

Abstract
The 1,2-diamine moiety is a ubiquitous structural motif present in a wealth of natural products, including non-proteinogenic amino acids and numerous alkaloids, as well as in pharmaceutical agents, chiral ligands and organic reagents. The biological activity associated with many of these systems and their chemical utility in general has ensured that the development of methods for their preparation is of critical importance. While a wide range of strategies for the preparation of 1,2-diamines have been established, the diamination of alkenes offers a particularly direct and efficient means of accessing these systems. The purpose of this review is to provide an overview of all methods of direct alkene diamination, metal-mediated or otherwise.
Keywords: alkene; aminoauration; aminocupration; aminomercuration; aminopalladation; aminothallation; bisnitration; catalysis; cycloguanidination; diaziridinone; bisazidonation; diamination; 1,2-diamine; 1,2-diaminoalkane; 1,2-diazide; dinitrogen tetroxide; dinitrogen trioxide; imidazolination; imidazolidinone; iodane; iodine azide; iodonium; guanidine; hypervalent iodine; nitroamination; nitrogen dioxide; nitrogen oxide; nitrosylation; organocatalysis; pseudonitrosite; sulfamide
1. Introduction
The 1,2-diamine moiety is a ubiquitous structural motif found in a wealth of natural products, including non-proteinogenic amino acids and alkaloids, in pharmaceutical agents, chiral ligands and bases, and organic reagents.1 The biological activity associated with many of these systems and their utility in general has ensured that the development of new methods for their preparation of is of critical importance. While a wide range of strategies for the preparation of 1,2-diamines have been established, the diamination of alkenes offers a particularly direct and efficient means of accessing these systems (Figure 1).
Figure 1.

General synthetic approaches to 1,2-diamines, including direct alkene difunctionalization.
Despite the importance of vicinal diamines, reviews concerning their preparation have, in the past, been rather infrequently published, in marked contrast to their 1,2-diol relatives.2 However, that a recent resurgence of interest in the metal-mediated diamination of alkenes is apparent from the number of reviews dedicated to this specific topic which have appeared over the last decade.3 These articles notwithstanding, the purpose of the current review is to provide an overview of all methods of alkene diamination, metal-mediated or otherwise. In this context, the term “direct alkene diamination” encompasses reactions that lead to the formation of vicinal Csp3-N bonds regardless of the substitution pattern or oxidation state of the nitrogen centers. Transformations in which carbon-nitrogen double bonds are formed, including the formation of α-amino-oximes during the photoaddition of N-nitroso compounds,4 will not be considered. Likewise, reactions in which 1,2-diamines are generated from alkenes with preexisting nitrogen-based substituents, e.g. the hydroamination of vinyl nitro compounds,5 will also not be discussed. Finally, since the hetero-Diels-Alder reaction of azo compounds has recently been reviewed in detail6 and involves 1,4- rather than 1,2-diamination of dienes, this subject will not be considered in the following article.
After an exposition of the occurrence and biological significance of 1,2-diamines, the review itself follows an essentially chronological path. Thus, the reaction of alkenes with binary nitrogen oxides and their surrogates is discussed first, followed by diamination processes that involve generation and addition of the azidyl radical. Methods employing haloamides, halogens, heavy metals and polyvalent iodine reagents then follow in sequence and finally, in the second half of the review, the metal-mediated diamination of alkenes under both stoichiometric and catalytic conditions will be discussed in depth.
2. Importance of 1,2-Diamines
2.1. Naturally Occurring 1,2-Diamines
1,2-Diamino carboxylic acids are widely distributed in the natural world, where they are found in an array of organisms in their both native state and as components of more complex natural products (Figure 2).7 Since they neither occur in proteins, or are coded for in the cellular genetic makeup, these amino acids are classified as non-proteinogenic.8 The simplest members of this group include 2,3-diaminopropionic (L-Dsp, 1) and 2,3-diaminobutanoic (Dap, 2) acid, which in addition to being found as components of non-ribosomal peptide antibiotics such as bleomycin9 and others,10 have also been isolated from an extraterrestrial source. Analysis of the Murchison meteorite has revealed the presence of a number of complex organic molecules, including 1 and 2, which may have participated in prebiotic polycondensation to form peptide nucleic acid material.11
Figure 2.

Naturally occurring, non-proteinogenic 1,2-diamino acids and their derivatives.
Much of the interest in native 1,2-diamino acids and in particular heterocyclic β-substituted alanine derivatives stems from their role as excitatory amino acids (EAA). EAA receptors are widely distributed in the mammalian central nervous system (CNS) and play a role in a range of neural functions and abnormalities, having been implicated in such disorders as Alzheimer’s disease, epilepsy, Parkinsonism and AIDS-related dementia.12 L-Quisqualic acid (3), isolated from the traditional Chinese medicine Shih-chun-tze, is a highly potent agonist of EAA receptors in both mammals and insects. Most recently, 3 was found as a component of the petals of zonal pelargoniums, and shown to act a potent antifeedant against the Japanese beetle, Popilla japonica.13 (-)-Dysibetaine (4), an unusual amino acid isolated from the marine sponge Dysidea herbacea, is also a neuroexcitotoxin, which may bind to the glutamate receptors present in the CNS of mice.14
As previously noted, 1,2-diamino acids are also found as components of non-ribosomal peptides. L-Capreomycidine (5), for example, is a key structural subunit of the tuberculostatic agent capreomycin 1B (6)15 while its β-epimer is found in the muraymycins, a family of uridylpeptide natural products that inhibit the peptidoglycan biosynthesis of Staphylococcus aureus.16,17 Although the broad spectrum antibiotic strephothricin (7) does not contain a capreomycidine residue, this bis-diamine may be biogenetically related to this amino acid.18 Diamino carboxylic acids also play an important role in glycobiology. DiN-acetylated uronic acid residues, for example, are found in the B-band O-antigen of the lipopolysaccharide (LPS) of a number of respiratory pathogens, where they are believed to play an important role in host colonization and maintenance of infection. In this regard, nucleotide sugar UDP-2,3-diacetamido-2,3-dideoxy-D-mannuronic acid (8) is a key building block in the biosynthesis of the LPS of Pseudomonas aeruginosa, an opportunistic pathogen.19
Many alkaloid natural products conspicuously contain the vicinal diamine motif and its presence is often associated with significant biological activity (Figure 3). Structural complexity runs the gamut from relatively simple systems such as the pyrrolizidine alkaloid loline (9),20 which despite it apparent simplicity presents a significant synthetic challenge,21 to the pentacycle citrinadin A (10)22,23 and its structural relatives PF1270 A-C, a group of histamine H3 receptor agonists.24 The 1,2-diamine functionality is also a unifying structural feature of the tetrahydroisoquinoline alkaloid family,25 of which as illustrated by the antibiotic lemonomycin (11),26,27 where it is embedded within the piperazine ring system at the core of all members of this large natural products class. In addition to terrestrial sources, marine organisms have also proven to be a rich source of biologically active 1,2-diamines, including the anti-tuberculosis agent manadomanzamine A (15),28,29 the antineoplastic agent agelastatin A (12)30 and eudistomin-K sulfoxide (13),31 a representative member of the eudistomin family that displays activity against both RNA and DNA viruses.32
Figure 3.

A representative selection of naturally occurring alkaloids that encompass the 1,2-diamine framework.
While a number of the alkaloids represented in Figure 2 have succumbed to total synthesis, most recently pactamycin (14),33,34 others including manadomanzamine A (15), remain unassailed and, a such, offer unique challenges for the development of new diamination methods. Despite the recent progress in diamination methodology, many of these targets present significant challenges to direct alkene amination methods and, as such, are an impetus to the continued investigation of diamine methods that offer enantiocontrol, differential N-protection and, importantly in the context of such complex targets, functional group compatibility.
2.2. 1,2-Diamine Pharmaceutical Agents
1,2-Diamines are found in wealth of non-natural, synthetic pharmacological tools and therapeutic agents, including several clinically approved drugs (Figure 4).35 For example, the presence of a diamine-based substituent at the 7-position has proven critical in the clinical efficacy of a number of fluoroquinoline antibiotics,36 including the fourth generation agent moxifloxacin (16) where this functionality is encompassed within a conformationally restricted 2,8-diazabicyclo[4.3.0]nonane ring system. Other notable anti-microbials bearing vicinal diamine moieties include the ethambutol analog SQ109 (17),37 which possesses potent activity against multi-drug resistant tuberculosis, and the viral neuraminidase inhibitors oseltamivir (18)38 and zanamivir (19),39 which are employed for the treatment and prophylaxis of influenza virus A and B infections. In further reference to drug development, 1,2-diamines have proven to be valuable scaffolds from which to rapidly build compound libraries: notable discoveries made in this manner include SQ109 (17)37 and stilbene diamine derivative 21, a potent inhibitor of hepatitis C virus RNA replication in the initial stage of infection.40
Figure 4.

Pharmaceutically active, synthetic and semi-synthetic 1,2-diamines.
In addition to anti-infectives, 1,2-diamines are also found within a array of other pharmaceutical agents, including the antiproliferative agent nutlin-3 (22),41 the anti-emetic agent and NK1-antagonist Sch 425078 (23),42 and the 2-substituted 6,8-diazabicyclo[3.2.2]nonane 20, which displays potent affinity for human σ- and δ-receptors, has a cytotoxic potency that exceeds cisplatin, and consequently has potential as an atypical anticancer agent.43
With regard to semi-synthetic pharmaceutical agents, the incorporation of the non-natural vicinal diamine framework into sphingolipids and the associated change in charge of the polar head unit has proven to have a significant impact on the biological profile and metabolic stability of these molecules.44 For example, α-galactosylceramide analog HS161 (24), which lacks a glycosidic linkage and bears an aminocyclitol as a carbohydrate surrogate, is a potent stimulator of invariant natural killer T cells,45 while the 1-amino-1-deoxy sphingoid analog 2546 is a specific inhibitor of human sphingosine kinase, an emerging target for cancer therapeutics.47
2.3. 1,2-Diamines as Tools for Organic Synthesis
The importance of 1,2-diamines extends significantly beyond their role in natural products and pharmaceutical agents since they have also proven to be invaluable scaffolds for the construction of novel metal ligands,48,49 including those that intercalate DNA,50 radiopharmaceuticals and imaging agents.51 Diamines also serve as organocatalysts,5253 chiral reagents and chiral lithium amide bases54 as well as organic receptors.55 In particular, trans-1,2-diaminocyclohexane56 and, to a lesser degree, it 5-membered congener57 have proven to be privileged structures in this regard.
3. Binary Nitrogen Oxides & Related Reagents
3.1. Dinitrogen Tetroxide-Nitrogen Dioxide
The earliest known examples of direct alkene diamination involve the addition reaction of nitrogen dioxide, a process which has been studied for over a hundred years and has played a historically important role in the development of organic synthesis.58,59 Among the various addition products formed in this process, including vicinal nitro-nitrato, nitro-nitrito and nitro-nitro compounds, it is the members of the latter group which are of interest since they are potential precursors of 1,2-diamines.
Longevity notwithstanding, the value of alkene dinitration has historically been limited by a number of practical difficulties, not least of which is the instability of the products formed in this process.60 The reaction of nitrogen dioxide with alkenes is also made complex by virtue of the delicate equilibrium that exists between this compound, dinitrogen tetroxide (N2O4; 28)61 and its nitrite isomer 26 (Scheme 1).62 Furthermore, in polar media, both 26 and 28 can undergo heterolytic dissociation to form nitrosonium-nitrate (29) and nitronium-nitrite (30) ion pairs, which can also participate in the addition process.63 As a consequence, reactions of N2O4-NO2 with alkenes are often complex, display significant solvent effects and lead to the formation of numerous products, as exemplified by 2,3-dimethyl-2-butene (31) (Scheme 2).64
Scheme 1.

Dyanamic equilibria of nitrogen dioxide.
Scheme 2.

Solvent dependancy in the reaction of nitrogen dioxide with 2,3-dimethyl-2-butene.
The mechanism of addition of N2O4-NO2 with alkenes has been extensively examined, both kinetically65 and spectroscopically (15N NMR),66 and found to be highly dependent upon the concentration of N2O4-NO2 as well as the nature of the reaction medium (Scheme 3).67 These studies have largely confirmed Schechter’s original proposal of a radical mechanism,68 in which NO2 undergoes addition to the less substituted alkene position to form a β-nitroalkyl radical 37. Trapping of this intermediate with a second molecule of NO2 then forms both dinitro 32 and nitro-nitronite 37 addition products. Oxidation of the latter species also gives rise to nitro-nitrate 34, which in common with 33 can undergo hydrolysis to form nitro alcohol 39. In more polar solvents, such as chloroform, addition can proceed through an ionic mechanism and, in this medium, formation of nitro-nitrato compound 34 becomes favored.
Scheme 3.

Mechanism of nitrogen dioxide-alkene addition.
At higher concentrations of NO2, N2O5 may also participate in the initial rate determining step through direct alkene addition to form nitronite product 34 or addition to form β-nitroalkyl radical intermediate 37 and NO2.69 Given the electron deficient nature of NO2 is it not surprising that the rate of addition of this radical is found to be highest with electron rich alkenes (Table 1).
Table 1.
| Substrate | Relative Rateb |
|---|---|
|
|
0.1 |
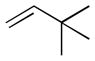
|
0.6 |
|
|
1.0 |
|
4.5 |
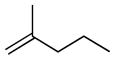
|
16 |
|
21.0 |
Values measured at ambient temperature in CCl4 with a N2O4-NO2 concentration of 0.1 M.
Values are reported relative to allylbenzene.
Although, in the case of non-symmetrically substituted alkenes, the regioselectivity of initial NO2 alkene addition is high, non-selective trapping of the resulting β-nitroalkyl radical often leads to the formation of dinitro and β-hydroxy-nitro compounds, which, in the later case, arise from hydrolysis of the first-formed β-nitro-nitrito compounds (R = NO) (Scheme 4). The absence of skeletal rearrangement during the reaction of camphene (43) has been cited as evidence for a lack of an ionic pathway.64b
Scheme 4.

Reaction of 1,2-disubstituted alkenes with N2O4.
From a practical standpoint, tetrasubstituted alkenes are the most suitable substrates for reaction with N2O4 since yields are generally high and bisnitration is the favored outcome. For example, reaction of Δ9,10-octalin (46) gives rise to the formation of trans-fused decaline 47 (Scheme 5). Jacobsen has also gainfully employed the addition of N2O4 to cyclohexene 49 as a means to access C2-symmetric trans-1,2-diamine 51.70 Reaction of N2O4 in this case generated compound 50 as a single, trans-diastereomer, albeit in relative low yield. Hydrogenation of this vicinal dinitro compound, under medium pressure in the presence of Pd(OH)2, proceeded in high yield to generate diamine 51, which was resolved by way of its mandelate salt. Jacobsen noted that slow addition of alkene to excess N2O4 was requisite for the success of this transformation since it served to avert polymerization between the tertiary radical intermediate and alkene. Müller-Bunz71 and Evans72 have reported closely related routes to diamine 51.
Scheme 5.

Reaction of cyclic tetrasubstituted alkenes with N2O4.
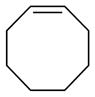
Other highly substituted cyclic alkene substrates which successfully undergo dinitration in the presence of N2O4, include 3-sulfolenes,73 3-phospholene oxides,74 and siliacyclopent-3-enes (Figure 5).75 In these cases, only the products of trans addition were observed.
Figure 5.

Dinitroalkane products resulting from the reaction of N2O4 with cyclic tetrasubstituted alkenes.
Electron deficient alkenes also undergo dinitration efficiently in the presence of nitrogen dioxide (Figure 6). Notable examples in this regard, include α,β-unsaturated nitriles76 and perhaloalkenes.77
Figure 6.

Dinitroalkane products resulting from the reaction of N2O4 with electron-deficient alkenes.
3.2. Nitric Oxide
Despite its possession of an unpaired single electron, nitric oxide (NO) in its pure state does not undergo addition to alkyl or aryl-substituted alkenes since this process is thermodynamically unfavorable. While this observation, first confirmed by Brown in 1957,78 holds true for reactions conducted in the absence of higher nitrogen oxides, such as NO2, this common impurity in NO catalyzes the addition process and leads to the formation of β-nitro-nitroso compounds (pseudonitrosites)79 and/or their dimers.80
Capitalizing on the fact that NO undergoes disproportionation to N2O and NO2 at high pressure, Wilkinson (Scheme 6)81 and others82 have successfully conducted the nitronitrosylation of a variety of alkene substrates under medium pressure, including perhaloalkenes.83 In cases where the β-nitro-nitroso addition products are unstable, other secondary processes can take place, including elimination to form nitro alkenes (Scheme 7).84
Scheme 6.

Addition of NO to alkenes is catalyzed by NO2
Scheme 7.

Formation and decomposition of a β-nitro-nitroso-compound.
3.3. Dinitrogen Trioxide
Generated by the combination of NO2 and NO at low temperature, through the aerial oxidation of NO, or by the treatment of metal nitrites with sulfuric acid, dinitrogen trioxide (N2O3) undergoes addition to alkenes to form β-nitro-nitroso compounds (pseudonitrosites) in high yield (Scheme 8).85 From a mechanistic perspective, this transformation has been interpreted as involving a radical process in which NO2 adds to the alkene to generate a β-nitro radical 63, which then traps NO. Despite some early confusion as to whether β-nitroso-nitrite products are also formed in this reaction, Pfab has unequivocally demonstrated that, in the case of 2-methylpropene (62), addition of N2O3 primarily generates 2-methyl-2-nitroso-1-nitropropane (64) and its trans-dimer 65.86
Scheme 8.

Reaction of N2O3 with 2-methylpropene.
The reaction between alkenes and nitrogen trioxide has historically played an important role in the structural determination studies of terpene natural products since their pseudonitrosite derivatives are often highly crystalline and thus amenable to qualitative and quantitative analysis. Humulene (66), for example, undergoes reaction with N2O3 to yield a mixture of humulene nitrosite (67a), dinitrohumulene (67b) and nitronitratohumulene (67c) (Scheme 9).87
Scheme 9.

Reaction of humulene with N2O3.
Extensive studies on the reaction of naturally-occurring propenylbenzenes, including asarone,88 isosafrole,89 cinnamyl acetate90 and related substrates91 with N2O3 have been carried out by Bruckner and others (Scheme 10). In these cases, addition occurs efficiently with high regioselectively, although the products are often unstable and, in addition to undergoing dimerization, rapidly tautomerize to the more stable β-nitro-oxime derivatives. That the stereochemical course of this transformation has rarely been determined is a further indication of the reactivity of the primary addition products.
Scheme 10.

Reaction of N2O3 with propenylbenzenes.
The reaction of dinitrogen trioxide with a range of cyclic alkenes and dienes, including cyclopentadiene,92 cycloctadiene,91,93 and indenes has also been reported (Figure 7).94
Figure 7.

Products of the reaction of N2O3 with cyclic alkenes
That allyl and vinylsilanes 71 undergo addition, rather than substitution,95 is indicative of a free radical mechanism (Scheme 11). A similar conclusion can be drawn from the regioselectivity observed during the addition of N2O3 to substituted chalcones 72.96
Scheme 11.

Reactions of N2O3 with unsaturated silanes and chalcones.
Despite the extensive body of literature concerning the preparation of pseudonitrosites, conversion of these alkene addition products to the corresponding 1,2-diamines remains a challenging undertaking. In large part, this is due to the propensity with which these systems undergo competitive elimination; e.g. treatment of compound 75 with LiAlH4 leads to the exclusive formation of monoamine 74 (Scheme 12).93 Vicinal diamine 78 can be accessed from 74, albeit indirectly, through a sequence of Lewis acid-mediated isomerization to the corresponding α-nitro-oxime 76 and stepwise hydrogenation.
Scheme 12.

Conversion of pseudonitrosites to the corresponding diamines.
3.4. Silver Nitrate & Trimethylsilyl Chloride
Most recently, Demir and Findik have a reported a convenient method for the generation of dinitrogen trioxide, through the action of AgNO3 on trimethylsilyl chloride (Scheme 13).97 Treatment of alkenes, such as cyclohexene (55) with this reagent in THF or acetonitrile yielded the corresponding β-nitroso-nitrite compounds and their dimers.
Scheme 13.

In-situ generation of N2O3 with AgNO3/TMSCl.
Since the focus of Demir’s study was the preparation of furoxanes (79), the initially formed addition products were directly treated with sulfuric acid to generate the desired heterocycles, presumably by way of the corresponding β-nitro-oximes. Nevertheless, the high yield of pseudonitrite (58a) and the furoxane products, indicate that the addition step in this case is efficient.
3.5. Nitrosyl Chloride & Dinitrogen Tetroxide
The reaction of nitrosyl chloride with alkenes has been extensively studied and almost exclusively leads to the formation of monomeric and dimeric β-nitroso chlorides. However, attempts by Adekenov and co-workers to nitrosochlorinate the guaianolide achillin (80a) led to the selective formation of cis-1,2-dinitro compound 81a (Scheme 14).98 A similar result was subsequently noted for the closely related terpene grossmisin (80b).99
Scheme 14.

Dinitration of unsaturated terpenes with the reagent combination NOCl/N2O4.
The unanticipated formation of compounds 81a and 81b was ascribed to the presence of N2O4 as an impurity in the NOCl employed in this transformation. N2O4 promotes a radical process and likely involves the formation of a β-nitroalkyl radical, which is trapped to form a nitrosonitrate, which undergoes oxidation 100 to form the observed products. Indeed, reaction of 80a with NOCl spiked with N2O4 led to increased yield and rate over purified NOCl (37 vs. 80%), while treatment with N2O4 alone also generated 81a, albeit in lower yield.
3.6. Photolysis of N-Nitroso Compounds
N-Nitroso compounds undergo photolysis under acidic conditions to generate nitric oxide and the corresponding aminium radical (Scheme 15).4 In the case of N-nitrosopiperidine (NNP; 82) photolysis in acidic aqueous solution (pH 2) generates piperidinium radical 85, which originates from the lowest singlet excited state (84) of the NNP-acid complex 83.101
Scheme 15.

