EtOAc = 1:0 to 9:1) gave the desired product 35a (403.8 mg, 0.533 mmol, 95% yield).
1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 6.56 (d, J = 8.4 Hz, 1H), 6.53 (s, 1H), 6.32 (d, J = 8.8 Hz, 1H), 5.98 (d, J = 9.6 Hz, 1H), 5.90 (d, J = 9.6 Hz, 1H), 5.11 (d, J = 6.0 Hz, 1H), 5.06 (d, J = 6.0 Hz, 1H), 4.20 – 4.11 (m, 1H), 4.11 – 4.03 (m, 1H), 3.72 (t, J = 8.0 Hz, 1H), 3.50 (s, 3H), 2.43 (s, 3H), 2.28 – 2.10 (m, 3H), 2.05 – 1.94 (m, 1H), 1.91 – 1.82 (m, 1H), 1.68 – 1.55 (m, 1H), 1.52 – 1.41 (m, 1H), 1.00 (s, 9H), 0.94 (s, 3H), 0.91 (s, 9H), 0.19 (s, 3H), 0.17 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 146.7, 144.4, 144.0, 142.1, 137.0, 135.2, 132.9, 129.7, 127.8, 125.2, 120.6, 118.4, 115.5, 111.2, 98.7, 77.8, 77.7, 67.0, 57.6, 50.2, 46.9, 34.1, 30.4, 25.7, 25.7, 21.5, 20.5, 18.1, 18.0, 13.8, −4.5, −4.9. [α]D19: +24.37 (c = 2.39, C1H2Cl2). IR (NaCl, cm−): 2954, 2929, 2887, 2857, 1472, 1386, 1363, 1252, 1178, 1124. HR-MS (FAB+) calcd for C40H60O8Si2S (M+): 756.3547, found 756.3535.
Synthesis of 35b
To a solution of 34b (215 mg, 0.376 mmol) and pyridine (304 μL, 3.76 mmol) in 15 mL CH2Cl2 was added MsCl (146 μL, 1.88 mmol) slowly at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 24 hours before it was quenched with aqueous saturated NH4Cl. The mixture was extracted with ether and washed with 1N HCl, water and brine, dried over MgSO4, filtered, and concentrated. The residue was purified by flash chromatography (hexane / EtOAc : 92 / 8) to give the desired product (220 mg, 90%).
1H NMR (400 MHz, CDCl3): δ 6.63 (d, J = 8.8 Hz, 1H), 6.50 (s, 1H), 6.48 (d, J = 8.4 Hz, 1H), 6.03 (d, J = 9.6 Hz, 1H), 5.95 (d, J = 9.2 Hz, 1H), 4.35-4.41 (m, 1H), 4.23-4.29 (m, 1H), 3.79 (s, 3H), 3.76 (t, J = 8.4 Hz, 1H), 2.83 (s, 3H), 2.27-2.38 (m, 3H), 1.94-2.06 (m, 2H), 1.69-1.75 (m, 1H), 1.49-1.56 (m, 1H), 1.07 (s, 3H), 1.00 (s, 9H), 0.91 (s, 9H), 0.17 (s, 3H), 0.16 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.0, 146.7, 142.8, 137.2, 135.5, 125.2, 120.9, 117.7, 115.0, 111.2, 77.9, 77.8, 66.3, 60.9, 50.3, 47.0, 37.2, 34.4, 30.5, 25.8, 25.7, 20.8, 18.2, 18.0, 14.0, −4.5, −4.6, −4.7, −4.9. FT-IR (NaCl, cm−1): 2955, 2926, 2858, 1475, 1361, 1254. HR-MS (FAB+) calcd for C33H54O7Si2S: 650.3129, found 650.3131.
Synthesis of 36a
To a vial with 2 mL THF solution of 35a (46.6 mg, 0.062 mmol) was added TBAF (1.0 M in THF, 0.065 mL). After stirring for 5 min, the reaction mixture was heated at 70 °C for 30 min. The vial was cooled to r.t. and all the volatiles were evaporated. Flash column chromatography (hexanes:EtOAc = 9:1) gave the desired product 36a (27.3 mg) in 94% yield.
1H NMR (400 MHz, CDCl3): δ 6.86 (d, J = 10.0 Hz, 1H), 6.53 (s, 1H), 6.25 (d, J = 9.6 Hz, 1H), 6.22 (d, J = 10.0 Hz, 1H), 6.05 (d, J = 9.6 Hz, 1H), 5.15 (d, J = 6.0 Hz, 1H), 5.10 (d, J = 6.0 Hz, 1H), 3.85 (t, J = 8.4 Hz, 1H), 3.51 (s, 3H), 2.31 – 1.96 (m, 5H), 1.93 – 1.86 (m, 1H), 1.80 – 1.65 (m, 2H), 1.60 – 1.50 (m, 1H), 0.95 (s, 3H), 0.91 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 181.8, 150.5, 147.1, 144.2, 142.4, 140.7, 129.1, 125.5, 115.5, 97.7, 85.2, 77.3, 77.1, 57.5, 47.5, 46.1, 37.7, 30.3, 29.4, 25.8, 19.2, 18.0, 15.0, −4.4, −4.9. [α]D20: +109 (c = 2.20, CH2Cl2). IR (NaCl, cm−1): 2954, 2928, 2896, 2855, 1659, 1633, 1583, 1149, 1119, 1018. HR-MS (FAB+) calcd for C27H39O5Si (M + 1): 471.2567, found 471.2576.
Synthesis of 36b
To a solution of mesylate 35b (300.8 mg, 0.462 mmol) in 18 mL THF was added TBAF (1.0 M in THF, 508 μL, 0.508 mmol). The reaction mixture was stirred for 15 min at room temperature, and then heated to 130 °C for 1 h (oil bath). After it was cooled to room temperature, it was quenched with aqueous saturated NH4Cl. The mixture was extracted with EtOAc three times. The combined organic layer was washed with water and brine, dried over MgSO4, filtered, and concentrated. The residue was purified by flash column chromatography (hexane / EtOAc : 90 / 10) to give the desired product 36b (194 mg, 95%).