Photolysis of N-nitrosopiperidine (NNP) under acidic conditions.
Aminium radicals generated in this manner are electron deficient and undergo a range of chemical reactions, including addition to alkenes (Scheme 16). In the case of cyclohexene (55), photolysis in the presence of an equimolar quantity of N-nitrosodimethylamine generates trans-addition product 86 in high yield.102 Depending on the reaction conditions employed, this compound, to varying degrees, undergoes dimerization to yield 87, rearranges to α-amino-oxime 88, or takes part in a secondary process with the monomer of hyponitrous acid (HNO) to form N-nitrosohydroxylamine 89a. In light of these multiple pathways, yields of this type of process are often impractically low, although Chow has reported conditions, involving extended photolysis and the use of excess nitrosoamine, that favor the formation of N-nitrosohydroxylamines (89a-c).103 In the case of cyclohexane derivative 86, reduction with LiAlH4 generates the corresponding 1,2-diamine, which was acetylated to provide 90.
Scheme 16.

Photoaddition of N-nitrosodimethylamine with cyclohexene and other alkenes.
While irreversible tautomerization of the initially formed β-amino-nitroso monomers, such as 86, is observed for most substrates that bear a hydrogen atom at the position alpha to the nitroso group, those systems that lack this feature react to generate the N-nitroso products, as in the case of methylcyclohexene (91) (Scheme 17).104 Unfortunately, while photoaddition with more substituted alkenes is highly regioselective, the addition products generated from these substrates are prone to fragmentation, as in the case of 92 which undergoes fragmentation and hydrolysis to form ketoaldehyde 94.
Scheme 17.

Formation and fragmentation of C-nitroso-β-amines.
3.7. Thermolysis of Tetramethyl-2-tetrazene-Lewis Acid Complexes
Prepared through the Hg(II)-mediated oxidation of 1,1-dimethylhydrazine,105 tetramethyl-2-tetrazene (TMT) forms 1:1 complexes with a range of Lewis acids, including zinc halides (Scheme 18).106 Taking advantage of the propensity of these compounds to readily undergo thermal decomposition to form dimethylamino radicals, Michejda and co-workers have developed a method for alkene diamination employing the zinc chloride complex 95.107
Scheme 18.

Diamination of styrenes via the thermolysis of the tetramethyl-2-tetrazene-zinc chloride complex.
Heating 95 in the presence of excess ZnCl2 and conjugated alkenes, such as indene (97) or α-methylstyrenes (100), leads to the formation of the corresponding bis(dimethylamino) adducts, albeit in low yield. That the reaction of 97 results in the exclusive formation of the trans addition product 99, was cited as evidence that addition of the two dimethylamino groups proceeds through a stepwise process rather than a concerted one. Detailed Hammett studies also suggest that the dimethylamino radical 96 generated upon the decomposition of 95 is intimately associated with zinc chloride.
3.8. Nitroamidation: Nitronium Salts & the Ritter Reaction
Alkenes undergo reaction with both nitrosonium and nitronium tetrafluoroborate in the presence of nitriles to generate carbocations 103 but whereas the former process leads to the formation N-hydroxyimidazolium salts,108 the reaction of nitronium ions can be employed as a means of diamination. In the presence of nitrile solvents, these intermediates are trapped to form nitrilium ions 104, which undergo hydrolysis to form the products of nitroamidation (Scheme 19). The first report of this type of process was by Scheinbaum, who in 1971, reported that the reaction of simple alkenes 102 with nitronium tetrafluoroborate in acetonitrile generated α-nitro amides 105.109,110
Scheme 19.

Nitroamidation alkenes with nitronium tetrafluoroborate in acetonitrile.
While Scheinbaum’s original report only encompassed three alkenes, Mellor and co-workers have subsequently studied this nitroamidation method in more detail, employing a wider range of substrates and found that, in the case of conjugated alkenes, higher yields can be obtained through use of CH2Cl2 as a co-solvent (Scheme 20).111 In all cases, addition was found to be rapid, highly regioselective and favored the Markovnikov products. That trans-β-methylstyrene (106) undergoes cis addition was confirmed by conversion of 107a to imidazoline 108. Notably, 1-phenylcyclohexene underwent trans addition to yield 107e. Nitroacetamidation of less nucleophilic alkenes, including hex-1-ene, oct-1-ene, cyclohexene, and cyclopentene, was found to be considerably less efficient.
Scheme 20.

Reaction of alkenes with nitronium tetrafluoroborate.
In light of the expense of nitronium tetrafluoroborate and its high moisture sensitivity, Mellor has developed a method for its in-situ electrogeneration from nitrogen dioxide, through anodic oxidation (Scheme 21).112 Yields, in most cases, are higher than those obtained with the non-electrogenerated reagent, an observation which was ascribed to the absence of acidic impurities present during electrolysis.
Scheme 21.

Nitroamidiation of conjugated alkenes with electrochemically generated nitronium tetrafluoroborate.
In this one-pot procedure, electrogeneration of the nitrogen electrophile must precede alkene addition since co-electrolysis failed to provide the nitroacetamide products.
3.9. Ceric Ammonium Nitrate-Sodium Nitrite-Acetonitrile
In an approach that avoids the use of nitronium salts, Vankar and co-workers have developed a nitroamidation method that entails the treatment of alkenes with ceric ammonium nitrate (CAN) and sodium nitrite in nitrile solvents (Scheme 22).113
Scheme 22.

Vankar’s alkene nitroamidation method.
Oxidation of nitrite under these conditions is proposed to generate nitrogen dioxide, which undergoes alkene addition to form a β-nitroalkyl radical 111. A second electron transfer to CAN then generates carbocation 112, which participates in a Ritter reaction to yield the observed products. Notably, use of benzonitrile and acrylonitrile offer access to benzamides (113b) and α,β-unsaturated amides (113e). That this process displays negligible diastereoselectivity in the substrates examined, reflects the intermediacy of the carbocation intermediate.
3.10. Acetyl Chloride-Silver Nitrate-Acetonitrile
Vankar has recently developed a reagent system comprising of acetyl chloride, silver nitrate and acetonitrile for the nitration and nitroamidation of glycals and simple alkenes (Scheme 23).114 The acetyl nitrate (115) generated under these conditions is posited to undergo reaction with the substrate to generate a β-nitro carbocationic intermediate 116 whose fate is highly dependent on both the reaction conditions employed and the nature of the substrate itself.115 In the case of tri-O-benzylated galactal 120, reaction at elevated temperature leads to proton loss from the intermediate glycosyl cation and formation of nitroglucal 119. Nitroamidation, on the other hand, is favored at lower temperatures and in the case of 120, leads to the formation of 121 with high diastereoselectivity. Routes to 2-nitro-1-acetamido sugars of this type are of importance in light of the biological significance of 2-amino-β-glycosylamines, which constitute the core structural motif of N-linked glycoproteins.
Scheme 23.

Vankar’s second-generation alkene nitroamidation method and its application to the preparation of 2-nitroglycals and 2-nitro-1-acetamido sugars.
A significant temperature dependence was also observed with E-stilbene (122), which underwent nitroamidation with complete cis selectivity (Scheme 24). Although cyclohexene and 1-methylcyclohexene undergo nitroamidation, the formation of 125a and 126a is accompanied by significant quantities of the elimination products 125b and 126b.
Scheme 24.

Nitroamidation of simple alkenes under Vankar’s conditions.
4. Alkene Bisazidonation via Redox
The azide anion (N3−) has a relatively low E0 (ca. −0.6 V)116 and consequently can be oxidized with a range of organic and metal-based oxidizing agents to the corresponding azidyl radical (N3·). This species is sufficiently electrophilic to undergo addition to a range of alkenes117,118 and trapping of the resulting β-azidoalkyl radical with a suitable azide donor, offers a convenient means of alkene diazidonation under mild reaction conditions. This “redox-chain” approach to diamination was first reported by Minisci and co-workers who utilized the reaction between tert-butyl hydroperoxide or hydrogen peroxide, and ferrous sulfate to effect the transformation (Scheme 25).119,120 From a mechanistic standpoint, Minisci has proposed that ferrous sulfate mediates the decomposition of hydroperoxide (1) and the resulting alkoxy radical interacts with an azido Fe(III) complex to generate the azidyl radical (2). Upon alkene addition, azide transfer between an iron(III) azide complex and the β-azido radical then generates the 1,2-diazide and completes the redox cycle.
Scheme 25.

Ferrous sulfate-mediated diazidonation of alkenes.
Minisci has also reported the use of a Fe(II)/Fe(III) system in the conjunction with a variety of oxidants, including hydrogen peroxide, permanganate,121 and Ce(IV) salts (Scheme 26).122
Scheme 26.

Ferric-Ferrous sulfate-mediated diazidonation of alkenes.
As shown in Scheme 27, the combination of ammonium peroxydisulfate and the Fe(II)/Fe(III) system also been employed for the diazidonation of styrene (127) (Scheme 27).123 In this case, the yield of diazide 128 is significantly improved over that obtained with hydrogen peroxide.
Scheme 27.

Ferric-Ferrous sulfate-mediated diazidonation of alkenes in the presence of ammonium peroxydisulfate.
Fristad has reported the use of Mn(III) acetate as a highly effective reagent for alkene diazidonation (Scheme 28).124,125 In this case, treatment of alkenes with Mn(OAc)3 and sodium azide in acetic acid at elevated temperatures leads to the formation of 1,2-diazides 133 in high yield. Efficiency not withstanding, this methodology necessitates a large excess of azide (15 equiv) in order to prevent the formation of monoazidination products, which presumably arise from hydrogen atom transfer to the β-azido radical intermediate. Although the mechanism of this transformation has yet to be fully delineated, the dramatic rate acceleration noted in the presence of alkenes was posited as evidence of a ligand-transfer oxidation, rather than the participation of the azide radical.
Scheme 28.

Mn(III)-mediated diazidonation of alkenes.
Snider has subsequently reported a modification of Fristad procedure in which replacement of acetic acid with a mixture of acetonitrile and trifluoroacetic acid as the reaction medium leads to significant improvements in yield (Scheme 29).126 That acid sensitive substrates, such as tetrahydropyran (134), are tolerant of these conditions is likely a reflection of the fact that reactions proceed at temperatures as low as −20 °C. Recent application of this methodology include the preparation of 1,2-cyclohexanediamines for oxaliplatin-type complexes127 and the synthesis of imine-based protein labels.128
Scheme 29.

Snider’s Mn(III)-mediated diazidonation of alkenes.
Alkene diazidonation can also be accomplished through electrochemical generation of the azidyl radical. Schäfer has reported that co-electrolysis of solutions of sodium azide in glacial acetic acid and alkenes (1:3, v/v) leads to the formation of 1,2-diazides 136 (Scheme 30).129
Scheme 30.

Alkene diazidonation via azide anion electrolysis.
While this method is reasonably efficient for electron-rich, styrene substrates, the yield for cyclic and acyclic alkenes is less satisfactory.
5. Heavy Metal-Mediated Bisazidonation
5.1. Lead(IV) Acetate-Trimethylsilyl Azide
Lead(IV) acetate azide [Pb(OAc)4-n(N3)n] (137) is an effective azide transfer reagent, which is generated by the reaction of lead(IV) acetate and trimethylsilyl azide.130 In light of its thermal instability (decomposition occurs rapidly above −20 °C), this reagent must be generated in situ.131 Zbiral has reported the reaction of this reagent with alkenes to form vic-diazides, albeit in a highly temperature and solvent dependent manner (Scheme 31). In the case of styrene (128), reaction of 137 in acetonitrile at −20 °C leads to the formation of phenacyl azide (138) while reaction at 20 °C generates vic-azide 139 in high yield. In dichloromethane, reaction of 137 and 128 leads to more complex mixtures of products whose composition is dependent on the order of reagent addition.
Scheme 31.

Generation of [Pb(OAc)4-n(N3)n] and its temperature dependent reaction with styrene.
The formation of elimination (143) and rearrangement (145) products during the reaction of camphene (142) and other bridged alkenes with 137, has been cited by Zbiral as persuasive evidence of a “positive” azide-transfer, in which aziridinium ion 141 and carbocation intermediate 142 are involved (Scheme 32).132 Unfortunately, in most substrates, diazidonation is accompanied by the formation of vicinal acetoxy-azido products. Furthermore, acyclic alkenes, such as trans-stilbene (122), undergo diazidonation in a non-stereospecific manner as a result of the cationic intermediate.
Scheme 32.

Reaction of [Pb(OAc)4-n(N3)n] with camphene and E-stilbene.
While the usefulness of Zbiral’s reagent with non-cyclic alkenes is limited by the intermediacy of cations, this is not the case with cyclic substrates. Draper has shown that steroidal 4,6-dien-3-ones 148 are suitable substrates for diamination and undergo reaction with 137 to form B-ring vic-diazides 150 with high stereoselectivity (Scheme 33).133 In this case, it is suggested that the lead-mediated azide transfer proceeds through silyl dienol ether 149, which is formed by the Lewis acid-mediated 1,4-addition of trimethylsilyl azide to 148.
Scheme 33.

Reaction of [Pb(OAc)4-n(N3)n] with steroidal 4,6-dien-3-ones.
The behavior of steroidal dienone substrates stands in contrast to that of the analogous trisubstituted Δ6-steroidal alkenes, which, depending on the reaction conditions employed, react with the lead tetraacetate-trimethylsilyl azide reagent to form allylic azides134 or seco keto nitriles.135
5.2. Thallium(III) Acetate-Trimethylsilyl Azide
Zbiral has also developed an analogous reagent to 137, generated from thallium(IV) acetate, which also mediates diazidonation, albeit in a less efficient manner (Scheme 34).136 Treatment of thallium(III) acetate with trimethylsilyl azide generates [Tl(OAc)3-n(N3)n] (151), which undergoes reaction with alkenes to generate the corresponding aziridinylazothallium compounds 152 and 154. While only putative intermediates in the analogous lead-mediated transformations, these organothallium compounds are sufficiently stable to permit isolation.
Scheme 34.

Reaction of [Tl(OAc)4-n(N3)n] with cyclohexene and 4-allylanisole.
Thermolysis of 152 and 154 leads to the formation of 1,2-diazido compounds 153 and 156, although, in both cases, the predominant products are the parent alkenes, which were proposed to arise through a cheletropic fragmentation reaction.
6. Heavy Metal-Mediated Diamination
6.1. Thallium Acetate-Amines
Barluenga has reported a method for the preparation of diamines using thallium(III) acetate. Treatment of alkenes with aromatic amines in the presence of this heavy metal salt leads to the efficient formation of the corresponding diamines 158 (Scheme 35).137 While primary and secondary aromatic amines participate in this process, primary aliphatic amines fail to react. This reaction is thought to proceed via aminothallation to generate an organothallium intermediate, 157, which subsequently undergoes substitution. Notably, 1,4-dienes, specifically 1,4-hexadiene and 1,4-cyclooctadiene, undergo double addition to form cyclic and bicyclic products 158f and 158g respectively. The relative stereochemistry of these products was not reported.
Scheme 35.

Addition of aromatic amines to alkenes in the presence of thallium(III) acetate.
6.2. Mercury Acetate-Amines
Barluenga and co-workers have demonstrated the ability of β-amino alkylmercury(II) salts, formed through alkene aminomercuration, to undergo substitution with a range of nucleophiles, including amines. Initial studies found that treatment of alkenes with the reagent generated from tetrafluoroboric acid and mercury(I) oxide and aromatic or primary amines led to the efficient formation of 1,2-diamines 160 and Hg(0) (Scheme 36).138 The efficiency of this process is highly dependent on the degree to which the C-Hg bond is polarized. For example, the treatment of the β-amino alkylmercury salts of acetate and halides with amines leads only to retromercuration and formation of alkenes.
Scheme 36.

Addition of aromatic amines to alkenes in the presence of mercury(II) tetrafluoroborate.
In a more recent mechanistic study, Barluenga has investigated the aminomercuration of 1,4-cyclooctadiene (161) under these conditions as a means of accessing 2,6-disubstituted-9-azabicyclo[3.3.l]nonanes (Scheme 37).139 Treatment of 161 with mercury(II) tetrafluoroborate in the presence of aniline leads directly to the formation of bicyclic triamine 163 as a single diastereomer. The reaction is thought to proceed via intermediate 162.140 The aminomercurials 162 (X = Cl, OAc) generated from mercury(II) chloride or acetate, proved to be less prone to substitution and their transformation to 163 requires more forcing conditions. Reagent notwithstanding, the authors propose that the formation of the observed products proceeds via an aziridinium ion intermediate(s) generated by the internal displacement of the mercury centers in 162.
Scheme 37.

Mercury(II)-mediated bis-diamination of 1,4-cyclooctadiene.
Barluenga has found that (+)-limonene (164) displays unexpected behavior in its diamination reactions with mercury(II) tetrafluoroborate (Scheme 38).141 While reaction of this diene at low temperature proceeded as anticipated, with Markovnikov selectivity at the more accessible exocyclic alkene, heating the resulting aminomercurial (165) at 80 °C leads to the formation of trans-diamine 167 (Ar = Ph, p-ClPh, p-MePh). The formation of this product was attributed to mercurinium ion exchange whereby 165, unable to undergo displacement to form an aziridinium ion, undergoes β-elimination and ion transfer to generate intermediate 166. Stepwise substitution with two equivalents of arylamine then leads to the observed trans-1,2-diamine.
Scheme 38.

Diastereoselective mercury(II)-mediated diamination of (+)-limonene proceeds with unanticipated regioselectivity.
7. N,N-Dihaloarylsulfonamides & N-Haloarylsulfonamides
7.1. N,N-Dihaloarylsulfonamides-Acetonitrile
Although a stepwise approach to diamination and thus beyond the purview of this article, the aziridination of alkenes and use of the strained products as substrates for ring-opening via a Ritter reaction is an appealing strategy for the stereocontrolled introduction of vicinal nitrogen functionality.142 In this regard, Li and co-workers, in 2003, first reported a novel method for the indolizidination of α,β-unsaturated carbonyl compounds which is thought to proceed via the Ritter-type reaction of an aziridinium ion intermediate (Scheme 39).143,144 Treatment of α,β-unsaturated ketones and esters, such as methyl cinnamate (168), with N,N-dichloro-p-toluenesulfonamide (TsNCl2, 167), 4 Å molecular sieves, and the complex generated from Rh2pfb4 and Ph3P in acetonitrile was found to generate trans-substituted 2-dichloromethyl-2-imidazolines 170 in moderate to high yield and with excellent diastereoselectivity. The formation of trans imidazolines in this case corresponds to a cis diamination process.
Scheme 39.

Direct imidazolination of α,β-unsaturated ketones and esters with the reagent combination TsNCl2/MeCN/Rh2pfb4·PPh3.
Li has also discovered a number of other catalysts that significantly accelerate the imidazolination process, including Rh2TFA4·PPh3,145 FeCl3·PPh3,146 MnO2,147 and most recently, triphenylphosphine.148 In the case of MnO2 and Rh2TFA4·PPh3, diamination proceeds to generate the trichloromethyl rather than dichloromethyl imidazolines. The copper-catalyzed (CuI·PPh3) addition of N,N-bromo-p-toluenesulfonamide (TsNBr2) to α,β-unsaturated ketones and esters has also been reported.149 In this case, the formation of dichloromethyl imidazolines is favored.
On the basis of their work on the related alkene aminohalogenation reaction,150 Li and co-workers have posited a general mechanistic interpretation of this remarkable transformation (Scheme 40). In a stereospecific process that is accelerated but not dependent upon catalyst additives (vide infra), diamination commences with the formation of N-sulfonyl-N-chloroaziridinium ion 172. In the presence of nitriles, participation of this intermediate in a Ritter-type reaction, involving nucleophilic ring opening at the more substituted or benzylic position, then generates 1N-(tosyl)imidazolium ion 174. Given the overall syn stereochemistry of the diamination process, Li has suggested that this process occurs via a [2+3] mechanism in which 174 is formed directly. Displacement of the 1N-chlorine group in this intermediate then gives rise to 3N-(tosyl)imidazolium ion 175, which undergoes proton loss and a second SN2′-type displacement to form 2-chloromethyl imidazoline 177. 3N-Chlorination of 177 and a repetition of the deprotonation and displacement steps are then proposed to lead to the observed product 178.
Scheme 40.

Proposed mechanism for Li’s alkene syn-imidazolination process.
Intriguingly, subsequent studies by Li have revealed that during the addition of TsNCl2 to α,β-unsaturated ketones the need for a catalyst can be obviated simply by raising the reaction temperature to 50 °C.151 Furthermore, in the case of the more reactive reagent N,N-dichloro-2-nitrobenzenesulfomamide (NsNCl2, 180), imidazolination of enones 179 proceeds in the absence of catalyst at room temperature to generate the dichloromethyl adducts (Scheme 41).152 Interestingly, when conducted in the absence of molecular sieves, addition of 180 proceeds at 50 °C to the corresponding trichloromethyl imidazoline products 181.
Scheme 41.

Direct imidazolination of α,β-unsaturated ketones with the reagent combinations NsNCl2/MeCN.
In order to avoid the inconvenience of handling N,N-dichlorosulfonamides 169 and 180, Li has a protocol for the in-situ generation of these unstable reagents. Treatment of enones and dienones with p-toluenesulfonamide (182) and N-chlorosuccinamide (NCS) at 50 °C generates the expected products in comparable yield to the parent reagent (Scheme 42).153 Notably, other nitrile partners, including isobutyronitrile and benzonitrile can be employed and, in the case of benzylideneacetone, leads to the formation of 183f and 183g, respectively. NsCl2 (180) can also be generated in this manner, although in the case of this more electrophilic reagent, esters as well as unsaturated ketones undergo imidazolination.154
Scheme 42.

Direct imidazolination of α,β-unsaturated ketones with the reagent combination TsNCl2/NCS/RCN.
Importantly, Li and co-workers have demonstrated that the imidazoline products 183 can be hydrolyzed to the corresponding open-chain diamines without epimerization at either chiral center (Scheme 43).155 Exposure of these heterocycles to aqueous hydrochloric acid at 70 °C mediates rapid hydrolysis to the differentially protected syn-1,2-diamines 184 in excellent yield. Stannic chloride (SnCl4/THF/H2O) has also been utilized as an effective promoter of imidazoline hydrolysis.149,148
Scheme 43.

Acidic hydrolysis of imidazolines to form differentially protected vicinal diamines.
7.2. N-Chlorosaccharin-Acetonitrile-KOEt
An elegant one-pot method for the cis-imidolizidination of alkenes has also been developed by Booker-Milburn and coworkers (Scheme 44).156 In the presence of acetonitrile, alkenes undergo a Ritter-type reaction with the electrophilic chlorinating agent N-chlorosaccharin (NCSacc, 185) to generate a putative nitrilium ion intermediate 187. Capture of 187 by the saccharin anion gives rise to β-chlorosulfonylamidines 188. Treatment of these reactive intermediates with potassium ethoxide then mediates ring-opening of the benzothiazoletrione ring to form an amidine anion 189, which cyclizes to form the corresponding imidazoline systems 186 in low to moderate yield. When isolable, by-products of this process include aziridines and allylic chlorides, which arise from the first-formed chloronium ion intermediate through eliminative ring opening and capture by saccharide, respectively. In common with Li, Booker-Milburn has found that the ring opening of the imidazoline products to differentially protected diamines under acidic conditions presents no difficulties.
Scheme 44.

Imidazolination of alkenes with the reagent combination NCSacc/MeCN/KOEt.
7.3. Chloramine-T-Iodine-Acetonitrile
In 2006, Ramesh and Kumar reported the use of chloramine T in a remarkably straightforward, one-pot method for the diamination of glycals (Scheme 45).157,158 Treatment of tri-O-acetyl-D-glucal (190) with 2.3 equivalents of chloramine T in the presence of a catalytic quantity of iodine (15 mol%) leads to the selective formation of β-D-gluco 1,2-disulfonamide 191a in good yield. This mild procedure is successful with a range of mono-, di- and trisaccharide glycals, is compatible with both O-acetate and O-benzyl protecting groups and, in most cases examined, proceeds with complete diastereoselectivity.
Scheme 45.

Iodine catalyzed one-pot diamination of glycals with chloramine-T.
On the basis of their own observations and studies by Komatsu on the iodine-catalyzed aziridination of alkenes using chloramine-T,159 Ramesh and Kumar have proposed a mechanism for the diamination process (Scheme 46). Rapid reaction of chloramine-T (192) with iodine is thought to generate iodine-chloramine-T complex 193, which reacts from the β-face of glycal 190 to form iodonium ion 194. Diaxial ring opening of this species at the anomeric center by chloramine-T then leads to 195, which, with the aid of iodide, undergoes cleavage of the N-Cl bond and ring closure to form glycal aziridine 196 and iodine monochloride (ICl). Ring opening of 196 by a second molecule of chloramine-T then yields 1,2-disulfonamide 191a, after protonation. That stoichiometric quantities of iodine monochloride were found to mediate this transformation in place
Scheme 46.

Proposed mechanism of Ramesh’s one-pot glycal diamination method.
In light of the central structural role that 2-amino-β-glycosylamines play in N-linked glycoproteins, the development of synthetic routes to this glycodomain is a goal considerable importance. In this regard, Ramesh has exploited the differential reactivity of the anomeric and C-2 sulfonamide groups within the glycal addition products to develop a general route to glycosyl amino acids and peptides (Scheme 47). For instance, N-acetylation of 197 proceeds only at the more nucleophilic C-2 position to provide 198 in excellent yield. Conversion of this material to N-Ala-Asp linked glycopeptide 200 was accomplished through a four-step sequence involving protection of the anomeric nitrogen, didetosylation, removal of the Alloc group from 199 and peptide coupling of the liberated anomeric amine.
Scheme 47.

Synthesis of N-Ala-Asp linked glycopeptide X.
8. Iodine Azide & Surrogates
8.1. Iodine Azide
The first reports of pseudohalogen-mediated diazidonation were made by Hassner and involve the use of iodine azide (IN3) prepared by the action of sodium azide on iodine monochloride (ICl) (Scheme 48).160 Although explosive in its pure state, iodine azide can be handled as a 0.25 M solution in polar organic solvents.161 In most cases, reaction with alkenes generates the products of anti-iodo-azidination (2), which are proposed to arise from the ring opening of an iodonium intermediate. In the presence of excess azide anion and with extended reaction times, displacement of the iodide group can take place to yield cis-1,2-diazides (3).162,163,164 In the absence of excess azide, diazides can also form since the iodide generated during displacement reacts with iodine azide to generate iodine and an azide anion (3).165
Scheme 48.
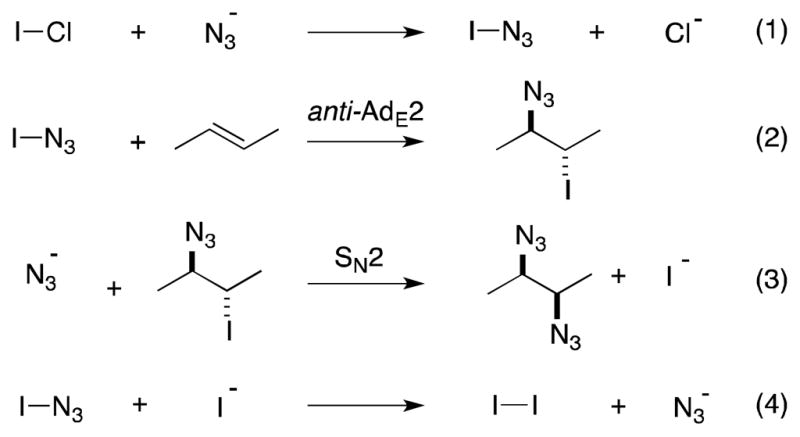
Generation, reaction and iodide-mediated disproportionation of iodine azide.
While acyclic 1,2-diazides have a tendency to undergo spontaneous elimination to form vinyl azides,166 Sasaki has demonstrated that medium-sized cyclic alkenes, including tropone ethyleneketal (201), 1-ethoxycarbonyl-1(1H)-azepine and cyclooctatetraene (205), undergo diazidonation successfully (Scheme 49).162 In light of the instability of these products, they were trapped as their respective 1,3-dipolar cycloadducts 204 and 208; treatment with dimethyl acetylenedicarboxylate (DAC) proceeded smoothly. In the case of 208, valence tautomerism of cyclooctatriene 206 generates bicyclo[4.2.0]octatriene 207 which undergoes cycloaddition.
Scheme 49.

Diazidonation of cyclic polyenes with iodine azide-sodium azide.
Tamura has also reported the reaction of benzo[b]furans and 1-acyl and 1-tosyl-indoles with IN3 in the presence of sodium azide to generate the corresponding 2,3-diazido-2,3-dihydrobenzo[a]furans and 2,3-diazidoindolines 212 (Scheme 50).167 In all cases, but compound 212d, mixtures of trans and cis stereoisomers were obtained reflecting the likely intermediacy of cationic intermediates, such as 211.162
Scheme 50.

Diazidonation of benzo[b]furans and 1-acyl and 1-tosyl-indoles.
Employing a modification of Hassner’s original conditions,161b Schönenberger has reported the preparation of 1,2-diaryl-1,2-diazidoethanes 213 through the addition of IN3 to E-stilbenes (Scheme 51).168 That in the case of 213c, anti rather syn addition occurs may indicate the intermediacy of a β-azidocarbocation, which would be trapped from the less hindered face.
Scheme 51.

Divergent reactions of 1,3-dienes with bromine and iodine azides.
Hassner has reported a single example of the 1,2-diazidonation of a 1,3-diene (Scheme 51).169 In this case, the reaction of E,E-diphenylbutadiene (215) with IN3 generates 1,2-diazide 216 while addition of BrN3 leads only to bromoazide 214; this likely reflects the diminished nucleofugality of the bromide group in comparison with iodide. Exposure of 215 to sodium azide in DMF generates compound 216.
In view of the difficulties associated with the handling of iodine azide, Kirschning and co-workers have developed a stable polymer-bound form of this useful reagent (Scheme 52).170 Sequential treatment of polymer-bound iodide 217 with (diacetoxyiodo)benzene (DIAB) and trimethylsilyl azide generates a resin formulated as bis(azido)iodate salt 219. While most substrates undergo only azido-iodination, prolonged reaction of electron-rich alkenes with this reagent leads to the formation of 1,2-diazides, albeit in low yield and without stereospecificity. In the case of 220, it seems probable that 222 arises from azido-iodide 221, through a non-concerted displacement process.
Scheme 52.

Preparation and reaction of polymer-bound iodine azide.
Generated by the reaction of DIAB (223) with Me3SiN3 and tetraethylammonium iodide, bis(azido)iodate salt 225 (Scheme 53),171 the solution phase variant of 219, has also been employed for alkene diazidonation, by Austin and co-workers in their synthesis of the marine natural product dibromophakellstatin (228).172
Scheme 53.

Generation of a solution-phase bis(azido)iodate salt and its use in the synthesis of (±)-dibromophakellstatin.
The key step is this endeavor involves installation of the syn-1,2-diamine functionality present on the piperidinone ring of 228. Treatment of alkene 226 with 225 provided syn-diazide 227b as the major product, albeit in modest yield, while reaction with iodine azide led to the exclusive formation of anti addition product 227a. The authors propose that while both diazides isomers may arise from a common haloazide intermediate, the cis isomer does so via bimolecular nucleophilic displacement. The trans isomer, on the other hand, appears to arise from an azido-iminium ion that undergoes ring opening from the less hindered face.
9. Halogen-Mediated Diamination & Cycloguanidination
9.1. Vicinal Diamination of 1,4-Dihydropyridines
The use of intermolecular alkene aminohalogenation as a means to accomplish the direct diamination of carbon-carbon double bonds is relatively uncommon since the competitive oxidation of alkyl and aryl amines presents a significant setback.173,174 However, in the case of highly reactive alkene partners, such as enamines, this undesirable process can be avoided. In this regard, Lavilla and co-workers have reported a highly effect method for the vicinal diamination of 1,4-dihydropyridines (Scheme 54).175 Treatment of N-alkyl-1,4-dihydropyridine 229 with iodine (3.5 equiv) in the presence of a range of cyclic amines (25 equiv) leads to the formation of the corresponding trans-2,3-diaminotetrahydropyridines 231 in high yield. The stereoselective generation of these products was rationalized in terms of the regioselective formation and trapping of a 3-iodo-3,4-dihydropyridinium ion to form intermediate 230. Internal displacement of the iodide then generates an aziridinium ion which undergoes ring opening at the 1-position with a second equivalent of amine giving rises to the trans-substituted products.
Scheme 54.

Vicinal diamination of 1,4-dihydropyridines in the presence of iodine.
While simple primary amines fail to undergo this reaction, N,N′-dimethylethylenediamine reacts with compound 229 to generate the cis-bicyclic adduct 231d in high yield.
9.2. Halogen-Mediated Cycloguanidination & Related Processes
While the direct cycloguanidination of alkenes via transition metal catalysis has recently garnered considerable attention (vide infra), the use of halogens and their synthetic equivalents to effect this transformation with 1,2-dihydropyridines has also met with considerable success. The first reports of this form of alkene diamination were made by Al-Mourabit and co-workers as part of their on-going study of 2-aminoimidazole alkaloid synthesis (Scheme 55). Treatment of carbomethoxydihydropyridine (232) with bromine or NBS in the presence of 3–4 equivalents of Boc-guanidine generated bicycles 233a and 233b, which upon exposure to HCl, where converted to cyclic guanidine 234.176 N-Protected tetrahydropyridines 235177 and 1,2-dihydropyridine imidates178 were also found to undergo this bromine-induced transformation. In both cases, the annulation process likely proceeds in stepwise fashion where bromoamination of the enamine precedes intramolecular ring closure through N-alkylation.
Scheme 55.

Bromine-mediated cycloguanidination of N-acylated dihydropyridines and tetrahydropyridines with Boc-guanidine.
Tepe and co-workers have gainfully employed a closely related aza-annulation in their recent synthesis of the oroidin-type alkaloid (±)-dibromophakellin (240) (Scheme 56).179 Treatment of a mixture of dipyrrolopyrazinone 238 and Boc-guanidine with NBS gave rise to the Boc-protected natural product 239 in low yield.
Scheme 56.

Key alkene cycloguanidination step in Tepe’s synthesis of dibromophakellin.
Further studies by Al-Mourabit have revealed that 2-aminopyrimidine (242), which is a stronger nucleophile than Boc-guanidine, can be used in place of this reagent (Scheme 57). For example, treatment of N-acylpyrrole tetrahydropyridine 241 with N-iodosuccinimide (NIS) in the presence of 242 afforded adduct 244 with moderate efficiency.180
Scheme 57.

NIS-mediated cycloguanidination of N-acylated dihydropyridines with 2-aminopyrimidine.
The aza-annulation of dipyrrolopyrazinone 245, albeit without the assistance of halogen reagents, has also been described by Lindel and co-workers in their synthesis of dibromophakellstatin (247) (Scheme 58).181 In this situation, the activated complex generated from ethyl-N-tosyloxycarbamate and calcium oxide is proposed to react with 247 to generate an acyliminium ion that traps excess ethyl-N-tosyloxycarbamate. Subsequent cyclization and loss of ethanol then generates the imidazolidinone ring of the natural product in a single step with reasonable yield.
Scheme 58.

Key alkene cycloguanidination step in Lindel’s synthesis of dibromophakellstatin.
Although the iodonium-induced cyclization of unsaturated carboxamides, sulfonamides and carbamates to form N-heterocycles is well documented,182 the participation of N-ω-alkenyl ureas in this type of cyclization, as exemplified by the conversion of 248 to 249,183 is comparatively rare (Scheme 59). Notwithstanding question of N vs. O selectivity, this is somewhat surprising given that 5-exo-tet cyclization of the iodoamination products in this case potentially offers an entry point to imidazolidin-2-ones, the formal products of alkene diamination. In this regard, Muñiz and Barluenga have successfully employed the iodonium source bis(pyridine)iodonium tetrafluoroborate (IPy2BF4) as a mediator of the direct intramolecular oxidative diamination of δ-alkenyl ureas 250 (Scheme 60).184
Scheme 59.

Illustrative example of the intramolecular iodoamidation of an N-δ-alkenyl urea.
Scheme 60.

Intramolecular oxidative diamination of alkenes with N-sulfonyl ureas in the presence of bis(pyridine)iodonium tetrafluoroborate.
Treatment of N-δ-alkenyl-N′-sulfonyl ureas 250 with IPy2BF4 in toluene at 120 °C was found to selectively generate, in the case of terminal and 1,1-disubstituted alkenes, bicyclic imidazolidin-2-ones 253 in consistently high yield (Scheme 60). A single N-alkyl-N′-sulfonyl guanidine derivative was also found to undergo cyclization to form compound 253g in near quantitative yield. Less successfully, the cyclization of internal alkene substrates, such as 250 (R = Me), gave rise to mixtures of diamination and oxamidation products 252. Notably, the formation of both bicyclic urea 253 (R = Me) and isourea 252 occur in a stereospecifically syn fashion with respect to alkene geometry.
From a mechanistic perspective, Muñiz and Barluenga have proposed that imidazolidinone formation occurs through a sequence of two anti C-N bond-forming steps: (a) formation and ring opening of an iodonium ion giving rise to intermediate 251 and, (b) selective SN2 displacement of the iodide group by the second nitrogen atom to form the products of a cis-diamination. Support for this hypothesis was gained from the deuterium labeling study outlined in Scheme 61.
Scheme 61.

Stereochemical course of N-sulfonyl ureas-alkene diamination reaction.
Windenhoefer and Li have recently reported a milder method for the intramolecular oxidative diamination of N-δ-alkenyl-N′sulfonyl ureas (Scheme 62).185, For example, treatment of 256 with iodosuccinimide (NIS; 2 equiv) and sodium bicarbonate (1 equiv) at room temperature gave rise to bicyclic imidiazolidin-2-one 258a in high yield. Notably, the formation of products 258f-h occurs with high diastereolselectivity. In contrast to the findings of Muñiz and Barluenga,184 the NIS-mediated cyclization of internal alkene substrates generates only diamination products, e.g. 258h, although, as before, this transformation proceeds in a stereospecific, syn manner.
Scheme 62.

Intramolecular oxidative diamination of alkenes with N-sulfonyl ureas in the presence of N-iodosuccinimide.
The oxidative cyclization of unsaturated sulfonylureas 256 has also been studied by Michael and co-workers, who employ iodosylbenzene in the presence of Lewis and Brønsted acids to mediate this process.186 Although alkene diamination was observed, the predominant outcome of these reactions proves to be the formation of cyclic isoureas through intramolecular oxamidation.
10. Hypervalent Iodine Reagents
In light of their ready availability, low toxicity and reduced environmental impact, hypervalent iodine reagents have largely replaced heavy metals, such as mercury, lead and thallium, as the reagents of choice for alkene diazidonation and diamination.187
10.1. Aryl-λ3-iodanes
The reaction of alkenes with aryl-λ3-iodanes has proven to be a particular effective and versatile method for the co-introduction of vicinal heteroatoms. However, in the case of alkene diamination use of these reagents is made impractical by the ease with which they oxidize primary and secondary amines. This however is not the case with azide ligands and the use of aryl-λ3-iodanes to effect alkene diazidonation has been reported by a number of groups. In 1972, Zbiral and Ehrenfreund reported that treatment of unsaturated esters 259 with PhI(OAc)2/TMSN3 leads to the formation of vic-diazides 260, albeit in low yield and with very limited substrate scope. (Scheme 63).188 Notably, electron-rich alkenes 261 display a different reactivity mode and are converted to α-azido ketones 262.189
Scheme 63.

Diazidonation of α,β-unsaturated esters with Zbiral’s hypervalent iodine reagent.
Moriarty and Khosrowshahi have reported a related, but considerable more effective diazidonation reagent generated by the action of sodium azide on iodosylbenzene (Scheme 64).190 A range of alkenes, including benzofuran and N-benzoylindole, undergo reaction to yield diazides 264 with variable diastereoselectivity. Although Moriarty proposed an ionic mechanism, involving the formation and displacement of an iodonium ion intermediate 263, subsequent studies by Magnus (vide infra) suggest that a radical pathway may also exist. That Δ5,6-steroids react with (PhIO)n/NaN3 to form the corresponding 7α-azidosteroids, rather than undergo diazidonation is further evidence of the presence of azide radicals in these reactions.191
Scheme 64.

Synthesis of vicinal diazides using Moriarty’s hypervalent iodine reagent.
Armimoto and co-workers have reported the diazidonation of allylsilanes using a mixture of iodosylbenzene and trimethylsilyl azide (TMSA) (Scheme 65).192 In this case, treatment of (PhIO)n with TMSA at −78 °C for 3 h generates a reagent formulated as 266 or (diazidoiodo)benzene (267). Reaction with allylsilanes then provides the corresponding vicinal diazides 268 in moderate to high yield. Although the diastereoselectivity of this process was not reported, the functional group tolerance is notable. Arimoto has proposed that diazide formation proceeds via a [2+3] cycloaddition to form a triazoline intermediate, which then undergoes ring opening with azide.
Scheme 65.

Diazidonation of allylsilanes with the reagent combination (PhIO)n/TMSN3.
While studying methods for the electrophilic amination of ketones and their derivatives,193 Magnus and co-workers found that treatment of triisopropyl (TIPS) enol ethers, such as 269, with the reagent combination (PhIO)n/TMSN3 led to dramatically differing results depending on the reaction temperature employed (Scheme 66). Reaction of 269 at 0 °C rapidly leads to the formation of β-azidonation product 270, while reduction at −78 °C favors the formation of 271a, the product of α-bis-azidonation.194 It was also found that addition of catalytic quantities of the stable radical TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl) served to suppress the β-azidonation pathway in favor of α-bis-azidonation. The α-azidonation process has wide substrate scope and, in most cases, proceeds with high stereoselectivity (Scheme 67).
Scheme 66.

Temperature dependent reactivity of TIPS enol ethers with (PhIO)2/Me3SiN3 and substrate scope of bis-azidonation.
Scheme 67.

Proposed mechanism of enol ether bis-azidonation.
The mechanisms of these divergent transformations have been studied in detail and while β-azidonation is thought to involve ionic dehydrogenation at the β-position and capture of the resulting enonium ion by azide, α-azidonation is an azide radical addition process (Scheme 67).195 Magnus has proposed that at low temperature, reaction between 265 and trimethylsilyl azide generates aryl-λ3-iodane 266 which is captured by TEMPO to form iodine(VI) species 272. Homolytic cleavage of 272 generates an azidyl radical, which then participates in the addition process to yield 271a by way of radical intermediate 274.
Although unsuitable precursors for the preparation of free 1,2-diamines, the α-bis-azidonation products shown in Scheme 67 undergo, with the aid of aluminum-based Lewis acids, substitution with a range of carbon nucleophiles to provide O-protected 1,2-azidoalcohols 275a and 275b (Scheme 68). Trapping of the intermediate onium ion in the case of bis-azide 271a proceeds with excellent diastereoselectivity.
Scheme 68.

Lewis acid-mediated reaction of bis-azide 271a with carbon nucleophiles.
Magnus has also employed the PhIO/Me3SiN3 reagent combination for the preparation of diamino pyrans (Scheme 69).196 For example, treatment of dihydropyran (276, R = H) provided trans-bis-azide 277a while bis-azidonation of unsaturated carbamate 276 (R = NHCO2Ad) proceeded with complete diastereoselectivity to yield 1,2-diaxial bis-azide 277b.
Scheme 69.

Bis-azidonation of glycals using the reagent combination PhIO/Me3SiN3.
In 2011, Muñiz and co-workers reported a breakthrough method for the iodine(III)-mediated intermolecular enantioselective diamination of styrenes (Scheme 70).197 This method is not only notable in that it is metal-free and practical, requiring only two components, but is the first example of intermolecular, enantioselective alkene diamination. Employing Ishihara’s C2-symmetric chiral iodane 280 (Scheme 71)198 and bismesylide (278) as the nitrogen source, alkenes underwent addition to form diamines 279 with good yields and high asymmetric induction. In all but a few cases, the crystallinity imparted by the bissulfonyl groups facilitates purification of these products to enantiomeric purity by a single recrystallization.
Scheme 70.