1H NMR (400 MHz, CDCl3): δ 6.83 (d, J = 10.0 Hz, 1H), 6.45 (s, 1H), 6.22 (d, J = 9.6 Hz, 1H), 6.19 (d, J = 10.0 Hz, 1H), 6.03 (d, J = 9.6 Hz, 1H), 3.83 (t, J = 8.4 Hz, 1H), 3.78 (s, 3H), 1.94-2.27 (m, 5H), 1.84-1.89 (s, 1H), 1.67-1.74 (m, 2H), 1.52-1.58 (m, 1H), 0.93 (s, 3H), 0.89 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 181.8, 150.2, 146.9, 143.7, 143.4, 142.2, 129.2, 125.4, 115.3, 85.1, 77.15, 77.09, 60.6, 47.4, 46.1, 37.6, 30.3, 29.4, 25.7, 19.1, 17.9, 14.9, −4.5, −4.9. FT-IR (NaCl, cm−1): 2955, 2858, 1775, 1731, 1659, 1631, 1582, 1462, 1441, 1399. [α]D19: −126.47 (c 0.45, CHCl3). HR-MS (FAB+) calcd for C26H37O4Si: 441.2461; found 441.2471.
Synthesis of 38a
To a stirred solution of 36a (51 mg, 0.11 mmol) in 2 mL THF was added L-Selectride (1.0 M, 0.12 mL) at −78 °C. 20 min later, 5,5-dibromo-Meldrum’s acid 37 (39.5 mg, 0.13 mmol) was added in one portion. 0.5 hr later, the reaction was quenched by adding NH4Cl (sat.), then EtOAc and brine were added, and the separated aqueous phase was re-extracted with EtOAc twice. The combined organic phase was dried over MgSO4 and filtered, and the filtrate was evaporated and flash column chromatography (hexanes:EtOAc = 9:1) gave the desired product 38a (38 mg, 0.069 mmol, 62% yield). It is recommended that the product be used in the next step as soon as possible.
Selected characterization of 38a: 1H NMR (400 MHz, CDCl3) key peaks: δ 6.46 (s, 1H, a-Br-38a), 6.41 (s, 1H, e-Br-38a), 4.80 (dd, J = 14.0, 5.2 Hz, 1H, e-Br-38a), 4.68 (dd, J = 4.8, 2.0 Hz, 1H, a-Br-38a). HR-MS (FAB+) calcd for C27H40O 795BrSi (M + 1): 551.1828, found 551.1854.
Synthesis of 3,4-dihydro-36b
To a cooled (−78 °C) solution of 36b (37.1 mg, 84.2μmol; azeotroped twice with benzene) in THF (4 mL) was added L-Selectride (1.0 M in THF, 92μL, 92μmol) dropwise. The clear, yellow solution was stirred for 22 min at −78 °C, then quenched by successive addition of MeOH (0.10 mL), NaOH (1.0 M in H2O, 0.50 mL), and H2O2 (30 wt % in H2O, 80μL). The mixture was allowed to warm to room temperature with vigorous stirring. The resulting clear, dull yellow solution was then diluted with EtOAc (12 mL) and brine (6 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 6 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated to give a cloudy yellow oil. Purification by flash chromatography (10% EtOAc/hexanes) afforded the desired trienone product 3,4-dihydro-36b as a clear, yellow oil in 80% yield (30.0 mg, 67.8 μmol).
1H NMR (400 MHz, CDCl3): δ 6.38 (s, 1H), 6.25 (d, J = 9.4 Hz, 1H), 6.04 (d, J = 9.4 Hz, 1H), 3.83 (t, J = 8.2 Hz, 1H), 3.71 (s, 3H), 2.67 – 2.58 (m, 1H), 2.52 (td, J = 14.3, 4.4 Hz, 1H), 2.43 (td, J = 14.3, 4.8 Hz, 1H), 2.24 – 2.11 (m, 3H), 2.10 – 1.99 (m, 3H), 1.96 – 1.87 (m, 1H), 1.79 – 1.63 (m, 2H), 1.59 – 1.48 (m, 1H), 0.93 (s, 3H), 0.90 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 193.2, 152.4, 145.7, 143.2, 142.7, 125.4, 115.4, 82.4, 79.2, 77.2, 60.6, 47.4, 46.2, 36.4, 35.1, 33.2, 31.5, 30.3, 25.8, 19.3, 18.0, 14.6, −4.4, −4.9. [α]D19: −70.28 (c = 0.88, CHCl3). IR (NaCl, cm−1): 2955, 2887, 2857, 1672, 1585, 1464, 1440, 1367, 1121. HR-MS (FAB+) calcd for C26H39O4Si (M+1): 443.2613, found 443.2602.
Synthesis of 3-bromo-3,4-dihydro-36b (38b)
To a cooled (−78 °C) solution of trienone 3,4-dihydro-36b (35.8 mg, 80.9 μmol; azeotroped twice with benzene) in THF (1.5 mL) was added LiHMDS (1.0 M in THF, 89 μL, 89 μmol) quickly down the side of the flask. The resulting clear, bright yellow solution was stirred at −78 °C for 20 min. A clear, colorless solution of 37 (1.0 M in THF, 100 μL, 100 μmol; freshly prepared) was then added quickly down the side of the flask. After stirring at −78 °C for 50 min (TLC indicated some starting material remained; some di-bromination is also generally observed), the reaction was quenched with Na2S2O3 (10% in H2O, 10 mL) and allowed to warm to room temperature with vigorous stirring. The mixture was then extracted with Et2O (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated to give a yellow solid. Purification by flash chromatography (5% EtOAc/hexanes) afforded 38b as a yellow foam in 73% yield (30.7 mg, 58.9 μmol, 1:1 mixture of diastereomers).
1H NMR (500 MHz, CDCl3, peaks for both diastereomers): δ 6.39 (s, 1H), 6.35 (s, 1H), 6.30 (d, J = 9.3 Hz, 1H), 6.29 (d, J = 9.3 Hz, 1H), 6.04 (d, J = 9.5 Hz, 1H), 6.04 (d, J = 9.4 Hz, 1H), 4.77 (dd, J = 14.1, 5.0 Hz, 1H), 4.66 (dd, J = 4.7, 2.2 Hz, 1H), 3.84 (t, J = 8.2 Hz, 1H), 3.83 (t, J = 8.2 Hz, 1H), 3.74 (s, 3H), 3.74 (s, 3H), 2.96 (dd, J = 15.0, 4.8 Hz, 1H), 2.91 (dd, J = 14.0, 12.3 Hz, 1H), 2.66 (dd, J = 12.2, 5.0 Hz, 1H), 2.48 (dd, J = 15.0, 2.0 Hz, 1H), 2.48 – 2.36 (m, 2H), 2.22 – 2.09 (m, 6H), 2.09 – 1.99 (m, 2H), 1.97 – 1.89 (m, 2H), 1.74 – 1.64 (m, 4H), 1.60 – 1.49 (m, 2H), 0.94 (s, 3H), 0.93 (s, 3H), 0.90 (s, 18H), 0.06 (s, 6H), 0.04 (s, 6H). 13C NMR (75 MHz, CDCl3, peaks for both diastereomers): δ 186.2, 186.1, 153.2, 153.1, 146.2, 145.8, 143.6, 143.5, 141.7, 141.5, 125.4, 125.3, 115.1, 115.0, 82.6, 81.5, 78.3, 77.6, 77.1, 60.7, 60.2, 48.4, 47.6, 47.5, 46.0, 45.9, 45.2, 44.8, 40.9, 38.2, 36.6, 31.4, 31.2, 30.3, 25.8, 19.2, 18.0, 14.6, −4.4, −4.9. [α]D21: −16.6 (c = 0.91, CH2Cl2).IR (NaCl, cm−1): 2954, 2930, 2884, 2856, 1671, 1580, 1471, 1462, 1439, 1367, 1312, 1302, 1283, 1258, 1213, 1193, 1169, 1120, 1093, 1046, 1031, 1013, 952, 903, 874, 860, 837, 776, 740. HR-MS (FAB+) calcd for C26H38O 794BrSi (M+1): 521.1723, found 521.1714.
Synthesis of 40b
To a clear, pale yellow solution of 38b (18.2 mg, 34.9 μmol; azeotroped twice with benzene) in DMF (0.70 mL) was added sodium azide (3.4 mg, 52.3 μmol). The resulting mixture was stirred at room temperature for 1 hr, then a second portion of sodium azide (3.4 mg, 52.3 μmol) was added. A reddish brown color was observed over time. After a total reaction time of 5hrs, TLC analysis indicated consumption of starting material. To this mixture was added paraformaldehyde (11.0 mg, 0.366 mmol, based on monomer), sodium triacetoxyborohydride (37.1 mg, 0.175 mmol), and glacial acetic acid (2 μL, 34.9μmol). The cloudy, orange-brown suspension was then heated in a 50 °C oil bath. Additional portions of paraformaldehyde were added at 2 hrs (11.0 mg, 0.366 mmol) and 5 hrs (5.6 mg, 0.186 mmol). After a total reaction time of 6 hrs, the reaction was quenched at room temperature by dropwise addition NaOH (1.0 M in H2O, 7 mL). The aqueous phase was saturated by the addition of solid NaCl and extracted with Et2O (3 × 14 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated to give a clear, yellow-orange oil. Purification by flash chromatography (8% EtOAc/hexanes with 1% Et3N) afforded 40b as a clear, yellow-orange oil in 69% yield (11.7 mg, 24.2 μmol).
1H NMR (400 MHz, CDCl3): δ 6.44 (s, 1H), 6.23 (d, J = 9.3 Hz, 1H), 6.05 (d, J = 9.5 Hz, 1H), 5.79 (s, 1H), 3.84 (dd, J = 8.5, 7.8 Hz, 1H), 3.78 (s, 3H), 2.72 (s, 6H), 2.28 (dd, J = 12.3, 7.4 Hz, 1H), 2.24 – 1.93 (m, 4H), 1.93 – 1.66 (m, 3H), 1.63 – 1.48 (m, 1H), 0.95 (s, 3H), 0.91 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 179.6, 150.4, 147.0, 143.8, 142.5, 142.0, 125.5, 122.1, 115.2, 84.6, 77.1, 76.4, 60.6, 47.4, 46.3, 42.0, 38.3, 30.4, 29.4, 25.8, 19.2, 18.0, 15.0, −4.4, −4.9. [α]D20: −148.3 (c = 0.515, CH2Cl2). IR (NaCl, cm−1): 2953, 2857, 2360, 2342, 1650, 1588, 1471, 1462, 1439, 1375, 1322, 1283, 1258, 1215, 1157, 1120, 1098, 1051, 1014, 968, 904, 887, 868, 837, 799, 776. HR-MS (FAB+) calcd for C28H42O4NSi (M+1): 484.2883, found 484.2893.