Enantioselective, intermolecular diamination of styrenes under metal-free conditions.
Scheme 71.

Structure of Ishihara’s C2-symmetric chiral iodane 280 and proposed mechanism for the enantioselective alkene diamination mediated by this reagent.
Muñiz has proposed a tentative mechanistic rationale for this transformation, in which 279 undergoes ligand exchange with 280 to generate unstable aryl-λ3-iodane 281 (Scheme 71). Reaction between 281 and the alkene then generates anti addition product 283 by way of iodonium ion 282. Ionization and formation of aziridinium ion 284 then precedes ring opening at the benzylic position to yield the observed product. The intermediacy of an aziridinium ion was invoked in order to rationalize the formation of the anti diamine product in the case of trans-β-methylstyrene (106).
As shown in Scheme 72, this methodology has been successfully applied to the preparation of the immunomodulator and veterinary anthelmintic (S)-levamisole (289). Removal of the four methanesulfonyl groups from styrene adduct 286 was accomplished in a four step sequence involving hydride reduction to bis-mono protected 287, N-benzoylation, radical N-desulfonylation under Parson’s conditions199 and acidic hydrolysis. After neutralization of salt 288, the free diamine was converted to target 289 by way of the corresponding mercaptoimidazoline.200
Scheme 72.

Muñiz’s enantioselective synthesis of the anthelmintic (S)-levamisole.
11. Transition Metal-Catalyzed Diamination
While non-radical, transition metal-mediated alkene diamination processes have been known since the early 1970s it is only in the last decade that the value of this approach has begun to be realized in earnest. Two general mechanistic pathways by which transition metal complexes can mediate alkene diamination can be envisioned (Scheme 73).201
Scheme 73.

General mechanistic pathways leading to the transition metal-mediated diamination of alkenes.
In the more classical manner, formation of a metal-alkene π complex precedes insertion, which generates a metal-alkyl species. Reductive elimination of this intermediate or nucleophilic displacement of the metal center then gives rise to the diamination product (eq. 1). Alternatively, metal complexes can undergo a ligand-based cis-addition reaction in which both carbon-nitrogen bonds are simultaneously generated (eq. 2).202 Although of this latter process also forms the basis of the catalytic, osmium(VIII)-mediated dihydroxylation and aminohydroxylation of alkenes and thus firmly established in the canon of organic synthesis,203 the preparation of diamines through this approach has proven to be considerably more challenging.
11.1. Metal Nitrosyl Complexes
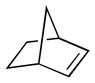
Brunner and Loskot first reported the ligand-based reaction of cobalt nitrosyl complex 291 with bicyclo[2.2.1]hep-2-enes in 1971.204 Generated from the reaction of cyclopentadienylcobalt dicarbonyl (290) and nitric oxide (NO) in hexanes,205 air-stable 291 undergoes addition to strained alkenes to form dinitrosoalkane complexes 292 (Scheme 74). This process is both diastereoselective; only the exo complexes are formed; and regioselective; in the case of dienes, i.e. in the case of 292, addition takes place only at the electron-rich alkene partner. Detailed mechanistic studies conducted by Bergman have subsequently revealed that the addition process proceeds through the intermediacy of CpCo(NO)2, which is generated by the reaction of NO with 291.206
Scheme 74.

Generation of cyclopentadienylnitrosylcobalt dimer and its reaction with strained alkenes.
Brunner’s early observations were subsequently developed by Bergman and co-workers, who employed this transformation as a method for the direct 1,2-diamination of alkenes (Scheme 75).207 In this case, in-situ reduction of the dinitrosoalkane ligands with LiAlH4 generates the corresponding 1,2-diamines 294 in fair to excellent yield. Notably, Bergman found that the reaction of 291 is not restricted to norbornyl systems and indeed undergoes stereospecific addition to a range of di-, tri- and tetra-substituted aliphatic alkenes. Unfortunately, despite the stereospecificity of the initial addition step, epimerization occurs to varying degrees during ligand reduction and mixtures of diamine isomers are obtained.
Scheme 75.

Two-step, one-pot 1,2-diamination of alkenes using cyclopentadienylnitrosylcobalt dimer/NO/LiAlH4.
Taking advantage of the ability of dinitrosyl cobalt complexes to undergo reversible exchange with alkenes and the reactivity of the dinitrosoalkane ligand system itself, Toste and Bergman have more recently employed cobalt nitrosoalkane adducts as vinyl anion equivalents for the C-H functionalization of alkenes208 and dienes.209 With regard to alkene diamination, a remarkable application of their strategy to the preparation of the polycyclic 1,2-diamine 300 is shown in Scheme 76.210
Scheme 76.

Cobalt-mediated [3+2]-annulation of alkenes with α,β-unsaturated ketones.
Most recently, Bergman and Toste have reported the first example of ruthenium-mediated alkene bis-nitrosylation (Scheme 77).211 Efficiently generated by the action of nitric oxide on [RuCl2(NO2)2(THF)2] (302), dinitrosyl complex 303 was found to undergo reaction with strained and tetrasubstituted alkenes in the presence of chelating ligands to form six-coordinate dinitrosoalkane complexes 304.
Scheme 77.

Reaction of alkenes with the dinitrosyl complex [RuCl2(NO)2(THF)].
In the case of 1,1-disubstituted and 1,1,2-trisubstituted alkenes addition is accompanied by tautomerization and complexes containing a nitrosoalkane and oxime functional group were isolated. In the case of 1-methylcyclohexene, reaction with 303 leads to the formation of compound 304d.
11.2. Imido-osmium(VIII) Reagents
Although first prepared in 1959,212 that imidoosmium(VIII) complexes undergo reaction with alkenes to form 1,2-diamines was not reported until 1977.213 These air and moisture-stable reagents are readily prepared by the condensation of amines, or their equivalents, with osmium tetroxide (305) (Scheme 78).214 In the case of N-trimethylsilyl-tert-butylamine, condensation proceeds to generate a mixture of compounds 306–308, which can be chromatographically separated.
Scheme 78.

Preparation of imido-osmium(VIII) complexes from osmium tetroxide.
Sharpless and co-workers were first to report the reaction of bis(tert-butylimido)osmium (307) and tris(tert-butylimido)osmium (308) with terminal and trans-disubstituted alkenes (Scheme 79).215 In all cases, addition took place in stereospecific and chemoselective fashion to form diimido complexes. In contrast to osmate(VI) esters, osmaimidazolidines display remarkable stability,216 although can be reduced to the corresponding 1,2-di-tert-butylamines 311 with LiAlH4. Regarding substrate reactivity, cis-disubstituted and trisubstituted alkenes react slowly with 307 and 308 while introduction of electron-withdrawing groups, as in the case of fumarate 309, increases the rate of addition. This reactivity reflects the increased nucleophilicity of the imido complexes in comparison with OsO4.217
Scheme 79.

Stoichiometric diamination of alkenes with oxotris(tert-butylimido)osmium(VIII)/LiAlH4.
The stoichiometric reaction of trisimidoosmium complexes with alkenes has also been studied by Schrock and co-workers, who reported the reaction of aryl imido complex 312 with simple alkenes, including ethylene and norbornene (Scheme 80).218 Despite substantial steric encumbrance at the metal center of 312, formation of the metallaimidazolidines 313 and 314 occurred smoothly at room temperature. To date, this chemistry has not been employed in the preparation of free 1,2-diamines.
Scheme 80.

Stoichiometric diamination of alkenes with a sterically encumbered oxotris(arylimido)osmium(VIII) complex.
During the past decade, Muñiz and co-workers have expanded the early studies of Sharpless and Schrock and extensively investigated various aspects of the osmium-mediated diamination reaction as well as optimized the yield of this process.219,220 In common with Sharpless, Muñiz has found that electron-deficient alkenes are the most favored substrates for diamination, but has also demonstrated that in addition to acrylate and cinnamate esters 315, α,β-unsaturated ketones, aldehydes, amides 318, nitriles and even 3-pyridyl acrylates are suitable substrates (Scheme 81).221 Liberation of the 1,2-diamines from their corresponding metallaimidazolidines can be accomplished through reduction with LiAlH4 or NaBH4.219
Scheme 81.

Stoichiometric diamination of electron-deficient alkenes with alkenes with dioxobis(tert-butylimido)osmium(VIII).
Although, to date, the inherent stability of the osmaimidazolidines has hampered the development of a fully catalytic, enantioselective variant of the diamination reaction, Muñiz has successfully deployed a number of strategies to control the absolute facial selectivity of the stoichiometric process. While efforts to effect enantioselection through the addition of Cinchona-based chiral ligands have largely been unsuccessful, as a result of the inability of 307 to complex these Lewis bases, use of chiral auxiliaries has proven more forthcoming. For example, reaction of (-)-8-phenylmenthyl acrylate derivatives 319 with 307 proceeds from the Re-face with good to excellent levels of diastereoselectivity (Scheme 82).219 An enantioselective catalytic variant has also been developed which employs a Ti-TADDOLate catalyst and 307 as the stoichiometric nitrogen source.222 Under these conditions, diamination of crotonyl oxazolidinone 321 (R = Me) takes place on the Re,Si-face with excellent levels of enantioinduction. The success of this process appears to stem from the comparatively low reactivity of this substrate class, which, in the absence of Lewis acids, reacts with 307 18 times slower than methyl crotonate.
Scheme 82.

Diastereo- and enantioselective diamination of electron-deficient alkenes with alkenes with dioxobis(tert-butylimido)osmium(VIII).
With regard to the reaction of the more nucleophilic tris(imido) osmium(VIII) reagent 308, Muñiz has found that, in the case of non-symmetrical olefins, formation of a configurationally stable stereogenic center at the osmium atom occurs concomitantly with diamination (Scheme 83).223 Methyl methacrylate (323), for example, undergoes reaction with near complete diastereoselectivity to favor formation of the osmaimidazoline 324a in which the ester group and bulky imino moiety are in a syn relationship. Compounds of this type represent rare examples of chiral-at-metal complexes with tetrahedral coordination. Diastereoselectivity was also observed in cinnamate and crotonate systems, albeit to a lesser degree.
Scheme 83.

Formation of stereogenic osmium centers during the diamination of alkenes with oxotris(tert-butylimido)osmium(VIII).
11.3. Stoichiometric Palladium(II)-Mediated Alkene Diamination
The first report of palladium-assisted alkene diamination was made in 1977 by Bäckvall, who employed stoichiometric quantities of trans-bis(benzonitrile)dichloropalladium(II) and various oxidants, including mCPBA, to mediate this one pot, two step transformation (Scheme 84). Diamine formation in this case occurs through a sequence of trans-aminopalladation,224 oxidation of the resulting β-amino σ-alkylpalladium(II) species 326 to a palladium(IV) species and reductive displacement by dimethylamine.225 Under these conditions, simple primary and 1,2-disubstituted alkenes, such as 325, underwent diamination with complete syn-selectivity in moderate to high yield. Although a range of oxidants, including Br2, Pb(OAc)4 and NBS, mediated the transformation of 326 to 327, mCPBA was consistently found to provide the highest yields. This observation may be attributable to the comparatively low reactivity of this reagent with alkenes, which were used in two to four-fold excess.
Scheme 84.

Bäckvall’s stoichiometric Pd-mediated 1,2-diamination of alkenes.
Bäckvall subsequently reported the stoichiometric palladium-assisted syn-1,4-diamination of 1,3-dienes with dimethylamine. 226 In this case, 1,2-diamination was not observed and the process is believed to involve the formation of a 4-dimethylamino π-allyl palladium complex that upon activation with AgBF4 or PPh3 undergoes displacement with excess amine to form the observed products.
11.4. Group 10 Metal-Catalyzed Diamination of Alkenes and Dienes with Urea Derivatives
Despite the promise of Bäckvall’s seminal work, catalytic variants of this diamination process were unknown until 2005, when the groups of Booker-Milburn (Scheme 86)227 and Muñiz (Scheme 86)228 respectively reported catalytic, inter- and intramolecular versions of this transformation. Although likely proceeding via differing catalytic cycles (vide infra), success in each case, was achieved through a realization that in order to develop a transition metal catalyzed diamination process, three major obstacles necessarily must be overcome, namely the avoidance of catalyst-product complexation, the attenuation of β-hydride elimination from the intermediate σ-alkylpalladium species and, in the case of 1,3-dienes, the circumvention of the 1,4-diamination process noted by Bäckvall.226 In both studies, undesirable product-Pd(II) ligation was avoided through the use of ureas as a source for both transferred nitrogen atoms. In the case of Booker-Milburn’s work on 1,3-dienes, use of N,N′-dialkylureas as a tethered nitrogen source also served to prevent the formation of 1,4-diamination products.
Scheme 86.

Proposed mechanism of Booker-Milburn’s Pd(II)-catalyzed diene diamination.
Booker-Milburn and co-workers found that treatment of 1,3-dienes with N,N′-diethylurea (328) in the presence of catalytic bis(acetonitrile)palladium dichloride and benzoquinone (p-BQ), as a stoichiometric oxidant, led to the efficient formation of vinylic cyclic ureas 329 (Scheme 85).227 Although the diamination of isoprene proceeded with marginal regioselectivity, addition takes place exclusively at the less substituted alkenes of 1-alkyl and aryl-substituted dienes. Noting, among other observations, that a chloride-bearing Pd(II) precatalyst is a prerequisite for the success of this transformation, Booker-Milburn proffered the mechanistic interpretation outlined in Scheme 86. In this case, anti-aminopalladation of the diene 330 generates electrophilic η3-allyl intermediate 332 (or its π-allyl isomer) which in preference to β-hydride elimination undergoes intramolecular reductive displacement of Pd(II) to generate cyclic urea 329a and a Pd(0) species which is reoxidized by p-BQ in the presence of HX.
Scheme 85.

Booker-Milburn’s Pd(II)-catalyzed intermolecular 1,2-diamination of 1,3-dienes via a “diverted” Wacker process.
In 2005, Muñiz and co-workers reported the first example of Pd(II)-catalyzed intramolecular alkene diamination employing ω-alkenyl-substituted ureas 334 (Scheme 87).228 In the presence of catalytic palladium(II) acetate, stoichiometric quantities of the hypervalent iodine oxidant PhI(OAc)2 and acetate as a base, these terminal alkenes underwent diamination to form cyclic ureas 335 with five-, six-, and seven-membered fused rings in excellent yield. Tricyclic systems, such as 335i, are also readily available under these remarkably mild conditions. This methodology is also notably robust: it can be conducted with a range of commercially available palladium catalysts and iodine(III) reagents; does not display significant solvent dependence; and proceeds under aerobic conditions.
Scheme 87.

Muñiz’s Pd(II)-catalyzed intramolecular 1,2-diamination of terminal alkenes.
Extensive kinetic, spectroscopic and labeling studies have led Muñiz to propose that diamination proceeds through a two-step mechanism whose catalytic cycle is outlined in Scheme 88. In this case, deprotonation and palladium coordination to the urea anio 337 is followed by syn-aminopalladation, which is found to be rate limiting. The resulting square-planar σ-alkylpalladium(II) complex 339 then undergoes oxidation with iodosobenzene diacetate to form cationic palladium(IV) intermediate 340.229 After dissociation of the urea from the octahedral palladium center, product formation takes place by way of an intramolecular SN2-type displacement, which proceeds with inversion and regenerates Pd(II) catalyst.230 Key to the success of this overall transformation is the observation that while σ-alkylpalladium(II) species 339 is susceptible to iodine(III)-mediated oxidation, palladium(II) complex is not.
Scheme 88.

Proposed catalytic cycle for Muñiz’s Pd(II)-catalyzed intramolecular 1,2-diamination of terminal alkenes.
While ω-alkenyl-substituted ureas which encompass internal alkenes are not suitable substrates for diamination with the palladium acetate-PhI(OAc)2 combination, Muñiz has shown that the transfer of two nitrogen atoms to homoallylic sulfonamide substrates such as 342 is not only feasible with these conditions but offers a convenient entry point to bisindolines (344a-c), bispyrrolidines (344d) and annulated indolines (344e) (Scheme 89).231 In this case, endo-selective anti-aminopalladation, palladium oxidation and amide dissociation is followed by internal displacement to generate the products of syn-diamination.
Scheme 89.

Muñiz’s route to bisindolines via Pd(II)-catalyzed intramolecular 1,2-diamination of internal alkenes.
Muñiz and Barluenga have also established that copper(II) salts can be employed as terminal oxidants in place of PhI(OAc)2 and that under these conditions, diamination of internal alkenes is feasible (Scheme 90).184 Notably, the stereochemical course of diaminations conducted in the presence of copper(II) bromide displays a distinct substrate dependence, which arises from variations in the manner by which the final C-N bond is generated. For example, cyclization of E-styrene derivative 345 proceeds with overall retention of alkene stereochemistry, while reaction of E-acrylate 350 takes place with inversion to yield the cis-configuration 351.232 In the former case, initial syn-aminopalladation is thought to be followed by anti-bromination/depalladation, giving rise to alkyl bromide 348. C-N bond formation then takes place through an intramolecular SN2-type displacement to generate both the anti-configured urea 346a and isourea 346b. While acrylate 350 also undergoes initial syn-aminopalladation, formation of the second C-N bond is proposed to take place through amide dissociation and oxidatively-induced SN2 ring closure of intermediate 354.
Scheme 90.

Stereochemically divergent diamination of non-terminal alkenes and its mechanistic origin.
The use of copper(II) salts as terminal oxidants has demonstrated excellent substrate generality and has also been employed in a remarkably straightforward construction of cyclic guanidines (Scheme 91).233 Cyclization of N-ω-alkenyl guanidines 355 generated a range of bicyclic products 356, including annulated piperidines, in excellent yield. That diamination in this case proceeds with overall syn-diastereoselectivity has been substantiated by deuterium labeling studies.
Scheme 91.

Direct synthesis of bicyclic guanidines via Pd(II)-catalyzed intramolecular cycloguanidination of terminal alkenes employing copper chloride as the terminal oxidant.
In a recent extension of their work on the diamination of tethered ureas, Muñiz and co-workers have developed an intermolecular variant of their methodology that features the regioselective transfer of nitrogen from two distinct sources, namely bistosylimide and saccharin, which exclusively takes part in the initial aminopalladation step (Scheme 92).234 Employing bis(benzonitrile)palladium dichloride as the precatalyst and iodosobenzene dipivalate as the terminal oxidant, a wide range of functionalized terminal alkenes 357 undergo addition to generate the corresponding diamines 358 with complete regioselectivity and in excellent yield. This methodology is not restricted to the use of bistosylimide and indeed diamination with bis(2-trimethylsilylethanesulfonyl)imide (SES2NH)235 offers the advantage that the saccharide residue and one of the SES groups can be simultaneously removed through treatment with cesium fluoride. In 2011, Muñiz also reported the intermolecular diamination of allylic ethers employing saccharine and N-fluoro-bis(phenylsulfonyl)imide (NSFI), which acts as both the oxidant and nitrogen source for the second carbon-nitrogen bond forming step.236
Scheme 92.

Intermolecular Pd(II)-catalyzed diamination of terminal alkenes.
In a complementary approach to Muñiz’s methodology, Michael and co-workers have developed an approach to the intra/intermolecular diamination of N-ω-alkenyl amides, carbamates and ureas which employs N-fluoro-bis(phenylsulfonyl)imide (NSFI) as an external electrophilic aminating agent (Scheme 93).237 In the presence of NSFI, catalytic palladium(II) trifluoroacetate and a triethylammonium benzenesulfonimide additive, these substrates, as exemplified by 360, undergo cyclization to form 2-aminomethyl pyrrolidine derivatives 361, in which benzenesulfonimide from NSFI is incorporated. The addition of TEMPO in this case serves to prevent alkene isomerization mediated by palladium hydride species. Michael’s methodology is notable for its generality, functional/protecting group tolerance and, importantly, the fact that the products of diamination can be differentially deprotected under relatively mild conditions.
Scheme 93.

N-Fluorobenzenesulfonamide-promoted alkene intra/intermolecular diamination of terminal alkenes.
Detailed mechanistic studies by Michael have revealed that diamination in this case proceeds via overall syn addition of both nitrogen centers through the proposed catalytic cycle outlined in Scheme 94.237b In this case, the initial step differs from the work of Muñiz in that it proceeds via an anti-aminopalladation step to form Pd(II)-alkyl complex 363. Oxidative addition of NFBS to 363 then generates highly reactive Pd(IV) species 364 from which benzenesulfonimide dissociates to form coordinatively unsaturated 365. Formation of the second C-N bond now occurs through intermolecular SN2 displacement of the C-Pd bond to form 366 and regenerate Pd(II).
Scheme 94.

Proposed catalytic pathway for the N-fluorobenzenesulfonamide (NSFI) promoted intra/intermolecular alkene diamination reaction.
11.5. Nickel-Mediated Alkene Diamination
Muñiz and co-workers have also employed nickel(II) salts for the intramolecular diamination of N-γ-alkenyl sulfamides, ureas and guanidines 367 (Scheme 95).238 In addition to the obvious economic advantages over palladium(II) complexes, nickel catalysts in this case are notable for their longevity under the reaction conditions and their ability to successfully mediate sulfamide transfer.239 While deuterium labeling studies have revealed that diamination is a stereospecific process with respect to alkene geometry and proceeds with syn stereochemistry whether this arises through a syn/anti or anti/syn process remains to be established. Nonetheless, that these reactions proceed through an initial aminometallation step is apparent from the isolation of 368a, which likely arises from protonation of a C-Ni bond. In general, substrates encompassing internal alkenes fail to undergo diamination in the presence of nickel catalysts.
Scheme 95.

Nickel(II)-catalyzed intramolecular diamination of N-γ-alkenyl sulfamides, ureas and guanidines.
11.6. Copper(II) Carboxylate-Mediated Alkene Diamination
Chemler and co-workers have pioneered the use of copper(II) carboxylates for the mediation and catalysis of alkene carboamination, aminooxygenation and diamination.240 From a synthetic perspective, this methodology represents a uniquely powerful confluence of organometallic and radical chemistry that has been applied to the preparation of a diverse range of heterocycles.241 In 2005, this group first reported the direct intramolecular diamination of γ-alkenyl and δ-alkenyl-substituted sulfamides, such as 369, which undergo cyclization in the presence of three equivalents of copper(II) acetate to form the corresponding five and six-membered sulfamides 370 (Scheme 96). In all cases examined, diamination proceeds with complete exo-regioselectively and, in the case of product 370e, with a high level of diastereoselectivity.
Scheme 96.