Synthesis of 42b
To a clear, yellow solution of 40b (2.9 mg, 6.0 μmol) in CH2Cl2 (0.30 mL) was added sodium triacetoxyborohydride (9.2 mg, 43μmol) and glacial acetic acid (3μL, 52μmol). After 36 hrs, the reaction was quenched by carefully adding it to another flask containing NaOH (1.0 M in H2O, 3 mL). The aqueous phase was saturated by the addition of solid NaCl, then the layers were separated, and the aqueous phase was extracted with EtOAc (3 × 3 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated to give a clear, golden yellow oil. The amine product has a tendency to undergo spontaneous oxidation back to the enamine (and decompose by other pathways), and thus the crude amine 42b was generally carried directly into the next step without further purification.
Synthesis of 43b
To a cooled (−78 °C) solution of the crude aminoketone (from the previous step) in THF (0.40 mL) was added DIBAL-H (1.0 M in toluene, 10 μL, 10 μmol). The clear, yellow solution was stirred at −78 °C for 30 min, then a second portion of DIBAL-H (1.0 M in toluene, 10 μL, 10 μmol) was added. After a total reaction time of 50 min, the reaction was quenched at −78 °C by the addition of EtOAc (50 μL). Saturated aqueous Rochelle salt (2 mL) was then added, and the mixture was stirred vigorously for ca. 1.5 hrs while allowing to warm to room temperature, resulting in a biphasic mixture with a clear, yellow layer on top, and a clear, colorless layer below. The aqueous phase was extracted with EtOAc (3 × 3 mL), then the combined organic layers were dried (MgSO4), filtered, and concentrated to give a clear, orange-yellow oil. Purification by flash chromatography (5–20% MeOH/CH2Cl2) afforded 43b as a minor product in < 20% yield and moderate purity (ca. 0.5 mg).
Selected characterization of 43b: 1H NMR (500 MHz, CDCl3) key peaks: δ 6.21 (s, 1H), 5.97 (d, J = 9.4 Hz, 1H), 5.93 (d, J = 9.4 Hz, 1H), 4.39 (d, J = 8.5 Hz, 1H), 3.79 (t, J = 8.2 Hz, 1H), 3.74 (s, 3H), 2.82 – 2.74 (m, 1H), 2.37 (s, 6H). HR-MS (FAB+) calcd for C28H46O4NSi (M+1): 488.3196, found 488.3181.
Synthesis of 42a
To a stirred solution of 38a (111.9 mg, 0.203 mmol) in 5 mL THF was added tetrabutylammonium azide (103.0 mg, 0.363 mmol) in one portion, and 50 min later, the solvent was evaporated and the crude mixture of 40a was covered by argon and directly used in the next step.
The crude mixture of 40a was re-dissolved in dichloromethane and NaBH3(CN) (36 mg) was added, followed by adding HOAc (0.40 mL) dropwise. 5 min later, 138 mg NaBH3(CN) was added in 3 portions, and the reaction mixture was stirred for 10 min before being quenched by adding NaHCO3 (sat.) slowly. The two phases were separated and the aqueous phase was re-extracted by dichloromethane, and the combined dichloromethane solution was evaporated and immediately used in the next step. The product 41a is not very stable to air, and rapid operation is recommended.
To a stirred solution of 41a in 10 mL methanol was added a methanol solution (10 mL) of CeCl3·7H2O (0.18 g), and stirring continued for 2 min at 0 °C. NaBH4 (0.20 g + 0.24 g + 0.21 g) was added in three portions with an interval of 15 min, and the total reaction lasted for 1 hr. Methanol was evaporated and Et2O, brine and NH3·H2O (28%) were added, and the organic phase was separated and the aqueous phase was re-extracted with Et2O twice. The combined organic phase was dried over Na2SO4, and prep-TLC (dichloromethane:methanol = 5:1, with a few drops of NH3·H2O) gave the desired product 42a (41.4 mg, 0.085 mmol, 42% yield from 38a).
42a
1H NMR (400 MHz, CDCl3): δ 6.13 (s, 1H), 6.00 (d, J = 9.2 Hz, 1H), 5.92 (d, J = 9.6 Hz, 1H), 4.98 (d, J = 6.4 Hz, 1H), 4.89 (d, J = 6.4 Hz, 1H), 4.06 (d, J = 8.0 Hz, 1H), 3.79 (t, J = 8.0 Hz, 1H), 3.52 (s, 3H), 3.01 (m, 1H), 2.21 – 1.45 (m, 11H), 0.90 (s, 12H, C18 methyl and tert-butyl), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 145.2, 144.5, 138.0, 126.1, 125.6, 114.9, 96.6, 82.1, 78.6, 77.5, 73.7, 56.8, 52.6, 47.0, 46.4, 41.4, 39.4, 33.3, 30.6, 25.7, 19.2, 18.0, 14.3, −4.5, −5.0. [α]D19: +2.18 (c = 1.51, CH2Cl2). IR (NaCl, cm−1): 3351, 3287, 2954, 2931, 2886, 2858, 1639, 1470, 1386, 1360, 1303, 1254, 1155, 1119, 1009. HR-MS (FAB+) calcd for C27H44O5NSi (M + 1): 490.2989, found 490.3006.
Synthesis of 45
To a stirred solution of 36a (27.3 mg, 0.057 mmol) in 1 mL THF was added 0.063 mL L-Selectride (1.0 M in THF) at −78 °C. 5 min later, 0.10 mL H2O was added and the whole reaction mixture was warmed to 0 °C. Next, 2 mL MeOH was added, followed by CeCl3·7H2O (36.3 mg) and then NaBH4 (15.3 mg). 5 min later, aqueous NH4Cl solution (sat.) was added, followed by Et2O and brine. After separation, the aqueous layer was re-extracted by Et2O twice. The combined organic layer was dried over Na2SO4, and after evaporation of the solvents, prep-TLC (hexanes : EtOAc = 2 : 1) gave the desired product 45 (18.0 mg, 65% yield from 36a).
1H NMR (400 MHz, CDCl3): δ 6.14 (s, 1H), 5.99 (d, J = 9.6 Hz, 1H), 5.92 (d, J = 9.6 Hz, 1H), 4.95 (d, J = 6.4 Hz, 1H), 4.89 (d, J = 6.4 Hz, 1H), 4.44 (t, J = 8.0 Hz, 1H), 3.79 (t, J = 8.0 Hz, 1H), 3.52 (s, 3H), 3.18 (s, 1H), 2.34 – 2.26 (m, 1H), 2.22 – 2.08 (m, 2H), 2.08 – 1.84 (m, 5H), 1.84 – 1.76 (m, 1H), 1.74 – 1.58 (m, 3H), 1.57 – 1.46 (m, 1H), 0.89 (s, 12H), 0.04 (s, 3H), 0.02 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 147.1, 145.5, 138.1, 125.6, 115.0, 97.1, 82.1, 79.5, 77.6, 66.2, 56.8, 47.1, 46.6, 39.0, 33.3, 32.4, 30.6, 28.8, 25.8, 19.3, 18.0, 14.4, −4.4, −4.9. [α]D20: −5.14 (c = 2.92, CH2Cl2). IR (NaCl, cm−1): 3424, 2954, 2886, 2859, 1637, 1462, 1388, 1361, 1256, 1156, 1120, 1100, 1006, 956, 869, 837, 776. HR-MS (FAB+) calcd for C27H42O5Si (M+): 474.2802, found 474.2812.
Acetylation of 45
Compound 45 (15.7 mg, 0.033 mmol) was dissolved in 1 mL CH2Cl2. 0.05 mL pyridine and 5.3 mg DMAP were added, followed by 0.03 mL Ac2O. The reaction was quenched 20 min later by adding MeOH, and aqueous NaHCO3 (sat.) was added 2 min later. The aqueous mixture was extracted with Et2O three times, and the combined organic layer was dried over MgSO4. After evaporation of the solvents, prep-TLC (hexanes : EtOAc = 3 : 1) gave the desired product (13.6 mg, 0.026 mmol) in 79% yield.
1H NMR (400 MHz, CDCl3): δ 6.20 (s, 1H), 6.01 (d, J = 9.6 Hz, 1H), 5.93 (d, J = 9.6 Hz, 1H), 5.66 (dd, J = 8.8, 7.2 Hz, 1H), 4.87 (d, J = 6.4 Hz, 1H), 4.76 (d, J = 6.0 Hz, 1H), 3.80 (t, J = 8.0 Hz, 1H), 3.43 (s, 3H), 2.43 – 2.34 (m, 1H), 2.09 (s, 3H), 2.26 – 1.46 (m, 12H), 0.90 (s, 3H), 0.90 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 170.6, 145.8, 142.3, 138.4, 130.1, 125.6, 115.0, 97.2, 82.2, 79.1, 77.6, 68.8, 56.9, 47.1, 46.5, 38.6, 33.1, 32.3, 30.6, 26.5, 25.8, 21.2, 19.3, 18.0, 14.4, −4.4, −4.9. [α]D17: +35.1 (c = 1.36, CH2Cl2). IR (NaCl, cm−1): 2954, 2926, 2856, 1739, 1636, 1472, 1369, 1246, 1157, 1120, 1102, 1050, 1013, 951, 895, 867, 837, 775. HR-MS (FAB+) calcd for C29H44O6Si (M+): 516.2907, found 516.2934.
Synthesis of 58
To a stirred solution of 43a (41.4 mg, 0.085 mmol) in 3 mL dioxane was added 0.50 mL 10% aq. Na2CO3, and then a solution of 125.0 mg Fmoc-OSu in 2 mL dioxane was added. 2 hrs later, the reaction mixture was evaporated and the residue was taken up by Et2O, brine and NH4Cl (sat.), and separated. The aqueous phase was re-extracted by Et2O twice, and the combined organic phase was dried over Na2SO4, and then evaporated. The crude mixture of 44a was directly used in the next step.
The mixture of 44a was dissolved in 20 mL dichloromethane, and 4-dimethylaminopyridine (48.0 mg) was added, followed by pyridine (0.40 mL). Acetic anhydride (0.25 mL) was then added dropwise. 10 min later, all volatiles were evaporated and the residue was purified by flash column chromatography (hexanes:EtOAc = 4:1) to give the desired product 58 (44.5 mg, 0.059 mmol) in 70% yield for 2 steps.
58
1H NMR (500 MHz, CDCl3): δ 7.76 (d, J = 7.5 Hz, 2H), 7.58 (d, J = 7.0 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.32 (d, J = 7.5 Hz, 2H), 6.16 (s, 1H), 6.05 (d, J = 9.0 Hz, 1H), 5.94 (d, J = 9.5 Hz, 1H), 5.68 (d, J = 8.5 Hz, 1H), 5.17 (d, J = 7.5 Hz, 1H), 4.86 (d, J = 6.0 Hz, 1H), 4.77 (d, J = 6.0 Hz, 1H), 4.40 – 4.28 (m, 2H), 4.22 (t, J = 7.5 Hz, 1H), 4.06 – 4.00 (m, 1H), 3.80 (t, J = 8.0 Hz, 1H), 3.43 (s, 3H), 2.28 – 1.48 (m, 14H), 0.91 (s, 3H), 0.90 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.6, 155.8, 146.3, 143,8, 141.2, 140.0, 139.0, 130.0, 127.7, 127.0, 125.4, 125.1, 119.9, 114.5, 97.5, 82.4, 77.9, 77.5, 71.7, 66.9, 57.0, 51.2, 47.2, 47.1, 46.4, 39.4, 38.5, 33.0, 30.5, 25.8, 21.1, 19.2, 18.0, 14.3, −4.4, −4.9. [α]D18: +65.27 (c = 2.95, CH2Cl2). IR (NaCl, cm−1): 3314, 2955, 2930, 2886, 2857, 1736, 1694, 1641, 1549, 1450, 1375, 1297, 1260, 1233, 1159, 1119, 1016. HR-MS (FAB+) calcd for C44H55O8NSi (M+): 753.3697, found 753.3690.