Copper(II) acetate-promoted intramolecular diamination of γ-alkenyl and δ-alkenyl-substituted sulfamides encompassing 1- and 1,2-substituted terminal alkenes.
From a mechanistic standpoint, Chemler has proposed that this diamination commences with base-assisted ligand exchange between unsaturated sulfamide 369 and copper(II) acetate to form intermediate 370 (Scheme 97). This species then undergoes rate-determining syn-aminocupration to form unstable alkylcopper(II) species 371 which subsequently suffers Cu-C bond homolysis giving rise to primary radical 372, whose existence is suggested by deuterium labeling studies. Interaction of this electron deficient intermediate with copper acetate would then form alkylcopper(III) complex 373 which, upon reductive elimination,242 would give rise to cyclic sulfamide 370a through formation of the second C-N bond.
Scheme 97.

Proposed mechanism of the copper(II) carboxylate promoted intramolecular alkene diamination reaction.
As a result of methodological limitations imposed by the low solubility of copper(II) acetate, Chemler has introduced copper(II) neodecanoate [Cu(nd)2] as a second-generation mediator of diamination (Scheme 98).243 Adoption of this organic soluble complex obviates the need for polar non-protic solvents such as DMSO and DMF and allows reactions to be conducted in 1,2-dichloroethane (DCE), albeit in a pressure tube since reaction temperatures are significantly higher than the boiling point (84 °C) of this solvent. Nonetheless, these conditions offer significantly higher yields and facilitate the generation of a broader range of bis(amino) products, including ureas (376f), bis(anilines) (376g) and α-amido-pyrroles (376h). Of particular note is the high selectivity for the formation of cis-2,5-disubstituted pyrrolidines observed during the cyclization of α-substituted pent-4-enyl sulfamides such as 376.
Scheme 98.

Copper(II) neodecanoate [Cu(nd)2]-promoted intramolecular diamination of terminal alkenes.
In a valuable extension of their original diamination methodology, Chemler and co-workers have most recently reported an intra/intermolecular variant of this chemistry in which external N-nucleophiles, including azide, sulfonamides, benzamide and a wide range of anilines, participate in the second C-N bond-forming step (Scheme 99).244 For example, in the presence of copper(II) 2-ethylhexanoate [Cu(eh)2] 2-substituted 1-allyl-1-benzyl ureas 378 undergo diamination with these external nucleophiles to form 4-substituted imidazolinones 379 in excellent yield. This strategy has also successfully been applied to the preparation of 2,5-disubstituted pyrrolidines such as 379e and 379f through the cyclization of N-γ-alkenyl sulfonamides.
Scheme 99.

Copper(II) 2-ethylhexanoate-promoted intra/intermolecular alkene diamination.
Through the use of the chiral bis(oxazoline) ligand (R,R)-Ph-box, Chemler has also successfully rendered the intermolecular diamination of N-mesyl-ortho-allylaniline (380) enantioselective (Scheme 100).244 While attempts to employ MnO2 as a terminal oxidant with unsaturated sulfamide and urea substrates failed, the cyclization of this substrate in the presence of p-toluenesulfonamide proceeds to form 381 with promising enantioselectivity and is catalytic in copper(II) triflate.
Scheme 100.

Enantioselective copper-catalyzed intra/intermolecular diamination of N-mesyl-ortho-allylaniline.
11.7. Gold-Mediated Alkene Diamination
Despite the substantial progress made over the last decade in the use of gold catalysts for alkene hydroamination245 and more recently oxidative coupling reactions,246 the exploitation of this chemistry towards alkene diamination is relatively unexplored. In this context, Muñiz’s 2009 report detailing the use a Au(I)/Au(III) cycle to intercept the hydroamination pathway in favor of the oxidative diamination of N-γ-alkenyl ureas 382 is significant (Scheme 101).247 Of several gold complexes examined, triphenylphosphinegold(I) acetate proved to be the most effective catalyst and, in the presence of PhI(OAc)2 and sodium acetate, mediates the conversion of these substrates to the corresponding imidazolinones 382 in excellent yield. In contrast to palladium catalysis, diamination under these conditions proceeds with overall syn-selectivity as revealed by the formation of tricycle 383h.
Scheme 101.

Gold-catalyzed intramolecular diamination of γ-alkenyl-substituted ureas encompassing 1-substituted and 1,2-disubstituted alkenes.
On the basis of a deuterium labeling study and investigation of the stoichiometric reactions of isolated model gold complexes, Muñiz has proposed the mechanism outlined in Scheme 102. Diamination is thought to commence with the formation of alkylgold(I) complex 385 through alkene anti-aminoauration. Oxidation of this intermediate then proceeds irreversibly giving rise to the corresponding gold(III) complex 386 which undergoes intramolecular displacement with inversion at the gold(III)-bearing carbon center to generate the reduced gold(I) complex and the observed product 388. In support of this proposal, Muñiz has demonstrated that while triphenylphosphinegold(I) acetate is inert to iodine(III)-mediated oxidation, a model alkylgold(I) complex is rapidly converted to the corresponding gold(III) species. Furthermore, it has been shown that this high-valent complex readily undergoes SN2-like displacement with the anion of an N-tosyl urea to form a C-N bond and generate triphenylphosphinegold(I) acetate.
Scheme 102.

Proposed mechanism for the gold-catalyzed, iodine(III)-promoted intramolecular diamination of γ-alkenyl-substituted ureas.
Nevado and de Haro have also employed Au(I)/Au(III) catalysis to achieve the oxidative difunctionalization of alkenes including the intra/intermolecular aminoamidation of N-tosyl-4-pentenyl alkenes 389 (Scheme 103).248 Using the cationic complex [(Ph3P)AuSbF6] and Selectfluor (391) as a bystanding oxidant,249 these substrates undergo 6-endo-trig cyclization in nitrile solvents to generate N-piperidin-3-yl carboxamides 390 in high yield. Several nitriles, including acetonitrile, propionitrile and butyronitrile act as external nucleophiles in this reaction and while currently limited to terminal alkenes, this diamination method represents an important complement to related Pd(II)/Pd(IV)-catalyzed processes which proceed with 5-exo selectivity; cf. Scheme 90.
Scheme 103.

Gold-catalyzed, oxidative intra/intermolecular diamination of N-tosyl-4-pentenyl amines in the presence of nitrile nucleophiles.
Nevado has suggested that diamination in this case can potentially occur through a number of manifolds, as outlined in Scheme 104. In the predominate pathway, 389 undergoes gold(I)-mediated 6-endo trans-aminoauration to form 392 which is oxidized by Selectfluor to the corresponding alkyl-gold(III) species 392. Substitution of fluoride by acetonitrile, hydrolysis of this now activated ligand and subsequent reductive elimination would then give rise to the major aminoamidation product 394. The formation five-membered amines, such as 390g, can be ascribed to the analogous 5-exo pathway involving intermediates 395 and 396. Nevado has also noted that diamination may also proceed through an alternative mechanism, namely one in which the common bicyclic aziridinium ion 398,250 formed through the intramolecular reductive elimination of 392 and/or 396, participates in a Ritter reaction with the nitrile solvents to form the observed products.251
Scheme 104.

Possible mechanistic pathways involved in the gold(III)-catalyzed intra/intermolecular alkene diamination reaction.
Although not strictly falling within the brief of this review as it involves alkene hydroamination rather diamination, Widenhoefer’s gold(I)-catalyzed cyclization of N-δ-allenyl ureas 399 is worthy of note since it offers diastereoselective access to bicyclic imidazolidinones 400 and related systems with high efficiency (Scheme 105).252 Cyclization in this case is promoted by a catalytic mixture of the gold(I) N-heterocyclic carbene complex (401)AuCl and AgPF6 at room temperature. In addition to displaying a tolerance for a range of urea N-substituents, incorporation of heteroatoms within the heptadienyl chain of 399 provides access to both morpholine (400b) and piperazine (400c) systems.
Scheme 105.

Preparation of bicyclic imidazolidin-2-ones through the gold(I)-catalyzed dihydroamination of N-δ-allenyl ureas.
11.8. Metal-Mediated Addition of Diaziridinones and Related Systems to Conjugated Alkenes
Arguably, the most well established and synthetically proven transition metal-mediated alkene diamination method is that developed Shi and co-workers, who have pioneered the use of strained diaziridinones and their analogs as unique and versatile nitrogen sources. In contrast to other metal-catalyzed diamination procedures reported to date, this approach to alkene oxidation is distinct in that catalytic turnover is mediated by the diaminating agent itself rather than an external oxidant. In this regard, Shi’s approach is conceptually related to the work of Donohue253 and Yoon254 who have respectively employed N-sulfonyloxycarbamates and N-sulfonyl oxaziridines as “preoxidized” nitrogen sources for the oxamination of alkenes. This general strategy holds a number of strategic advantages, not least of which are operational simplicity and the increased functional group tolerance associated with the absence of an external oxidizing agent.
In 2007, Shi first reported the use of di-tert-butyldiaziridinone (402)255 as a nitrogen source in the diamination of conjugated dienes (403) (Scheme 106).256 Employing 402 and Pd(PPh3)4 as a catalyst (10 mol%), a range of substrates, including acyclic and cyclic dienes, dienol ethers, dienoates and trienes, underwent diamination to form imidazolidinones 404 with high yield and excellent regioselectivity for the internal alkene. By adding diaziridinone 402 slowly in the absence of solvent, Shi has subsequently established that catalytic loading can be reduced to 1–2 mol%. That the regioselectivity observed by Shi is different from that previously found by Booker-Milburn and Lloyd-Jones227 is an indication that a different mechanism is involved in this case. Importantly from a practical perspective, removal both partial and complete, of the N-tert-butyl groups from the diamination products 404 can be accomplished cleanly through solvolysis with strong acids, such as TFA (vide infra). Alternatively, treatment with aqueous acid under forcing conditions (conc. HCl, reflux, 30 h) mediates complete deprotection to the corresponding 1,2-diamines.
Scheme 106.

Palladium(0)-catalyzed diamination of conjugated dienes and trienes using di-tert-butyldiaziridinone as the nitrogen source.
On the basis of detailed spectroscopic studies and accompanying kinetic data, Shi has proposed that diamination in this case proceeds through the mechanism outlined in Scheme 107. The reaction commences with the rate-determining oxidative insertion of a Pd(0) species, thought to be Pd(PPh3)2, into the N-N bond of 402 to form symmetrical four-membered Pd(II) complex 405. Reversible ligand exchange with the dienyl substrate 403 then gives rise to complex 406, which undergoes migratory insertion to form π-allyl species 407. In the final step of the cycle, reductive elimination of the product 408 regenerates the Pd(0) catalyst.
Scheme 107.

Proposed catalytic cycle for palladium(0)-catalyzed diamination of 1,3-dienes using di-tert-butyldiaziridinone.
With regard to asymmetric diamination, Shi and co-workers have successfully identified a number ligand types that effectively mediate this intermolecular process. To date, the most successful ligand identified has been the BINOL-based phosphorous amidite 410, which when employed in a 1:2.2 ratio with Pd2(dba)3 offers good yields and high enantioselectivity with a range of substrates (Scheme 108).257 Asymmetric internal diamination of 1,3-dienes has also been achieved with N-heterocyclic carbene-Pd(0) complexes, albeit with slightly lower enantioselectivity (62–78% ee) than that found with 410.258
Scheme 108.

Asymmetric palladium(0)-catalyzed diamination of conjugated dienes and trienes with di-tert-butyldiaziridinone.
In addition to dienes and trienes, Shi has also found that monosubstituted and 1,1-disubstituted olefins 411 undergo Pd(0)-catalyzed diamination at the allylic and homoallylic positions in the presence of excess di-tert-butyldiaziridinone (402) (Scheme 109). Under solvent free conditions, slow addition of 402 to these terminal alkenes leads to the formation of diamination products 412 in good yield and complete regio- and stereoselectivity. In addition to displaying good functional tolerance, this transformation is also successful with substrates that bear two terminal alkenes thus providing access to fully protected tetramines such as 412h.
Scheme 109.

Palladium-catalyzed dehydrogenative diamination of terminal alkenes at allylic and homoallylic positions: use of di-tert-butyldiaziridinone as the nitrogen source and terminal oxidant.
From a mechanistic standpoint, Shi has suggested that this remarkable transformation proceeds through a catalytic cycle in which diaziridinone 413 plays a dual role as both hydrogen acceptor in the oxidation of alkene 411 to 1,3-diene 417 and the nitrogen source for the diamination of this intermediate (Scheme 110). After formation via oxidative insertion of Pd(0) into the N-N bond of 402, Pd(II) species 414 complexes olefin 411 and mediates formation of π-allyl complex 416 through removal of the allylic hydrogen. β-Hydride elimination from 416 then regenerates the Pd(0) catalyst and gives rise to urea 418 and diene 417, which undergoes diamination through the mechanism previously discussed. Although proceeding through a different mechanism, diaziridinone 413 (R = t-Bu) has also been used as a dehydrogenating agent in its own right, under copper catalysis.259
Scheme 110.

Proposed catalytic cycle for palladium-catalyzed dehydrogenative diamination of terminal alkenes with di-tert-butyldiaziridinone.
Shi has also developed a catalytic asymmetric variant of the allylic/homoallylic diamination reaction that employs the chiral H8-BINOL-derived phosphorus amidite 422 in combination with Pd2(dba)3 (Scheme 111).260 Success in this case was found to depend significantly upon the palladium/ligand ratio with a 1:2.2 mixture offering excellent levels of enantioselectivity with most substrates. In the case of bis-diamination (421e) and those substrates with existing stereogenic centers, the stereochemical outcome of diamination was found to be primarily controlled by the chiral catalyst.
Scheme 111.

Palladium-catalyzed asymmetric dehydrogenative diamination of terminal alkenes at allylic and homoallylic positions.
Having successfully established conditions for asymmetric alkene diamination, Shi and co-workers have subsequently demonstrated the value of this methodology in its application to the synthesis of (+)-CP-99,994 (427), a human neurokinin 1 (NK1) substance P receptor antagonist (Scheme 112).261 Obtained through the diamination of 4-phenyl-1-butene, building block 423 was converted to ester 423 by way of a 3-step sequence featuring oxidative cleavage of the alkene, olefination of the resulting aldehyde and hydrogenation. Treatment of 423 with trifluoroacetic acid then effected selective mono-de-N-alkylation to provide compound 424 in excellent yield and suitably differentiated for introduction of the N-benzyl group found in target 427. After N-nosylation of 424, the remaining N-tert-butyl group was cleaved by treatment with methanesulfonic acid to yield 425. Saponification of this imidazolidinone and recyclization under acidic conditions then provided δ-lactam 426, which was converted to (+)-CP-99,994 (427) through a sequence of N-alkylation, with 2-methoxybenzyl chloride, removal of the N-nosyl group and lactam reduction.
Scheme 112.

Shi’s asymmetric synthesis of (+)-CP-99,994.
While thiadiaziridine 1,1-dioxide (428)262 also participates in the palladium-catalyzed allylic/homoallylic diamination reaction, it does so with complementary regioselectivity to that displayed by diaziridinone 402 (Scheme 113).263 For example, alkenes 429 in the presence of 2 equivalents of 428 undergo dehydrogenative diamination at the termination position to form mono-substituted sulfamides 430 in moderate to good yield. The observation that 1,3-pentadiene undergoes diamination with 428 at the internal position has led Shi to propose that rather than involving the formation of a diene intermediate, diamination actually occurs through the Pd(II)-mediated formation and cyclization of an allylic sulfamide.
Scheme 113.

Palladium-catalyzed dehydrogenative diamination of terminal alkenes at allylic and homoallylic positions: use of N,N-di-tert-butylthiadiaziridine 1,1-dioxide as nitrogen source and oxidant.
The search of alternative diamination catalyst systems with increased substrate scope and complementary regioselectivity to that offered by palladium has lead Shi and co-workers to investigate the use of copper(I) salts.264 Among a various systems evaluated, a 1:1 combination of CuCl and triphenyl phosphite (P(OPh)3) was found to effectively mediate the reaction of 402 with a wide range of conjugated dienes and trienes 403 at the terminal position (Scheme 114). Reactions in this case proceed at room temperature and do so with good to high yield.
Scheme 114.

Copper(I)-catalyzed terminal diamination of conjugated dienes and trienes with di-tert-butyldiaziridinone.
Despite the challenges presented by the involvement of radicals species in the copper-catalyzed, internal diamination reaction (vide infra), Shi has successfully developed an asymmetric variant of this process which employs a combination of CuCl and the bisphosphine ligand (R)-DTBM-SEGPHOS.265,266 In this case, diamination of conjugated dienes and a triene with 402 proceeded in 59–93% yield and with enantioselectivities between 62 and 74%. Shi has also shown that copper(I) catalysts imbued with a chiral, BINOL-based phosphate ligand can also mediate asymmetric, external diamination, albeit with somewhat lower ee’s (49–61%).267
In more recent studies, Shi has found that, in the absence of phosphine ligands, copper(I) salts and in particular inexpensive CuBr promote diene diamination at the internal alkene position, in common with palladium-based catalysts (Scheme 115).268 Using di-tert-butyldiaziridinone (402) as the nitrogen source, a diverse range of substrates, including 1-substituted, 1,2-disubstitiuted, 1,3-disubstituted, cyclic and even 1,2,3-trisubstituted dienes undergo reaction to form imidazolidinones 421 in high yield and regioselectivity. Notably, diamination under these conditions can also be conducted on large scale; e.g. using 5 mol% CuBr, compound 421a was prepared on a 38 g scale.
Scheme 115.

Copper(I)-catalyzed internal diamination of conjugated dienes and trienes with di-tert-butyldiaziridinone.
The remarkable switch in selectivity for external and internal diamination observed with the use of CuCl-P(OPh)3 and CuBr catalysts has been attributed, by Shi, to the existence of two competing reaction mechanisms respectively involving Cu(II) and Cu(III) species (Scheme 116). In both cases, diamination is proposed to commence with the Cu(I)-mediated reductive cleavage of diaziridinone 402 to form Cu(III) complex 433 and/or the corresponding open-chain Cu(II) amidyl radical 434. In the presence of phosphine ligands, generation of 434 appears to be favored and addition of this species for reasons of sterics and product stability takes place at the terminal position of diene 410 to generate allyl radical 436 and/or its organocopper(III) analog 47. Reductive elimination the generates the second C-N bond and gives rise to the terminal diamination product 431 and the Cu(I) catalyst. Internal diamination, on the other hand, is thought to involve the intermediacy of four-membered Cu(III) complex 433 that, in the absence of phosphine ligands, coordinates the diene to form 437. In an analogous fashion to the Pd-catalyzed process, migratory insertion of the nitrogen atom to the internal double bond of this system then gives rise to π-allyl species 438. Final reductive elimination then gives rise to internal diamination product 432 and returns the Cu(I) catalyst.
Scheme 116.

Proposed dual mechanisms for the copper(I)-catalyzed internal and external diamination of conjugated dienes.
With regard to the copper-catalyzed diamination of alkenes, Shi has found that, if activated by the presence of an aryl group, 1,1-disubstituted olefins 439 are suitable substrates for this process (Scheme 117).269 Using 5 mol% of a 1:1 combination of CuCl and PPh3, a wide range of styrene and acrylate derivatives undergo diamination with di-tert-butyldiaziridinone (402) at 65 °C to form the corresponding imidazolidinones 440 in high yield. Of particular note has been Shi’s application of this methodology to synthesis of the potent NK1 antagonist Sch 425078 (443) from allyl ether 421.269
Scheme 117.

Copper(I)-catalyzed diamination of disubstituted terminal alkenes; synthesis of Sch 425078.
While monosubstituted terminal alkenes fail to undergo diamination with di-tert-butyldiaziridinone (402) under palladium catalysis, N,N-di-tert-butylthiadiaziridine 1,1-dioxide (428) in the presence of CuCl-P(n-Bu)3 undergoes addition to a range of activated systems, including styrenes, 3-vinylindoles, enynes and enol ethers to form cyclic sulfamides 446 (Scheme 118).270 Most recently, in 2011, Shi and co-workers has reported that while conjugated dienes also undergo terminal diamination with reagent 428 in the presence of CuCl-P(n-Bu)3, the use of CuBr leads to a near complete change in regioselection for internal diamination.271
Scheme 118.

Copper(I)-catalyzed intermolecular diamination of activated terminal alkenes with di-tert-butyldiaziridinone.
The common occurrence of cyclic guanidines in pyrrole-2-aminoimidazole alkaloids and the intense synthetic interest that this marine natural product family has evoked ensures that the development of methods to access this functional group is of considerable importance.272 In this regard, the cycloguanidation of alkenes offers a particularly appealing approach for the late-stage introduction of these highly polar ring systems in fully protected form. Shi and co-workers have found that di-tert-butyl diaziridinimine 402 readily undergoes Cu(I)-catalyzed addition at the terminal positions of conjugated dienes, trienes, enynes and monosubstituted alkenes 447 to yield the cyclic N-cyano guanidines 448 in good to excellent yield (Scheme 119).273 Deprotection of these products is readily accomplished in high yield by solvolysis in acid and neutralization with NaOH.
Scheme 119.

Copper(I)-catalyzed cycloguanidation of alkenes, dienes and trienes using di-tert-butyldiaziridimine.
12. Diamination via Pericyclic Processes
12.1. Imido Selenium Compounds
While the use of pericyclic reactions is perhaps the least well developed approach to direct alkene 1,2-diamination, a select number of notable and compelling studies have been reported. Sharpless and Singer, for example, have employed the reaction of selenium dioxide bis(imide) 451 with 1,3-dienes to accomplish diamination with exclusive cis selectivity (Scheme 120).274 Generated in situ by the oxidation of selenium powder in the presence of p-toluenesulfonamide (449) and it sodium salt 450, compound 451 undergoes regioselective reaction with cyclic and acyclic 1,3-dienes to generate 1,2-diaminoalkanes 456 in moderate to low yield. In the case of 452, this transformation is believed to proceed through the intermediacy of [4+2] cycloadduct 453, which undergoes ring opening with p-toluenesulfonamide to form 453. [2,3]-Sigmatropic rearrangement of this intermediate then gives rise to the observed cis products, after desulfurization of the selenium(II) amide 455.
Scheme 120.