Synthesis of 61
To a solution of 58 (43.8 mg, 0.058 mmol) in 2 mL THF was added 0.5 mL water, and the reaction mixture was cooled to 0 °C. Then 0.10 mL solution of Br2 in THF (made by adding 0.033 mL Br2 into 1.0 mL THF) was added dropwise, and the whole reaction mixture was stirred for 2 min before quenching by 10% aqueous solution of Na2S2O3. Et2O and brine were added, and the two layers were separated. The organic layer was re-extracted by Et2O twice. The combined organic phase was dried over MgSO4, and after filtration, the solvent was evaporated. The crude mixture 59 (about 49.1 mg) was directly used in the next step. This procedure was used for other bromine-induced MOM deprotection reactions.
The crude mixture of 59 was evenly divided into three batches. In each batch, 16.4 mg crude 59 was dissolved in 0.8 mL C6D6 in a J-Young NMR tube, and then a C6D6 solution of AIBN (2 mg in 0.20 mL C6D6) was added. The whole mixture was degassed twice (freeze-pump-thaw) and refilled with Ar. Then 0.030 mL n-Bu3SnH was added, and the reaction mixture was further degassed four times before being immersed in a 70 °C oil bath. The reaction was carefully monitored by 1H NMR, and 4 hrs later, the reaction mixture was cooled and combined. After evaporating all the volatiles, prep-TLC (hexanes:EtOAc = 3 :2) gave 11.5 mg crude product 60 which was used directly in the next step.
Intermediate 60 was dissolved in 1 mL dichloromethane/MeOH (1:1) solution, and the reaction mixture was cooled to 0 °C. CeCl3•7H2O (7.6 mg) was added, and NaBH4 (5.2 mg) was added to this stirred mixture 2 min later. The reaction was quenched by NH4Cl (sat.) after 5 min, and brine and dichloromethane were added. The separated aqueous layer was re-extracted with dichloromethane twice, and the combined organic phase was dried over MgSO4. After filtration, the solvent was evaporated and prep-TLC gave 4.4 mg crude reduction product, which was directly used in the next step.
The Luche reduction product was dissolved in 1 mL dichloromethane, and 5.5 mg DMAP and 0.040 mL pyridine were added, followed by dropwise addition of 0.030 mL Ac2O. MeOH was added 10 min later and after 2 min, all volatiles were evaporated and prep-TLC (hexanes:EtOAc = 3:2) gave the desired product 61 (2.9 mg, 7% from 58).
61
1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 6.8 Hz, 2H), 7.40 (t, J = 7.2 Hz, 2H), 7.31 (d, J = 7.6 Hz, 2H), 5.81 (s, 1H), 5.61 (d, J = 9.2 Hz, 1H), 5.44 (m, 1H), 5.17 (d, J = 8.4 Hz, 1H), 4.92 (t, J = 8.8 Hz, 1H), 4.34 (m, 2H), 4.20 (t, J = 7.2 Hz, 1H), 3.93 (m, 1H), 3.77 (t, J = 8.4 Hz, 1H), 2.15 (s, 3H), 2.02 (s, 3H), 2.36 – ca. 1.20 (m, 13H), 0.89 (s, 9H), 0.74 (s, 3H), 0.04 (s, 6H). IR (NaCl, cm−1): 2954, 2928, 2857, 1741, 1540, 1450, 1376, 1278, 1246, 1143, 1106, 1021. HR-MS (FAB+) calcd for C44H56O8NSi (M + 1): 754.3775, found 754.3773.
Supplementary Material
Acknowledgement
This work is generously supported by National Institute of Health (HL 25848). Z. W. thanks Eli Lilly for a helpful graduate fellowship (2008 - 2009), M. D. thanks Bristol-Myers Squibb, Guthikonda family, and Roche for financial support. P. K. P. thanks Amgen for a postdoctoral fellowship (2009 - 2010). We thank John Hartung Jr., Feng Peng and Dr. Ping Wang for helpful discussions, and Ms. Rebecca Wilson for preparation of this manuscript.
References
- i(a).Wilson RM, Danishefsky SJ. J. Org. Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]; (b) Wilson RM, Danishefsky SJ. Angew. Chem. Int. Ed. 2010;49:6032–6056. doi: 10.1002/anie.201000775. [DOI] [PubMed] [Google Scholar]
- ii(a).Rivkin A, Chou T-C, Danishefsky SJ. Angew. Chem. Int. Ed. 2005;44:2838–2850. doi: 10.1002/anie.200461751. [DOI] [PubMed] [Google Scholar]; (b) Chou T-C, Zhang X, Zhong Z-Y, Li Y, Feng L, Eng S, Myles DR, Johnson R, Wu N, Yin YI, Wilson RM, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2008;105:13157–13162. doi: 10.1073/pnas.0804773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iii(a).Gaul C, Njardarson JT, Danishefsky SJ. J. Am. Chem. Soc. 2003;125:6042–6043. doi: 10.1021/ja0349103. [DOI] [PubMed] [Google Scholar]; (b) Njardarson JT, Gaul C, Shan D, Huang XY, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:1038–1040. doi: 10.1021/ja039714a. [DOI] [PubMed] [Google Scholar]; (c) Oskarsson T, Nagorny P, Krauss IJ, Perez L, Mandal M, Ouerfelli O, Xiao D, Moore MAS, Massague J, Danishefsky SJ. J. Am. Chem. Soc. 2010;132:3224–3228. doi: 10.1021/ja9101503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shan D, Chen L, Njardarson JT, Gaul C, Ma X, Danishefsky SJ, Huang XY. Proc. Natl. Acad. Sci. USA. 2005;102:3772–3776. doi: 10.1073/pnas.0500658102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lecomte N, Njardarson JT, Nagorny P, Yang G, Downey RJ, Ouerfelli O, Moore MAS, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:15074–15078. doi: 10.1073/pnas.1015247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iv(a).Yun HD, Danishefsky SJ. J. Org. Chem. 2003;68:4519–4522. doi: 10.1021/jo0341665. [DOI] [PubMed] [Google Scholar]; (b) Yun HD, Chou TC, Dong H, Tian Y, Li YM, Danishefsky SJ. J. Org. Chem. 2005;70:10375–10380. doi: 10.1021/jo0515475. [DOI] [PubMed] [Google Scholar]; (c) Chou TC, Dong H, Zhang X, Lei X, Hartung J, Zhang Y, Lee JH, Wilson RM, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:14336–14341. doi: 10.1073/pnas.1111332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- v(a).Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. J. Am. Chem. Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h. Watanabe Y, Aoki S, Tanabe D, Setiawan A, Kobayashi M. Tetrahedron. 2007;63:4074–4079. Aoki S, Watanabe Y, Tanabe D, Setiawan A, Arai M, Kobayashi M. Tetrahedron Lett. 2007;48:4485–4488. Unless otherwise stated, all numberings are based on the numbering system of cortistatin A.