One-pot 1,2-diamination of 1,3-dienes with the selenium dioxide bis(imide) reagent 451.
In order to facilitate the deprotection of the diamine products, Sharpless has also developed reagent 459 where the tosyl groups are replaced by o-nitro benzenesulfonyl substituents (Scheme 121).275 Prepared under milder conditions, from the corresponding N,N′-dichlorosulfonamide 457 and sodium sulfonamide 458, this compound, with the notable exception of 1,3-cyclopentadiene, reacts less efficiency with 1,3-dienes than 451. Electron-withdrawing substituents were found to reduce substrate reactivity towards this highly electrophilic species: reaction of 1,3-cyclohexadiene carboxylate failed to yield diamine and provided only traces of 460d, the product of an apparent ene reaction.
Scheme 121.

1,2-Diamination of 1,3-dienes with the modified selenium dioxide bis(imide) reagent 459.
The primary advantage of reagent 459 is the degree to which N-nosyl groups facilitate the N-alkylation of the vicinal sulfonamides and the ease with which this protecting group can subsequently be cleaved (Scheme 122). For example, bis-N-alkylation of 460a and deprotection using Fukuyama’s conditions,276 provides a route to mono-N-alkylated diamines 461 with reasonable overall efficiency.
Scheme 122.

12.2. Imido Sulfur Compounds
Weinreb has reported a related, but stepwise method for the stereocontrolled 1,2-diamination of 1,3-dienes which employs sulfur dioxide bis(imides) 463a and 463b (Scheme 123).277 In comparison to their selenium congeners (i.e. 453), the 3,6-dihydrothiazin-imines resulting from the [4+2] cycloaddition of these reagents are less reactive. Consequently, conversion to the diamination products requires a distinct ring-opening step with an organometallic nucleophile to generate an allylic sulfilimine prior to its [2,3]-sigmatropic rearrangement. In the case of (E,E)-2,4-hexadiene (462), reaction with bis(sulfonamide) 463a gave rise to a separable 1:1 mixture of S-epimers 464a and 464b. Sequential treatment of cia-trans isomer 464b with phenylmagnesium bromide and trimethyl phosphite provided vicinal sulfonamide 466 in excellent yield. Although cis-cis adduct 464a failed to react with the Grignard reagent because of steric hindrance, its conversion to 466 was accomplished with methyllithium. Notably, simple heating cis-cis-thiazinimine 464a promotes [2,3]-sigmatropic rearrangement to thiadiazolidine 467, which can be reduced with NaBH4 to the unsaturated disulfonamide 466.
Scheme 123.

Weinreb’s three-step, two-pot 1,2-diamination of 1,3-dienes with the sulfur dioxide bis(imides).
13. Diamination via Organocatalysis
Despite the volume of published work concerning organocatalysis and its use for both the α- and β-amination of saturated and unsaturated carbonyl compounds,52 there are relative few reports concerning the exploitation of this powerful synthetic strategy for the direct 1,2-diamination of alkenes. In 2007, Jørgensen and co-workers disclosed the first example of the organocatalyzed asymmetric diamination of α,β-unsaturated aldehydes (Scheme 124).278 Use of chiral secondary amine 471, in this case, promotes the regioselective and enantioselective addition of succinamide (468) to enals 469. Upon subsequent addition of diethyl azodicarboxylate (412), intermediate enamine 474 underwent amination giving rise to the syn-diamination products 470 with excellent enantiomeric excess, albeit in rather low conversion and moderate diastereoselectivity. Difficulties faced in the development of this process include the tendency of the electrophilic nitrogen-source to undergo decomposition in the presence of succinamide as well as its ability to act as a dienophile towards α,β-unsaturated aldehydes.
Scheme 124.

Jørgensen’s organocatalyzed asymmetric multicomponent syn-selective diamination of α,β-unsaturated aldehydes.
More recently, MacMillan and co-workers have successfully brought to bear their cycle-specific organocascade catalysis strategy to this problem and in doing so have accomplished the stepwise, one-pot diamination of crotonaldehyde (477) in high yield and near complete enantioselectivity (Scheme 125).279 In this case, while imidazolidinone 476 is employed as a LUMO-lowering iminium catalyst to promote the 1,4-addition of silyloxycarbamate 478, (R) or (S)-proline are used as catalysts for the subsequent enamine amination reaction with dibenzylazodicarboxylate (479). A strategic advantage of this approach is that through the appropriate choice of catalyst combination, both the absolute and relative stereochemistry of the addition process can be controlled. From a practical standpoint, diamination is conducted in a one-pot, stepwise manner where proline, 479 and water are introduced after the completion of the 1,4-addition and the resulting α-amino aldehydes are reduced to the corresponding primary alcohols 480 in situ.
Scheme 125.

Application of cycle-specific organocatalysis to the enantio- and diastereoselective diamination of crotonaldehyde.
14. Summary and Outlook
Although the direct diamination of alkenes has been known for more than a century, the last decade has heralded a renaissance of interest in this important yet traditionally problematic reaction. In large part, success in this area has stemmed from the development of new metal-catalyzed and iodine(III)-mediated processes. However, since no single method currently offers complete stereo- and/or regiocontrol for all systems, there remains much to be accomplished in this field. For example, despite the promise of diaminations that are reliant on binary nitrogen oxides, these methods are limited by the difficultly associated with handling these toxic reagents and complexity of the transformations they mediate. In this context, the development of catalytic metal-mediated dinitrosylation methods is clearly a goal of considerable importance. Despite the promising work of Muñiz in the area of osmium-mediated alkene diamination, a methodology gap remains between this stoichiometric chemistry and a practical, catalytic diamination variant of the Sharpless asymmetric dihydroxylation reaction. In this regard, the use of pre-oxidized nitrogen sources has proven to be a highly productive approach for Shi and co-workers and invites other developments of this elegant approach to alkene diamination. With regard to organocatalytic methods of diamination, although initial studies by Jørgensen and MacMillan have confirmed the feasibility of this approach, future developments in this area will likely be dependent on the identification of nucleophilic and electrophilic nitrogen species that are compatible with one another. Notwithstanding the notable examples noted in the current article, the application of metal-mediated diamination to the synthesis of complex targets and in particular natural products remains in its infancy. Undoubtedly, the deployment of new alkene diamination reactions in preparation of such complex substrates will not only clarify the scope and limitation of existing processes but also spur the development of new methodologies.
15. Note Added in Proof
While this paper was in review, Chiba and co-workers reported the development of a novel method for the synthesis of bi- and tricyclic amidines. This alkene diamination process proceeds through the intramolecular, copper-catalyzed aerobic [3+2]-annulation of N-alkenyl amidines. The resulting cyclic amidines are readily converted to the corresponding 1,2-diamines by reduction with alane (Wang, Y. F., Zhu, X., Chiba, S. J. Am. Chem. Soc. 2012, 134, 3679–3682).
Figure 8.

1,2-Diazides generated by the reaction of iodine azide with trans-stilbenes.
Acknowledgments
I thank my colleague Professor Tom Driver for sharing his mechanistic insights and stimulating discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For a review detailing the biological relevance of 1,2-diamines and their derivatives, see: Saibabu Kotti SRS, Timmons C, Li G. Chem Biol Drug Des. 2006;67:101–114. doi: 10.1111/j.1747-0285.2006.00347.x.See also references 7 and 35.
- 2.a) Lucet D, Le Gall T, Mioskowski C. Angew Chem, Int Ed. 1998;37:2581–2627. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (b) Kemp JEG. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Ley SV, editors. Vol. 7. Pergamon; Oxford: 1991. p. 488. [Google Scholar]; (c) Gasc MB, Lattes A, Perie JJ. Tetrahedron. 1983;39:703–731. [Google Scholar]
- 3.For reviews detailing the metal-mediated synthesis of 1,2-diamines, see Muñiz K, Iesato A, Nieger M. Chem Eur J. 2003;9:5581–5596. doi: 10.1002/chem.200305011.Muñiz K. Chem Soc Rev. 2004;33:166–174. doi: 10.1039/b307102m.Cardona F, Goti A. Nat Chem. 2009;1:269–275. doi: 10.1038/nchem.256.de Figueiredo RM. Angew Chem Int Ed. 2009;48:1190–1193. doi: 10.1002/anie.200804362.Chemler SR. Org Biomol Chem. 2009;7:3009–3019. doi: 10.1039/B907743J.Majumdar KC, Roy B, Debnath P, Taher A. Curr Org Chem. 2010;14:846–887.Chemler SR. J Organomet Chem. 2011;696:150–158. doi: 10.1016/j.jorganchem.2010.08.041.
- 4.Chow YL. Acc Chem Res. 1973;6:354–3603. d60. [Google Scholar]
- 5.(a) Imagawa K, Hata E, Yamada T, Mukaiyama T. Chem Lett. 1996:291–292. [Google Scholar]; (b) Wang L, Shirakawa S, Maruoka K. Angew Chem Int Ed. 2011;50:5327–5330. doi: 10.1002/anie.201101307. [DOI] [PubMed] [Google Scholar]
- 6.(a) Yamamoto H, Kawasaki M. Bull Chem Soc Jpn. 2007;80:595–607. [Google Scholar]; (b) Nair V, Biju AT, Mathew SC, Babu BP. Chem Asian J. 2008;3:810–820. doi: 10.1002/asia.200700341. [DOI] [PubMed] [Google Scholar]; (c) Molina CL, Chow CP, Shea KJ. J Org Chem. 2007;72:6816–6823. doi: 10.1021/jo070978f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For comprehensive discussions of the occurrence, biologically activity and synthesis of α,β-amino acids, see: Viso A, Fernández de la Pradilla R, García A, Flores A. Chem Rev. 2005;105:3167–3196. doi: 10.1021/cr0406561.Viso A, Fernández de la Pradilla R, García A, Flores A. Chem Rev. 2011;111:PR1-PR42. doi: 10.1021/cr100127y.
- 8.Marahiel MA, Stachelhaus T, Mootz HD. Chem Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 9.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem Rev. 2005;105:739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kimura K-I, Bugg TDH. Nat Prod Rep. 2003;20:252–273. doi: 10.1039/b202149h. [DOI] [PubMed] [Google Scholar]; (b) Winn M, Goss RJ, Kimura K, Bugg TD. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 11.(a) Meierhenrich UJ, Muñoz Caro GM, Bredehöft JH, Jessberger EK, Thiemann WH. Proc Natl Acad Sci U S A. 2004;101:9182–9186. doi: 10.1073/pnas.0403043101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Strasdeit H. ChemBioChem. 2005;6:801–803. doi: 10.1002/cbic.200400435. [DOI] [PubMed] [Google Scholar]
- 12.Monaghan DT, Bridges RJ, Cotman CW. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- 13.Ranger CM, Winter RE, Singh AP, Reding ME, Frantz JM, Locke JC, Krause CR. Proc Natl Acad Sci U S A. 2011;108:1217–1221. doi: 10.1073/pnas.1013497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakai R, Oiwa C, Takaishi K, Kamiya H, Tagawa M. Tetrahedron Lett. 1999;40:6941–6944. (isolation) [Google Scholar]
- 15.DeMong DE, Williams RM. J Am Chem Soc. 2003;125:8561–8565. doi: 10.1021/ja0351241. and references therein. [DOI] [PubMed] [Google Scholar]
- 16.McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. J Am Chem Soc. 2002;124:10260–10261. doi: 10.1021/ja017748h. (isolation) [DOI] [PubMed] [Google Scholar]
- 17.For the total synthesis of muraymycin D2, see: Tanino T, Ichikawa S, Shiro M, Matsuda A. J Org Chem. 2010;75:1366–1377. doi: 10.1021/jo9027193.Tanino T, Ichikawa S, Matsuda A. Org Lett. 2011;13:4028–4031. doi: 10.1021/ol201527k.
- 18.(a) Kusumoto S, Imaoka S, Kambayashi Y, Shiba T. Tetrahedron Lett. 1982;23:2961–2964. (synthesis) [Google Scholar]; (b) Jackson MD, Gould SJ, Zabriskie TM. J Org Chem. 2002;67:2934–2941. doi: 10.1021/jo016182c. (biosynthesis) [DOI] [PubMed] [Google Scholar]
- 19.For leading references, see: Wenzel CQ, Daniels C, Keates RAB, Brewer D, Lam JS. Mol Microbiol. 2005;57:1288–1303. doi: 10.1111/j.1365-2958.2004.04767.x.Larkin A, Imperiali B. Biochemistry. 2009;48:5446–5455. doi: 10.1021/bi900186u.
- 20.For an comprehensive introduction to the loline family, see: Schardl CL, Grossman RB, Nagabhyru P, Faulkner JR, Mallik UP. Phytochemistry. 2007;68:980–996. doi: 10.1016/j.phytochem.2007.01.010.
- 21.For recent synthetic efforts, see: Cakmak M, Mayer P, Trauner D. Nat Chem. 2011;3:543–545. doi: 10.1038/nchem.1072.Hovey MT, Eklund EJ, Pike RD, Mainkar AA, Scheerer JR. Org Lett. 2011;13:1246–1249. doi: 10.1021/ol200155p.Blakemore PR, Kim SK, Schulze VK, White JD, Yokochi AFT. J Chem Soc, Perkin Trans 1. 2001;15:1831–1845.
- 22.(a) Tsuda M, Kasai Y, Komatsu K, Sone T, Tanaka M, Mikami Y, Kobayashi J. Org Lett. 2004;6:3087–3089. doi: 10.1021/ol048900y. [DOI] [PubMed] [Google Scholar]; (b) Mugishima T, Tsuda M, Kasai Y, Ishiyama H, Fukushi E, Kawabata J, Watanabe M, Akao K, Kobayashi J. J Org Chem. 2005;70:9430–9435. doi: 10.1021/jo051499o. [DOI] [PubMed] [Google Scholar]
- 23.For recent synthetic efforts towards the citrinadins, see: Guerrero CA, Sorensen EJ. Org Lett. 2011;13:5164–5167. doi: 10.1021/ol2020362.McIver AL, Deiters A. Org Lett. 2010;12:1288–1291. doi: 10.1021/ol100177u.Pettersson M, Knueppel D, Martin SF. Org Lett. 2007;9:4623–4626. doi: 10.1021/ol702132v.
- 24.Kushida N, Watanabe N, Okuda T, Yokoyama F, Gyobu Y, Yaguchi T. J Antibiot. 2007;60:667–673. doi: 10.1038/ja.2007.85. [DOI] [PubMed] [Google Scholar]
- 25.For excellent overviews of the biology, chemistry and synthesis of tetrahydroisoquinoline antitumor alkaloids, see: Scott JD, Williams RM. Chem Rev. 2002;102:1669–1730. doi: 10.1021/cr010212u.Siengalewicz P, Rinner U, Mulzer J. Chem Soc Rev. 2008;37:2676–2690. doi: 10.1039/b804167a.
- 26.(a) Whaley HA, Patterson EL, Dann M, Shay AJ, Porter JN. Antimicrob Agents Chemother. 1964;8:83–86. [PubMed] [Google Scholar]; (b) He H, Shen B, Carter GT. Tetrahedron Lett. 2000;41:2067–2071. [Google Scholar]
- 27.Ashley ER, Cruz EG, Stoltz BM. J Am Chem Soc. 2003;125:15000–15001. doi: 10.1021/ja039223q. [DOI] [PubMed] [Google Scholar]
- 28.Isolation: Peng J, Hu J-F, Kazi AB, Li Z, Avery M, Peraud O, Hill RT, Franzblau SG, Zhang F, Schinazi RF, Wirtz SS, Tharnish P, Kelly M, Wahyuono S, Hamann MT. J Am Chem Soc. 2003;125:13382–13386. doi: 10.1021/ja030087z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.For synthetic efforts towards the manadomanzamines, see: Allin SM, Duffy LJ, Towler JMR, Page PCB, Elsegood MRJ, Saha B. Tetrahedron. 2009;65:10230–10234.Allin SM, Duffy LJ, Page PCB, McKee V, McKenzie MJ. Tetrahedron Lett. 2007;48:4711–4714.
- 30.(a) Faulkner DJ. Nat Prod Rep. 2002;19:1. doi: 10.1039/b009029h. [DOI] [PubMed] [Google Scholar]; (b) Hoffmann H, Lindel T. Synthesis. 2003:1753. [Google Scholar]; (b) Weinreb SM. Nat Prod Rep. 2007;24:931. doi: 10.1039/b700206h. [DOI] [PubMed] [Google Scholar]; (c) Arndt HD, Riedrich M. Angew Chem Int Ed. 2008;47:4785. doi: 10.1002/anie.200801793. [DOI] [PubMed] [Google Scholar]
- 31.For synthetic efforts towards members of the eudistomin family, see: McNulty J, Still IWJ. Curr Org Chem. 2000;4:121–121.Yamashita T, Kawai N, Tokuyama H, Fukuyama T. J Am Chem Soc. 2005;127:15038–15039. doi: 10.1021/ja055832h.van Maarseveen JH, Scheeren HW, Declercq E, Balzarini J, Kruse CG. Bioorg Med Chem. 1997;5:955–970. doi: 10.1016/s0968-0896(97)00034-5.
- 32.Lake RJ, Brennan MM, Blunt JW, Munro MHG, Pannell LK. Tetrahedron Lett. 1988;29:2255–2256. [Google Scholar]
- 33.(a) Argoudelis AD, Jahnke HK, Fox JA. Antimicrob Agents Chemother. 1962:191–198. [Google Scholar]; (b) Bhuyan BK, Dietz A, Smith CG. Antimicrob Agents Chemother. 1962:184–190. [Google Scholar]
- 34.Hanessian S, Vakiti RR, Dorich S, Banerjee S, Lecomte F, Delvalle JR, Zhang J, Descheênes-Simard B. Angew Chem Int Ed. 2011;50:3497–3500. doi: 10.1002/anie.201008079. [DOI] [PubMed] [Google Scholar]
- 35.For a comprehensive review of the preparation of conformationally restricted diamines and their importance in drug design, see: Grygorenko OO, Radchenko DS, Volochnyuk DM, Tolmachev AA, Komarov IV. Chem Rev. 2011;111:5506–5568. doi: 10.1021/cr100352k.
- 36.(a) De Souza MVN, Vasconcelos TRA, De Almeida MV, Cardoso SH. Curr Med Chem. 2006;13:455–463. doi: 10.2174/092986706775527965. [DOI] [PubMed] [Google Scholar]; (b) Da Silva AD, De Almeida MV, De Souza MVN, Couri MRC. Curr Med Chem. 2003;10:21–39. doi: 10.2174/0929867033368637. [DOI] [PubMed] [Google Scholar]; (c) Renau TE, Sanchez JP, Gage JW, Dever JA, Shapiro MA, Gracheck SJ, Domagala JM. J Med Chem. 1996;39:729–735. doi: 10.1021/jm9507082. [DOI] [PubMed] [Google Scholar]
- 37.(a) Protopopova M, Hanrahan C, Nikonenko B, Samala R, Chen P, Gearhart J, Einck L, Nacy CA. J Antimicrob Chemother. 2005;56:968–974. doi: 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]; (b) Lee RE, Protopopova M, Crooks E, Slayden RA, Terrot M, Barry CE., III J Combin Chem. 2003;5:172–187. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 38.Magano J. Tetrahedron. 2011;67:7875–7899.Shibasaki M, Kanai M, Yamatsugu K. Isr J Chem. 2011;51:316–328.Jianzhi G, Wenfang X. Curr Med Chem. 2008;15:3145–3159. doi: 10.2174/092986708786848497.Shibasaki M, Kanai M. Eur J Org Chem. 2008:1839–1850.(e) Reference 39.
- 39.Magano J. Chem Rev. 2009;109:4398–4438. doi: 10.1021/cr800449m. [DOI] [PubMed] [Google Scholar]
- 40.Gastaminza P, Pitram SM, Dreux M, Krasnova LB, Whitten-Bauer C, Dong J, Chung J, Fokin VV, Sharpless KB, Chisari FV. J Virol. 2011;85:5513–5523. doi: 10.1128/JVI.02116-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]; (b) Davis TA, Johnston JN. Chem Sci. 2011;2:1076–1079. doi: 10.1039/C1SC00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Shih N-Y, Shue H-J, Reichard GA, Paliwal S, Blythin DJ, Piwinski JJ, Xiao D, Chen X. 0144200. PCT Int Appl WO. 2001; (b) Reichard GA, Stengone C, Paliwal S, Mergelsberg I, Majmundar S, Wang C, Tiberi R, McPhail AT, Piwinski JJ, Shih NY. Org Lett. 2003;5:4249–4251. doi: 10.1021/ol030104p. [DOI] [PubMed] [Google Scholar]
- 43.Geiger C, Zelenka C, Weigl M, Fröhlich R, Wibbeling B, Lehmkuhl K, Schepmann D, Grünert R, Bednarski PJ, Wünsch B. J Med Chem. 2007;50:6144–61. doi: 10.1021/jm070620b. [DOI] [PubMed] [Google Scholar]
- 44.For leading references to this approach, see: Schick A, Kolter T, Giannis A, Sandhoff K. Tetrahedron. 1996;52:2945–2956.Hakogi T, Taichi M, Katsumura S. Org Lett. 2003;5:2801–2804. doi: 10.1021/ol034771u.Harrak Y, Llebaria A, Delgado A. Eur J Org Chem. 2008;2008:4647–4654.
- 45.(a) Harrak Y, Barra CM, Bedia C, Delgado A, Castaño RA, Llebaria A. ChemMedChem. 2009;4:1608–1613. doi: 10.1002/cmdc.200900193. [DOI] [PubMed] [Google Scholar]; (b) Harrak Y, Barra CM, Delgado A, Castaño AR, Llebaria A. J Am Chem Soc. 2011;133:12079–12084. doi: 10.1021/ja202610x. [DOI] [PubMed] [Google Scholar]
- 46.Kim JW, Kim YW, Inagaki Y, Hwang YA, Mitsutake S, Ryu YW, Lee WK, Ha HJ, Park CS, Igarashi Y. Bioorg Med Chem. 2005;13:3475–3485. doi: 10.1016/j.bmc.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 47.Pyne NJ, Pyne S. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 48.(a) Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (b) Douthwaite RE. Coord Chem Rev. 2007;251:702–717. [Google Scholar]; (c) Hems WP, Groarke M, Zanotti-Gerosa A, Grasa GA. Acc Chem Res. 2007;40:1340–1347. doi: 10.1021/ar7000233. [DOI] [PubMed] [Google Scholar]; (d) Kizirian JC. Chem Rev. 2008;108:140–205. doi: 10.1021/cr040107v. [DOI] [PubMed] [Google Scholar]
- 49.(a) Larrow JF, Jacobsen EN. Top Organomet Chem. 2004;6:123–152. [Google Scholar]; (b) Cozzi P Giorgio. Chem Soc Rev. 2004;33:410–421. doi: 10.1039/b307853c. [DOI] [PubMed] [Google Scholar]; (c) Katsuki T. Chem Soc Rev. 2004;33:437–444. doi: 10.1039/b304133f. [DOI] [PubMed] [Google Scholar]; (d) Matsumoto K, Saito B, Katsuki T, Rivers EC. Chem Commun. 2007:3619–3627. doi: 10.1039/b701431g. [DOI] [PubMed] [Google Scholar]
- 50.(a) Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]; (b) Wong E, Giandomenico CM. Chem Rev. 1999;99:2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 51.(a) Liu S, Edwards DS. Chem Rev. 1999;99:2235–2268. doi: 10.1021/cr980436l. [DOI] [PubMed] [Google Scholar]; (b) Bartholomä MD, Louie AS, Valliant JF, Zubieta J. Chem Rev. 2010;110:2903–2920. doi: 10.1021/cr1000755. [DOI] [PubMed] [Google Scholar]
- 52.For reviews, see: Saito S, Yamamoto H. Acc Chem Res. 2004;37:570–579. doi: 10.1021/ar030064p.Notz W, Tanaka F, Barbas CF., III Acc Chem Res. 2004;37:580–591. doi: 10.1021/ar0300468.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016.Guillena G, Nájera C, Ramón DJ. Tetrahedron: Asymmetry. 2007;18:2249–2293.Melchiorre P, Marigo M, Carlone A, Bartoli G. Angew Chem Int Ed. 2008;47:6138–6171. doi: 10.1002/anie.200705523.Ting A, Goss JM, McDougal NT, Schaus SE. Top Curr Chem. 2009:145–200. doi: 10.1007/978-3-642-02815-1_23.Bertelsen S, Jørgensen KA. Chem Soc Rev. 2009;38:2178–2189. doi: 10.1039/b903816g.Wang M, Lin L, Shi J, Liu X, Kuang Y, Feng X. Chem Eur J. 2011;17:2365–2368. doi: 10.1002/chem.201002961.
- 53.For a selection of recent 1,2-diamine-based organocatalysts, see: Wang J, Qi C, Ge Z, Cheng T, Li R. Chem Commun. 2010;46:2124–2126. doi: 10.1039/b923925a.Basak AK, Shimada N, Bow WF, Vicic DA, Tius MA. J Am Chem Soc. 2010;132:8266–8267. doi: 10.1021/ja103028r.Moteki SA, Xu S, Arimitsu S, Maruoka K. J Am Chem Soc. 2010;132:17074–17076. doi: 10.1021/ja107897t.Fotaras S, Kokotos CG, Tsandi E, Kokotos G. Eur J Org Chem. 2011:1310–1317.Wang M, Lin L, Shi J, Liu X, Kuang Y, Feng X. Chem Eur J. 2011;17:2365–2368. doi: 10.1002/chem.201002961.
- 54.For reviews, see: Cox PJ, Simpkins NS. Tetrahedron: Asymmetry. 1991;2:1–26.Hodgson DM, Gibbs AR, Lee GP. Tetrahedron. 1996;52:14361–14384.Magnus A, Bertilsson SK, Andersson PG. Chem Soc Rev. 2002;31:223–229. doi: 10.1039/b104372m.O’Brien PJ. Chem Soc, Perkin Trans. 1998;1:1439.
- 55.(a) Savoia D, Gualandi A. Curr Org Syn. 2009;6:102–118. [Google Scholar]; (b) Savoia D, Gualandi A. Curr Org Syn. 2009;6:119–142. [Google Scholar]; (c) Alfonso I. Curr Org Syn. 2010;7:1–23. [Google Scholar]
- 56.Bennani YL, Hanessian S. Chem Rev. 1997;97:3161–3195. doi: 10.1021/cr9407577. [DOI] [PubMed] [Google Scholar]
- 57.González-Sabín J, Rebolledo F, Gotor V. Chem Soc Re. 2009;38:1916–1925. doi: 10.1039/b818679k. [DOI] [PubMed] [Google Scholar]
- 58.For a review of these early studies, see: Riebsomer JL. Chem Rev. 1945;36:157–233.
- 59.For reviews of the organic reactions of dinitrogen tetroxide, see: Larson HO. In: The Chemistry of the Nitro and Nitroso Groups, Part 1. Feuer H, editor. Interscience; New York, N.Y: 1969. pp. 316–324.Olah GA, Malhotra R, Narang SC. Nitration: Methods and Mechanisms. VCH; New York: 1989. pp. 243–269.Stepanov AV, Veselovsky VV. Russ Chem Rev. 2003;72:327–341.Shiri M, Zolfigol MA, Kruger H, Gerhardus, Tanbakouchian Z. Tetrahedron. 2010;66:9077–9106.
- 60.Wieland H, Garbsch P, Chavan JJ. Liebigs Ann Chem. 1928;461:29. [Google Scholar]
- 61.(a) Redmond TF, Wayland BB. J Phys Chem. 1968;72:1626–1629. [Google Scholar]; (b) Brunning J, Frost MJ, Smith IWM. Int J Chem Kinet. 1988;20:957–965. [Google Scholar]
- 62.Pinnick DA, Agnew SF, Swanson BI. J Phys Chem. 1992;96:7092–7096. [Google Scholar]
- 63.Addison CC. Chem Rev. 1980;80:21–39. [Google Scholar]
- 64.(a) Brand JCD, Stevens IDR. J Chem Soc. 1958:629–638. [Google Scholar]; (b) Stevens TEJ. Am Chem Soc. 1959;81:3593–3597. [Google Scholar]; (b) Shechter H, Gardikes JJ, Cantrell TS, Tiers GVD. J Am Chem Soc. 1967;89:3005–3014. [Google Scholar]
- 65.(a) Pryor WA, Lightsey JW, Church DF. J Am Chem Soc. 1982;104:6685–6692. [Google Scholar]; (b) Giamalva DH, Kenion GB, Church DF, Pryor WA. J Am Chem Soc. 1987;109:7059–7063. [Google Scholar]; (c) Bryant DK, Challis BC, Iley J. Chem Commun. 1989:1027–1028. [Google Scholar]; (d) Chatterjee J, Coombes RG, Barnes JR, Fildes MJJ. Chem Soc, Perkin Trans. 1995;2:1031–1032. [Google Scholar]; (e) Golding P, Powell JL, Ridd JH. J Chem Soc, Perkin Trans 2. 1996:813–819. [Google Scholar]
- 66.Powell JL, Ridd JH, Sandall JPB. Chem Commun. 1990:402–403. [Google Scholar]
- 67.Bonetti GA, Desavigny CB, Michalski C, Rosenthal R. J Org Chem. 1968;33:237–243. [Google Scholar]
- 68.Shechter H. Record Chem Progr. 1964;25:55–76. [Google Scholar]
- 69.Suzuki H, Mori T. J Org Chem. 1997;62:6498–6502. [Google Scholar]
- 70.Zhang W, Jacobsen EN. Tetrahedron Lett. 1991;32:1711–1714. [Google Scholar]
- 71.Kerrigan NJ, Müller-Bunz H, Gilheany DG. J Mol Catal A: Chem. 2005;227:163–172. [Google Scholar]
- 72.Bowyer WJ, Evans DH. J Org Chem. 1988;53:5234–5239. [Google Scholar]
- 73.Berestovitskaya VM, Speranskii EM, Perekalin VV. Russ J Gen Chem. 1977;13:1934–1944. [Google Scholar]
- 74.(a) Efremov DA, Berestovitskaya VM, Perekalin VV. Russ J Gen Chem. 1979;49:947. [Google Scholar]; (b) Berestovitskaya VM, Efremov DA, Berkova GA, Perekalin VV, Zakharov VI. Russ J Gen Chem. 1980;50:2680–2690. [Google Scholar]
- 75.(a) Trukhin EV, Berestovitskaya VM, Perekalin VV. Russ J Gen Chem. 1982;52:1167–1176. [Google Scholar]; (b) Trukhin EV, Berestovitskaya VM, Perekalin VV. Russ J Gen Chem. 1982;52:1022–1030. [Google Scholar]
- 76.Dore J-C, Viel C. Eur J Med Chem. 1974;9:673–680. [Google Scholar]
- 77.Haszeldine RN. J Chem Soc. 1953:2075–2081. [Google Scholar]
- 78.Brown JF. J Am Chem Soc. 1957;79:2480–2488. [Google Scholar]
- 79.(a) Gowenlock BG, Richter-Addo GB. Chem Rev. 2004;104:3315–3340. doi: 10.1021/cr030450k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartung J. Chem Rev. 2009;109:4500–4517. doi: 10.1021/cr900085j. [DOI] [PubMed] [Google Scholar]
- 80.(a) Phillips LV, Coyne DM. J Org Chem. 1964;29:1937–1942. [Google Scholar]; (b) Burkhard CA, Brown JF. J Org Chem. 1964;29:2235–2239. [Google Scholar]; (c) Rockenbauer A, Győr M, Tüdős F. Tetrahedron Lett. 1986;27:3425–3428. [Google Scholar]; (d) Park JSB, Walton JC. J Chem Soc Perkin Trans 2. 1997:2579–2583. [Google Scholar]; (e) Kelly DR, Jones S, Adigun JO, Koh KSV, Hibbs DE, Hursthouse MB, Jackson, Tetrahedron SK. 1997;53:17221–17234. [Google Scholar]
- 81.Chiu KW, Savage PD, Wilkinson G, Williams DJ. Polyhedron. 1985;4:1941–1945. [Google Scholar]
- 82.Tuaillon J, Perrot R. Helv Chim Acta. 1978;61:558–566. [Google Scholar]
- 83.(a) Park JD, Stefani AP, Crawford GH, Lacher JR. J Org Chem. 1961;26:3316–3319. [Google Scholar]; (b) Birchall JM, Bloom AI, Haszeldine RN, Willis CJ. J Chem Soc. 1962:3021–3032. [Google Scholar]
- 84.(a) Hata E, Yamada T, Mukaiyama T. Bull Chem Soc Jpn. 1995;68:3629–3636. [Google Scholar]; (b) Kelly DR, Jones S, Adigun JO, Koh KSV, Hibbs DE, Hursthouse MB, Jackson SK. Tetrahedron. 1997;53:17221–17234. [Google Scholar]
- 85.For a review of the reaction of N2O3 with alkenes, see: reference 79a.
- 86.Pfab J. Chem Commun. 1977:766–767. [Google Scholar]
- 87.(a) Chapman AC. J Chem Soc. 1895;67:54. [Google Scholar]; (b) Chapman AC. J Chem Soc. 1895;67:780. [Google Scholar]; (c) MacAlpine DK, Porte AL, Sim GA. J Chem Soc, Perkin Trans 1. 1981:999–1005. [Google Scholar]; (d) MacAlpine DK, Porte AL, Sim GA. J Chem Soc, Perkin Trans 1. 1981:2533–2538. [Google Scholar]; (e) MacAlpine DK, Porte AL, Sim GAJ. Chem Soc, Perkin Trans. 1982;1:1385–1388. [Google Scholar]
- 88.Bruckner V. J Prakt Chem. 1933;138:268–270. [Google Scholar]
- 89.Bruckner V. Liebigs Ann Chem. 1935;518:226–241. [Google Scholar]
- 90.Kurihara Y, Yamagishi K. Bull Chem Soc Jpn. 1965;38:1327–1330. [Google Scholar]
- 91.(a) Klamann D, Koser W, Weyerstahl P, Fligge M. Chem Ber. 1965;98:1831–1836. [Google Scholar]; (b) Govindachari TR, Pai BR. J Org Chem. 1953;18:1253–1262. [Google Scholar]
- 92.(a) Wieland H, Stenzel H. Liebigs Ann Chem. 1908;860:299. [Google Scholar]; (b) Rule A. J Chem Soc. 1908;93:1508. [Google Scholar]
- 93.Scheinbaum ML. J Org Chem. 1970;35:2785–2790. [Google Scholar]
- 94.Colette M, Perrot R. Helv Chim Acta. 1977;60:2089–2098. [Google Scholar]
- 95.(a) Jolibois H, Doucet A, Perrot R. Helv Chim Acta. 1975;58:1801–1804. [Google Scholar]; (b) Jolibois H, Doucet A, Perrot R. Helv Chim Acta. 1976;59:1352–1356. [Google Scholar]
- 96.Hauff J-P, Tuaillon J, Perrot R. Helv Chim Acta. 1978;61:1207–1212. [Google Scholar]
- 97.Demir AS, Findik H. Lett Org Chem. 2005;2:602–604. [Google Scholar]
- 98.Adekenov SM, Alebastrov OV, Raldugin VA, Bagryanskaya IY, Gatilov YV, Shakirov MM, Kulyyasov AT, Tolstikov GA. Chem Nat Compd. 2001;37:228–233. [Google Scholar]
- 99.(a) Alebastrov OV, Raldugin VA, Bagryanskaya IY, Gatilov YV, Shakirov MM, Kulyyasov AT, Adekenov SM. Chem Nat Compd. 2003;39:362–365. [Google Scholar]; (b) Mynzhasarovich AS, Bagdatovna RB, Tabylovich KA, Zareen S, Choudhary MI, Atta-ur-Rahman Heterocycles. 2003;60:1053–1063. [Google Scholar]
- 100.Harrison WA, Jones ERH, Meakins GD, Wilkinson PA. J Chem Soc. 1964:3210–3214. [Google Scholar]
- 101.(a) Chow YL, Lau MP, Cessna AJ, Yip RW. J Am Chem Soc. 1971;93:3808–3809. [Google Scholar]; (b) Wagner BD, Ruel G, Lusztyk J. J Am Chem Soc. 1996;118:13–19. [Google Scholar]
- 102.(a) Chow YL, Chen SC, Chang DWL. Can J Chem. 1971;49:3069–3071. [Google Scholar]; (b) Chow YL, Chen SC, Chang DWL. Can J Chem. 1970;48:157–162. [Google Scholar]