- vi.Figg WD, Folkman J. Angiogenesis – An Integrative Approach from Science to Medicine. Springer Science + Business Media, LLC; 2008. pp. v–vi.pp. 1–14. [Google Scholar]
- vii.Folkman J. Nat. Rev. Drug Discovery. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- viii.Nicolaou KC, Peng X-S, Sun Y-P, Polet D, Zou B, Lim CS, Chen DY-K. J. Am. Chem. Soc. 2009;131:10587–10597. doi: 10.1021/ja902939t. [DOI] [PubMed] [Google Scholar]
- ix.Aoki S, Watanabe Y, Tanabe D, Arai M, Suna H, Miyamoto K, Tsujibo H, Tsujikawa K, Yamamoto H, Kobayashi M. Bioorg. Med. Chem. 2007;15:6758–6762. doi: 10.1016/j.bmc.2007.08.017. [DOI] [PubMed] [Google Scholar]
- x.Cee VJ, Chen DY-K, Lee MR, Nicolaou KC. Angew. Chem. Int. Ed. 2009;48:8952–8957. doi: 10.1002/anie.200904778. [DOI] [PubMed] [Google Scholar]
- xi(a).Shenvi RA, Guerrero CA, Shi J, Li C-C, Baran PS. J. Am. Chem. Soc. 2008;130:7241–7243. doi: 10.1021/ja8023466. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shi J, Manolikakes G, Yeh C-H, Guerrero CA, Shenvi RA, Shigehisa H, Baran PS. J. Am. Chem. Soc. 2011;133:8014–8027. doi: 10.1021/ja202103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xii(a).Nicolaou KC, Sun Y-P, Peng X-S, Polet D, Chen DY-K. Angew. Chem. Int. Ed. 2008;47:7310–7313. doi: 10.1002/anie.200803550. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Peng X-S, Sun Y-P, Polet D, Zou B, Lim CS, Chen DY-K. J. Am. Chem. Soc. 2009;131:10587–10597. doi: 10.1021/ja902939t. [DOI] [PubMed] [Google Scholar]
- xiii.Lee HM, Nieto-Oberhuber C, Shair MD. J. Am. Chem. Soc. 2008;130:16864–16866. doi: 10.1021/ja8071918. [DOI] [PubMed] [Google Scholar]
- xiv.Flyer AN, Si C, Myers AG. Nat. Chem. 2010;2:886–892. doi: 10.1038/nchem.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xv(a).Yamashita S, Iso K, Hirama M. Org. Lett. 2008;10:3413–3415. doi: 10.1021/ol8012099. [DOI] [PubMed] [Google Scholar]; (b) Yamashita S, Kitajima K, Iso K, Hirama M. Tetrahedron Lett. 2009;50:3277–3279. [Google Scholar]; (c) Yamashita S, Iso K, Kitajima K, Himuro M, Hirama M. J. Org. Chem. 2011;76:2408–2425. doi: 10.1021/jo2002616. [DOI] [PubMed] [Google Scholar]
- xvi.Nilson MG, Funk RL. J. Am. Chem. Soc. 2011;133:12451–12453. doi: 10.1021/ja206138d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvii(a).Simmons EM, Hardin AR, Guo X, Sarpong R. Angew. Chem. Int. Ed. 2008;47:6650–6653. doi: 10.1002/anie.200802203. [DOI] [PubMed] [Google Scholar]; (b) Simmons EM, Hardin-Narayan AR, Guo XL, Sarpong R. Tetrahedron. 2010;66:4696–4700. doi: 10.1016/j.tet.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xviii(a).Liu L, Gao Y, Che C, Wu N, Wang DZ, Li CC, Yang Z. Chem. Commun. 2009:662–664. doi: 10.1039/b817376a. [DOI] [PubMed] [Google Scholar]; (b) Fang L, Chen Y, Huang J, Liu L, Quan J, Li C-C, Yang Z. J. Org. Chem. 2011;76:2479–2487. doi: 10.1021/jo102202t. [DOI] [PubMed] [Google Scholar]
- xix(a).Baumgartner C, Ma S, Liu Q, Stoltz BM. Org. Biomol. Chem. 2010;8:2915–2917. doi: 10.1039/c004275g. [DOI] [PubMed] [Google Scholar]; (b) Magnus P, Littich R. Org. Lett. 2009;11:3938–3941. doi: 10.1021/ol901537n. [DOI] [PubMed] [Google Scholar]; (c) Craft DT, Gung BW. Tetrahedron Lett. 2008;49:5931–5934. doi: 10.1016/j.tetlet.2008.07.155. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yu F, Li G, Gao P, Gong H, Liu Y, Wu Y, Cheng B, Zhai H. Org. Lett. 2010;12:5135–5137. doi: 10.1021/ol102058f. [DOI] [PubMed] [Google Scholar]; (e) Liu LL, Chiu P. Chem. Commun. 2011;47:3416–3417. doi: 10.1039/c1cc00087j. [DOI] [PubMed] [Google Scholar]
- xx(a).Shi J, Shigehisa H, Guerrero CA, Shenvi RA, Li C-C, Baran PS. Angew. Chem. Int. Ed. 2009;48:4328–4331. doi: 10.1002/anie.200901116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Czakó B, Kürti L, Mammoto A, Ingber DE, Corey EJ. J. Am. Chem. Soc. 2009;131:9014–9019. doi: 10.1021/ja902601e. [DOI] [PubMed] [Google Scholar]
- xxi.Chapelon A-S, Moraléda D, Rodriguez R, Ollivier C, Santelli M. Tetrahedron. 2007;63:11511–11616. [Google Scholar]
- xxii.Kametani T, Nemoto H. Tetrahedron. 1981;37:3–16. [Google Scholar]
- xxiii.Ananchenko SN, Torgov IV. Tetrahedron Lett. 1963;4:1553–1558. [Google Scholar]
- xxiv.Hajos ZG, Parrish DR. Org. Syn., Coll. Vol. 1990;7:363–368. [Google Scholar]
- xxv.Wilson RM, Danishefsky SJ. J. Org. Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- xxvi.Dai M, Danishefsky SJ. Heterocycles. 2009;77:157–161. doi: 10.3987/com-08-s(f)6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxvii.Tang Y, Oppenheimer J, Song Z, You L, Zhang X, Hsung RP. Tetrahedron. 2006;62:10785–10813. [Google Scholar]
- xxviii(a).Winstein S, Baird R. J. Am. Chem. Soc. 1957;79:756–757. [Google Scholar]; (b) Masamune S. J. Am. Chem. Soc. 1961;83:1009–1010. [Google Scholar]; (c) Lalic G, Corey EJ. Org. Lett. 2007;9:4921–4923. doi: 10.1021/ol702323s. [DOI] [PubMed] [Google Scholar]
- xxix(a).Dai M, Danishefsky SJ. Tetrahedron Lett. 2008;49:6610–6612. doi: 10.1016/j.tetlet.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dai M, Wang Z, Danishefsky SJ. Tetrahedron Lett. 2008;49:6613–6616. doi: 10.1016/j.tetlet.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxx.Matsumoto T, Sohma T, Hatazaki S, Suzuki K. Synlett. 1993:843–846. [Google Scholar]
- xxxi(a).Chauder BA, Kalinin AV, Taylor NJ, Snieckus V. Angew. Chem. Int. Ed. 1999;38:1435–1438. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1435::AID-ANIE1435>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]; (b) Chauder BA, Kalinin AV, Snieckus V. Synthesis. 2001:140–144. [Google Scholar]
- xxxii.Alvarez-Manzaneda EJ, Chahboun R, Pérez IB, Cabrera E, Alvarez E, Alvarez-Manzaneda R. Org. Lett. 2005;7:1477–1480. doi: 10.1021/ol047332j. [DOI] [PubMed] [Google Scholar]
- xxxiii.Li C, Johnson RR, Porco JA., Jr. J. Am. Chem. Soc. 2003;125:5095–5106. doi: 10.1021/ja021396c. [DOI] [PubMed] [Google Scholar]
- xxxiv.Smith AB, III, Sperry JB, Han Q. J. Org. Chem. 2007;72:6891–6900. doi: 10.1021/jo071146k. [DOI] [PubMed] [Google Scholar]
- xxxv.Seyden-Penne J. Reductions by the Alumino- and Borohydrides in Organic Synthesis. 2nd ed Wiley-VCH, Inc.; 1997. [Google Scholar]
- xxxvi.Ricci A. Modern Amination Methods. Wiley-VCH; Weinheim: 2000. [Google Scholar]
- xxxvii.O’Brien C. Chem. Rev. 1964;64:81–89. [Google Scholar]
- xxxviii.Evans DA, Faul MM, Bilodeau MT. J. Org. Chem. 1991;56:6744–6746. [Google Scholar]
- xxxix.Shawakfeh KQ, Al-Said NH, Al-Zoubi RM. Steroids. 2008;73:579–584. doi: 10.1016/j.steroids.2008.01.012. [DOI] [PubMed] [Google Scholar]
- xl.Luche JL. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]
- xli.Compound 45 was made from 36a by L-Selectride reduction followed by Luche reduction (one pot, 65% yield).
- xlii.Compound 50 was made by acetylation of 45 (Ac2O/py/DMAP in dichloromethane, 80% yield) followed by bromine-induced MOM deprotection (Br2/H2O-THF, 0 °C).
- xliii.Gavrilan M, André-Barrès C, Baltas M, Tzedakis T, Gorrichon L. Tetrahedron Lett. 2001;42:2465–2468. [Google Scholar]
- xliv.Liu L-G, Su X-D, Li Z-S. Syn. Comm. 1995;25:3113–3124. [Google Scholar]
- xlv.Najjar F, André-Barrès C, Lauricella R, Gorrichon L, Tuccio B. Tetrahedron Lett. 2005;46:2117–2119. [Google Scholar]
- xlvi.Wasserman HH, Lipshutz BH. Tetrahedron Lett. 1975;16:1731–1734. [Google Scholar]
- xlvii.Gersmann HR, Bickel AF. J. Chem. Soc., (B) 1971:2230–2237. [Google Scholar]
- xlviii.Li C, Johnson RR, Porco JA., Jr. J. Am. Chem. Soc. 2003;125:5095–5106. doi: 10.1021/ja021396c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.