- 103.(a) Reference 102b Chow YL, Perry RA, Menon BC, Chen SC. Tetrahedron Lett. 1971;12:1545–1548.Pillay KS, Chen SC, Mojelsky T, Chow YL. Can J Chem. 1975;53:3014–3021.
- 104.Chow YL, Colon CJ, Chen SC. J Org Chem. 1967;32:2109–2115. [Google Scholar]
- 105.Bull WE, Seaton JA, Audrieth LF. J Am Chem Soc. 1958;80:2516–2518. [Google Scholar]
- 106.Noltes JG, van den Hurk JWG. J Organomet Chem. 1964;1:377–383. [Google Scholar]
- 107.(a) Michejda CJ, Campbell DH. Tetrahedron Lett. 1977;18:577–580. [Google Scholar]; (b) Michejda CJ, Campbell DH. J Am Chem Soc. 1974;96:929–930. [Google Scholar]; (c) Michejda CJ, Campbell DH. J Am Chem Soc. 1979;101:7687–7693. [Google Scholar]
- 108.Scheinbaum ML, Dines MB. Tetrahedron Lett. 1971;12:2205–2208. [Google Scholar]
- 109.Scheinbaum ML, Dines M. J Org Chem. 1971;36:3641–3642. [Google Scholar]
- 110.Bach RD, Holubka JW, Badger RC, Rajan SJ. J Am Chem Soc. 1979;101:4416–4417. [Google Scholar]
- 111.Bloom AJ, Fleischmann M, Mellor JM. J Chem Soc, Perkin Trans 1. 1984:2357–2362. [Google Scholar]
- 112.(a) Bloom AJ, Fleischmann M, Mellor JM. Tetrahedron Lett. 1984;25:4971–4974. [Google Scholar]; (b) Bloom AJ, Fleischmann M, Mellor JM. J Chem Soc, Perkin Trans 1. 1986:79–82. [Google Scholar]; (c) Bloom AJ, Fleischmann M, Mellor JM. Electrochim Acta. 1987;32:785–790. [Google Scholar]
- 113.Reddy MVR, Mehrotra B, Vankar YD. Tetrahedron Lett. 1995;36:4861–4864. [Google Scholar]
- 114.Kancharla PK, Reddy Y, Dharuman S, Vankar YD. J Org Chem. 2011;76:5832–5837. doi: 10.1021/jo200475h. [DOI] [PubMed] [Google Scholar]
- 115.For a discussion of the reaction of acetyl nitrate with alkenes, see: Borisenko AA, Nikulin AV, Wolfe S, Zefirov NS, Zyk NV. J Am Chem Soc. 1984;106:1074–1079.
- 116.Kochi JK. Organometallic Mechanisms and Catalysis: The Role of Reactive Intermediates in Organic Processes. Academic Press; New York: 1978. p. 18. [Google Scholar]
- 117.Workentin MS, Wagner BD, Lusztyk J, Wayner DDM. J Am Chem Soc. 1995;117:119–126. [Google Scholar]
- 118.For a recent review of the radical chemistry of the azide anion and organoazides, see: Jimeno C, Renaud P. Radical Chemistry with Azides. In: Braäse S, Banert K, editors. Organic Azides: Syntheses and Applications; John Wiley & Sons, Ltd; Chichester, U.K: 2010. pp. 239–267.
- 119.Minisci F, Galli R. Tetrahedron Lett. 1962;3:533–538. [Google Scholar]
- 120.For a review of free-radical redox addition to alkenes, including diazidonation, see: Minisci F. Acc Chem Res. 1975;8:165–171.
- 121.Potter GWH, Coleman MW, Monro AMJ. Heterocycl Chem. 1975;12:611–614. [Google Scholar]
- 122.(a) Minisci F, Galli R, Cecere, Gazz M. Chim Ital. 1964;94:67–90. [Google Scholar]; (b) Minisci F, Galli R. Procédé le Preparation de Diazides et Produits Obtenus. 1,350,360. FR Patent. 1964 Mar 11;
- 123.Galli R, Malatesta V. Org Prep Proced Int. 1971;3:231–233. [Google Scholar]
- 124.Fristad WE, Brandvold TA, Peterson JR, Thompson SR. J Org Chem. 1985;50:3647–3649. [Google Scholar]
- 125.For a recent application of Fristad’s methodology, see: Suzuki T, Shibata A, Morohashi N, Ohba Y. Chem Lett. 2005;34:1476–1477.
- 126.Snider BB, Lin H. Synth Commun. 1998;28:1913–1922. [Google Scholar]
- 127.Habala L, Dworak C, Nazarov AA, Hartinger CG, Abramkin SA, Arion VB, Lindner W, Galanski M, Keppler BK. Tetrahedron. 2008;64:137–146. [Google Scholar]
- 128.(a) Minakawa M, Guo HM, Tanaka F. J Org Chem. 2008;73:8669–8672. doi: 10.1021/jo8017389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guo HM, Minakawa M, Ueno L, Tanaka F. Bioorg Med Chem Lett. 2009;19:1210–1213. doi: 10.1016/j.bmcl.2008.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schäfer H. Angew Chem Int Ed. 1970;9:158–159. [Google Scholar]
- 130.For a review, see: Zbiral E. Synthesis. 1972:285–302.
- 131.(a) Zbiral E, Kischa K. Tetrahedron Lett. 1969;10:1167–1168. [Google Scholar]; (b) Zbiral E, Stütz A. Monatsh Chem. 1973;104:249–262. [Google Scholar]
- 132.Zbiral E, Stütz A. Tetrahedron. 1971;27:4953–4963. [Google Scholar]
- 133.Draper RW. J Chem Soc, Perkin Trans 1. 1983:2787–2791. [Google Scholar]
- 134.Kischa K, Zbiral E. Tetrahedron. 1970;26:1417–1426. doi: 10.1016/s0040-4020(01)92971-0. [DOI] [PubMed] [Google Scholar]
- 135.Zbiral E, Nestler G. Tetrahedron. 1971;27:2293–2302. [Google Scholar]
- 136.(a) Maxa E, Zbiral E, Schulz G, Haslinger E. Liebigs Ann Chem. 1975;1975:1705–1720. [Google Scholar]; (b) Emmer G, Zbiral E, Brill G, Musso H. Liebigs Ann Chem. 1979;1979:796–802. [Google Scholar]
- 137.Gómez Aranda V, Barluenga J, Aznar F. Synthesis. 1974:504–505. [Google Scholar]
- 138.Barluenga J, Alonsocires L, Asensio G. Synthesis. 1979:962–964. [Google Scholar]
- 139.Barluenga J, Perez-Prieto J, Asensio G, Garcia-Granda S, Salvado MA. Tetrahedron. 1992;48:3813–3826. [Google Scholar]
- 140.Barluenga J, Pérez-Prieto J, Bayón AM, Asensio G. Tetrahedron. 1984;40:1199–1204. [Google Scholar]
- 141.Barluenga J, Aznar F, de Mattos MCS, Kover WB, Garcia-Granda S, Pérez-Carreño E. J Org Chem. 1991;56:2930–2932. [Google Scholar]
- 142.For representative examples of this approach, see: Gandhi S, Bisai A, Bhanu Prasad BA, Singh VK. J Org Chem. 2007;72:2133–2142. doi: 10.1021/jo062564c.Wu J, Sun X, Xia HG. Tetrahedron Lett. 2006;47:1509–1512.Voronkov MV, Kanamarlapudi RC, Richardson P. Tetrahedron Lett. 2005;46:6907–6910.Concellón JM, Riego E, Suárez JR, García-Granda S, Díaz MR. Org Lett. 2004;6:4499–4501. doi: 10.1021/ol048176j.
- 143.Li G, Wei H-X, Kim SH, Carducci MD. Angew Chem Int Ed. 2001;40:4277–4280. doi: 10.1002/1521-3773(20011119)40:22<4277::AID-ANIE4277>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 144.For a recent review of methods for imidazoline synthesis, see: Liu H, Du DM. Adv Synth Catal. 2009;351:489–519.
- 145.Wei H-X, Siruta S, Li G. Tetrahedron Lett. 2002;43:3809–3812. [Google Scholar]
- 146.Wei H-X, Kim SH, Li G. J Org Chem. 2002;67:4777–4781. doi: 10.1021/jo0200769. [DOI] [PubMed] [Google Scholar]
- 147.Timmons C, McPherson LM, Chen D, Wei H-X, Li G. J Peptide Res. 2005;66:249–254. doi: 10.1111/j.1399-3011.2005.00294.x. [DOI] [PubMed] [Google Scholar]
- 148.Wu H, Ji X, Sun H, An G, Han J, Li G, Pan Y. Tetrahedron. 2010;66:4555–4559. [Google Scholar]
- 149.Han J, Li T, Pan Y, Kattuboina A, Li G. Chem Biol Drug Des. 2008;71:71–77. doi: 10.1111/j.1747-0285.2007.00604.x. [DOI] [PubMed] [Google Scholar]
- 150.For an overview of this mechanism and the aminohalogenation reaction, see: Li G, Kotti SRSS, Timmons C. Eur J Org Chem. 2007:2745–2758.
- 151.Pei W, Wei H-X, Chen D, Headley AD, Li G. J Org Chem. 2003;68:8404–8408. doi: 10.1021/jo030193j. [DOI] [PubMed] [Google Scholar]
- 152.Chen D, Timmons C, Wei H-X. J Org Chem. 2003;68:5742–5745. doi: 10.1021/jo030098a. [DOI] [PubMed] [Google Scholar]
- 153.Timmons C, Chen D, Xu X, Li G. Eur J Org Chem. 2003:3850–3854. [Google Scholar]
- 154.Timmons C, Chen D, Barney CE, Kirtane S, Li G. Tetrahedron. 2004;60:12095–12099. [Google Scholar]
- 155.Pei W, Timmons C, Xu X, Wei H-X, Li G. Org Biomol Chem. 2003;1:2919–2921. doi: 10.1039/b305149h. [DOI] [PubMed] [Google Scholar]
- 156.Booker-Milburn KI, Guly DJ, Cox B, Procopiou PA. Org Lett. 2003;5:3313–3315. doi: 10.1021/ol035374m. [DOI] [PubMed] [Google Scholar]
- 157.(a) Kumar V, Ramesh NG. Chem Commun. 2006:4952–4954. doi: 10.1039/b612151a. [DOI] [PubMed] [Google Scholar]; (b) Kumar V, Ramesh NG. Org Biomol Chem. 2007;5:3847–3858. doi: 10.1039/b712841j. [DOI] [PubMed] [Google Scholar]
- 158.For use of this methodology in the preparation of iminocyclitols, see: Ganesan M, Madhukarrao RV, Ramesh NG. Org Biomol Chem. 2010;8:1527–1530. doi: 10.1039/b926123k.
- 159.(a) Ando T, Kano D, Minakata S, Ryu I, Komatsu M. Tetrahedron. 1998;54:13485–13494. [Google Scholar]; (b) Jeong JU, Tao B, Sagasser I, Henniges H, Sharpless KB. J Am Chem Soc. 1998;120:6844–6845. [Google Scholar]
- 160.Hassner AA. cc Chem Res. 1971;4:9–16. [Google Scholar]
- 161.(a) Hassner A, Levy LA. J Am Chem Soc. 1965;87:4203–4204. [Google Scholar]; (b) Fowler FW, Hassner A, Levy LA. J Am Chem Soc. 1967;89:2077–2082. [Google Scholar]
- 162.Sasaki T, Kanematsu K, Yukimoto Y. J Org Chem. 1972;37:890–894. [Google Scholar]
- 163.For a recent method for the formation of IN3 from sodium azide and iodine, see: Terent’ev AO, Krylov IB, Kokorekin VA, Nikishin GI. Synth Commun. 2008;38:3797–3809.
- 164.For diamination through sequential azidoiodination and displacement of the iodide with azide, see: Pfaendler HR, Klingl A. Helv Chim Acta. 2005;88:1486–1490.
- 165.Hantzsch A. Ber Dtsch Chem Ges. 1900;33:522–527. [Google Scholar]
- 166.L’Abbé G, Hassner A. J Org Chem. 1971;36:258–260. [Google Scholar]
- 167.(a) Tamura Y, Kwon S, Tabusa F, Ikeda M. Tetrahedron Lett. 1975;16:3291–3294. [Google Scholar]; (b) Kwon S, Okada T, Ikeda M, Tamura Y. Heterocycles. 1977;6:33–36. [Google Scholar]; (c) Tamura Y, Chun MW, Kwon S, Bayomi SM, Okada T, Ikeda M. Chem Pharm Bull. 1978;26:3515–3520. [Google Scholar]
- 168.(a) Müller R, Gust R, Schönenberger H, Klement U. Chem Ber. 1991;124:2381–2389. [Google Scholar]; (b) Gust R, Bernhardt G, Spruss T, Krauser R, Koch M, Schöenenberger H, Bauer KH, Schertl S, Lu Z. Arch Pharm. 1995;328:645–653. doi: 10.1002/ardp.19953280904. [DOI] [PubMed] [Google Scholar]
- 169.Hassner A, Keogh J. Tetrahedron Lett. 1975;16:1575–1578. [Google Scholar]
- 170.Kirschning A, Monenschein H, Schmeck C. Angew Chem Int Ed. 1999;38:2594–2596. doi: 10.1002/(sici)1521-3773(19990903)38:17<2594::aid-anie2594>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 171.Kirschning A, Hashem MA, Monenschein H, Rose L, Schöning K-U. J Org Chem. 1999;64:6522–6526. [Google Scholar]
- 172.Chung R, Yu E, Incarvito CD, Austin DJ. Org Lett. 2004;6:3881–3884. doi: 10.1021/ol0490532. [DOI] [PubMed] [Google Scholar]
- 173.For a review of alkene co-halogenation, see: Rodriguez J, Dulcére JP. Synthesis. 1993:1177–1205.
- 174.This is also not the case for the intramolecular variant of this transformation. For selected examples, see: Knight DW, Redfern AL, Gilmore J. J Chem Soc, Perkin Trans 1. 2001:2874–2883.Kim JH, Curtis-Long MJ, Seo WD, Ryu YB, Yang MS, Park KH. J Org Chem. 2005;70:4082–4087. doi: 10.1021/jo050079w.Davis FA, Song M, Augustine A. J Org Chem. 2006;71:2779–2786. doi: 10.1021/jo052566h.Minakata S, Morino Y, Oderaotoshi Y, Komatsu M. Org Lett. 2006;8:3335–3337. doi: 10.1021/ol061182q.Diaba F, Ricou E, Bonjoch J. Org Lett. 2007;9:2633–2636. doi: 10.1021/ol070770g.
- 175.(a) Lavilla R, Kumar R, Coll O, Masdeu C, Bosch J. Chem Commun. 1998:2715–2716. [Google Scholar]; (b) Lavilla R, Kumar R, Coll O, Masdeu C, Spada A, Bosch J, Espinosa E, Molins E. Chem Eur J. 2000;6:1763–1772. doi: 10.1002/(sici)1521-3765(20000515)6:10<1763::aid-chem1763>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 176.Abou-Jneid R, Ghoulami S, Martin MT, Dau ET, Travert N, Al-Mourabit A. Org Lett. 2004;6:3933–3936. doi: 10.1021/ol048529e. [DOI] [PubMed] [Google Scholar]
- 177.Salvatori MD, Abou-Jneid R, Ghoulami S, Martin MT, Zaparucha A, Al-Mourabit A. J Org Chem. 2005;70:8208–8211. doi: 10.1021/jo050862o. [DOI] [PubMed] [Google Scholar]
- 178.Picon S, Zaparucha A, Al-Mourabit A. Tetrahedron Lett. 2009;50:6826–6829. [Google Scholar]
- 179.Hewlett NM, Tepe JJ. Org Lett. 2011;13:4550–4553. doi: 10.1021/ol201741r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.(a) Picon S, Tran HD, Martin MT, Retailleau P, Zaparucha A, Al-Mourabit A. Org Lett. 2009;11:2523–2526. doi: 10.1021/ol900745c. [DOI] [PubMed] [Google Scholar]; (b) Schroif-Gregoire C, Travert N, Zaparucha A, Al-Mourabit A. Org Lett. 2006;8:2961–2964. doi: 10.1021/ol0608451. [DOI] [PubMed] [Google Scholar]
- 181.(a) Zöllinger M, Mayer P, Lindel T. J Org Chem. 2006;71:9431–9439. doi: 10.1021/jo061813u. [DOI] [PubMed] [Google Scholar]; (b) Zöllinger M, Mayer P, Lindel T. Synlett. 2007:2756–2758. [Google Scholar]
- 182.For reviews, see: Cardillo G, Orena M. Tetrahedron. 1990;46:3321–3408.Robin S, Rousseau G. Tetrahedron. 1998;54:13681–13736.
- 183.Gataullin RR, Minnigulov FF, Fatykhov AA, Spirikhin LV, Abdrakhmanov IB. Russ J Org Chem. 2001;37:1289–1296.See also: Shen M, Li C. J Org Chem. 2004;69:7906–7909. doi: 10.1021/jo0488006.
- 184.Muñiz K, Hövelmann CH, Campos-Gómez E, Barluenga J, González JM, Streuff J, Nieger M. Chem Asian J. 2008;3:776–788. doi: 10.1002/asia.200700373. [DOI] [PubMed] [Google Scholar]
- 185.Li H, Widenhoefer RA. Tetrahedron. 2010;66:4827–4831. doi: 10.1016/j.tet.2010.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cochran BM, Michael FE. Org Lett. 2008;10:5039–5042. doi: 10.1021/ol8022165. [DOI] [PubMed] [Google Scholar]
- 187.For a review of iodine-mediated alkene diazidonation, see: Koser G. Top Curr Chem. 2003;224:137–172.
- 188.Ehrenfreund J, Zbiral E. Tetrahedron. 1972;28:1697. [Google Scholar]
- 189.(a) Ref. 188. Ehrenfreund J, Zbiral E. Liebigs Ann Chem. 1973;2:290–300.
- 190.Moriarty RM, Khosrowshahi JS. Tetrahedron Lett. 1986;27:2809–2812. [Google Scholar]
- 191.Moriarty RM, Khosrowshahi JS. Synth Commun. 1987;17:89–94. [Google Scholar]
- 192.Arimoto M, Yamaguchi H, Fujita E, Nagao Y, Ochiai M. Chem Pharm Bull. 1989;37:3221–3224. [Google Scholar]
- 193.(a) Magnus P, Lacour J. J Am Chem Soc. 1992;114:767–769. [Google Scholar]; (b) Magnus P, Evans PA, Lacour J. Tetrahedron Lett. 1992;33:2933–2936. [Google Scholar]
- 194.Magnus P, Roe MB, Hulme C. Chem Commun. 1995:263–265. [Google Scholar]
- 195.Magnus P, Lacour J, Evans PA, Roe MB, Hulme C. J Am Chem Soc. 1996;118:3406–3418. [Google Scholar]
- 196.Magnus P, Roe MB. Tetrahedron Lett. 1996;37:303–306. [Google Scholar]
- 197.Roben C, Souto JA, Gonázlez Y, Lishchynskyi A, Muñiz K. Angew Chem Int Ed. 2011;50:9478–9482. doi: 10.1002/anie.201103077. [DOI] [PubMed] [Google Scholar]
- 198.Uyanik M, Yasui T, Ishihara K. Angew Chem Int Ed. 2010;49:2175–2177. doi: 10.1002/anie.200907352. [DOI] [PubMed] [Google Scholar]
- 199.Parsons AF, Pettifer RM. Tetrahedron Lett. 1996;37:1667–1670. [Google Scholar]
- 200.Raeymaekers AHM, Roevens LFC, Janssen PAJ. Tetrahedron Lett. 1967;8:1467–1470. doi: 10.1016/s0040-4039(00)90983-3. [DOI] [PubMed] [Google Scholar]
- 201.Scheme adapted from ref. 207b
- 202.Frühauf H-W. Coord Chem Rev. 2002;230:79–96. [Google Scholar]
- 203.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 204.(a) Brunner H, Loskot S. Angew Chem Int Ed. 1971;10:515–516. [Google Scholar]; (b) Brunner H, Loskot S. J Organomet Chem. 1973;61:401–414. [Google Scholar]
- 205.Brunner H. J Organomet Chem. 1968;12:517–522. [Google Scholar]
- 206.Becker P, Bergman NRG. J Am Chem Soc. 1983;105:2985–2995. [Google Scholar]
- 207.(a) Becker PN, White MA, Bergman RG. J Am Chem Soc. 1980;102:5676–5677. [Google Scholar]; (b) Becker PN, Bergman RG. Organometallics. 1983;2:787–796. [Google Scholar]
- 208.(a) Schomaker JM, Boyd WC, Stewart IC, Toste FD, Bergman RG. J Am Chem Soc. 2008;130:3777–3779. doi: 10.1021/ja800738d. [DOI] [PubMed] [Google Scholar]; (b) Zhao C, Toste FD, Bergman RG. J Am Chem Soc. 2011;133:10787–10789. doi: 10.1021/ja204564z. [DOI] [PubMed] [Google Scholar]
- 209.Boyd WC, Crimmin MR, Rosebrugh LE, Schomaker JM, Bergman RG, Toste FD. J Am Chem Soc. 2010;132:16365–16367. doi: 10.1021/ja107968c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schomaker JM, Toste FD, Bergman RG. Org Lett. 2009;11:3698–3700. doi: 10.1021/ol901542n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Crimmin MR, Bergman R, Toste GFD. Angew Chem Int Ed. 2011;50:4484–4487. doi: 10.1002/anie.201100816. [DOI] [PubMed] [Google Scholar]
- 212.Milas NA, Iliopulos MI. J Am Chem Soc. 1959;81:6089–6089. [Google Scholar]
- 213.For an excellent overview of the preparation, structure and reactivity of imido osmium complexes, see: Muñiz K. Chem Soc Rev. 2004;33:166–174. doi: 10.1039/b307102m.
- 214.Nugent W, Harlow AR, McKinney LRJ. J Am Chem Soc. 1979;101:7265–7268. [Google Scholar]
- 215.Chong AO, Oshima K, Sharpless KB. J Am Chem Soc. 1977;99:3420–3426. [Google Scholar]
- 216.For an illustration of this chemical stability, see: Muñiz K. Tetrahedron Lett. 2003;44:3547–3549.
- 217.Muñiz K. Eur J Org Chem. 2004;2004:2243–2252. [Google Scholar]
- 218.(a) Schofield MH, Kee TP, Anhaus JT, Schrock RR, Johnson KH, Davis WM. Inorg Chem. 1991;30:3595–3604. [Google Scholar]; (b) Anhaus JT, Kee TP, Schofield MH, Schrock RR. J Am Chem Soc. 1990;112:1642–1643. [Google Scholar]
- 219.For a detailed account of these efforts, see: Muñiz K. New J Chem. 2005;29:1371–1385.
- 220.For theoretical studies, see: Deubel DV, Muñiz K. Chem Eur J. 2004;10:2475–2486. doi: 10.1002/chem.200305467.
- 221.(a) Muñiz K, Nieger M. Synlett. 2003:211–214. [Google Scholar]; (b) Muñiz K, Iesato A, Nieger M. Chem Eur J. 2003;9:5581–5596. doi: 10.1002/chem.200305011. [DOI] [PubMed] [Google Scholar]
- 222.(a) Muñiz K, Nieger M. Chem Commun. 2005:2729–2731. doi: 10.1039/b502150b. [DOI] [PubMed] [Google Scholar]; (b) Almodovar I, Hövelmann CH, Streuff J, Nieger M, Muñiz K. Eur J Org Chem. 2006:704–712. [Google Scholar]
- 223.Muñiz K, Nieger M, Mansikkamäki H. Angew Chem Int Ed. 2003;4(2):5958–5961. doi: 10.1002/anie.200352244. [DOI] [PubMed] [Google Scholar]
- 224.For an informative introduction to the process of alkene aminopalladation, see: Minatti A, Muñiz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j.See also McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y.
- 225.(a) Bäckvall JE. Tetrahedron Lett. 1978;19:163–166. [Google Scholar]; (b) Bäckvall JE. Acc Chem Res. 1983;16:335–342. [Google Scholar]
- 226.Akermark B, Bäckvall J-E, Löwenborg A, Zetterberg K. J Organomet Chem. 1979;166:C33–C36. [Google Scholar]
- 227.Bar GLJ, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2005;127:7308–7309. doi: 10.1021/ja051181d. [DOI] [PubMed] [Google Scholar]
- 228.(a) Streuff J, Hövelmann CH, Nieger M, Muñiz K. J Am Chem Soc. 2005;127:14586–14587. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (b) Muñiz K, Hövelmann CH, Streuff J. J Am Chem Soc. 2008;130:763–773. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]
- 229.For recent reviews of high-oxidation-state-palladium catalysis as it pertains to alkene diamination, see: Muñiz K, Hövelmann CH, Streuff J, Campos-Gómez E. Pure Appl Chem. 2008;80:1089–1096.Muñiz K. Angew Chem Int Ed. 2009;48:9412–9423. doi: 10.1002/anie.200903671.Sehnal P, Taylor RJ, Fairlamb IJ. Chem Rev. 2010;110:824–889. doi: 10.1021/cr9003242.
- 230.DFT calculations conducted by Lin and co-workers suggest, contrary to experimental evidence, that diamination in this case may proceed through a sequence of anti-aminopalladation and direct reductive elimination from the Pd(IV) intermediate: Yu HZ, Fu Y, Guo QX, Lin ZY. Organometallics. 2009;28:4507–451.
- 231.Muñiz K. J Am Chem Soc. 2007;129:14542–14543. doi: 10.1021/ja075655f. [DOI] [PubMed] [Google Scholar]
- 232.Muñiz K, Streuff J, Chávez P, Hövelmann CH. Chem Asian J. 2008;3:1248–1255. doi: 10.1002/asia.200800148. [DOI] [PubMed] [Google Scholar]
- 233.Hövelmann CH, Streuff J, Brelot L, Muñiz K. Chem Commun. 2008:2334–2336. doi: 10.1039/b719479j. [DOI] [PubMed] [Google Scholar]
- 234.Iglesias A, Pérez EG, Muñiz K. Angew Chem Int Ed. 2010;49:8109–8111. doi: 10.1002/anie.201003653. [DOI] [PubMed] [Google Scholar]
- 235.Dastrup DM, VanBrunt MP, Weinreb SM. J Org Chem. 2003;68:4112–4115. doi: 10.1021/jo020754r. [DOI] [PubMed] [Google Scholar]
- 236.Muñiz K, Kirsch J, Chávez P. Adv Synth Catal. 2011;353:689–694. [Google Scholar]
- 237.(a) Sibbald PA, Michael FE. Org Lett. 2009;11:1147–1149. doi: 10.1021/ol9000087. [DOI] [PubMed] [Google Scholar]; (b) Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]
- 238.Muñiz K, Streuff J, Hövelmann C, Núñez A. Angew Chem Int Ed. 2007;46:7125–7127. doi: 10.1002/anie.200702160. [DOI] [PubMed] [Google Scholar]
- 239.Muñiz co-workers have most recently reported the use of palladium catalysis for the intramolecular diamination of acrylic esters using sulfamates as the nitrogen source: Chávez P, Kirsch J, Streuff J, Muñiz K. J Org Chem. 2012;77:1922–1930. doi: 10.1021/jo202507d.
- 240.For an account of these efforts, see: Reference 3f.
- 241.Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250–11251. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]
- 242.Kochi JK. Acc Chem Res. 1974;7:351–360. [Google Scholar]
- 243.Zabawa TP, Chemler SR. Org Lett. 2007;9:2035–2038. doi: 10.1021/ol0706713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sequeira FC, Turnpenny BW, Chemler SR. Angew Chem Int Ed. 2010;49:6365–6368. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.For reviews of gold-catalyzed alkene hydroamination, see: Widenhoefer RA, Han X. Eur J Org Chem. 2006;2006:4555–4563.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788.Krause N, Winter C. Chem Rev. 2011;111:1994–2009. doi: 10.1021/cr1004088.Alcaide B, Almendros P. Adv Synth Catal. 2011;353:2561–2576.
- 246.For an introduction to this topic, see: Hopkinson MN, Gee AD, Gouverneur V. Chem Eur J. 2011;17:8248–8262. doi: 10.1002/chem.201100736.Tkatchouk E, Mankad NP, Benitez D, Goddard WA, III, Toste FD. J Am Chem Soc. 2011;133:14293–14300. doi: 10.1021/ja2012627.
- 247.Iglesias A, Muñiz K. Chem Eur J. 2009;15:10563–10569. doi: 10.1002/chem.200901199. [DOI] [PubMed] [Google Scholar]
- 248.De Haro T, Nevado C. Angew Chem Int Ed. 2011;50:906–910. doi: 10.1002/anie.201005763. [DOI] [PubMed] [Google Scholar]
- 249.For recent review of this concept, see: Engle KM, Mei T-S, Wang X, Yu J-Q. Angew Chem Int Ed. 2011;50:1478–1491. doi: 10.1002/anie.201005142.
- 250.(a) Wardrop DJ, Bowen EG, Forslund RE, Sussman AD, Weerasekera SL. J Am Chem Soc. 2009;132:1188–1189. doi: 10.1021/ja9069997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bowen EG, Wardrop DJ. Org Lett. 2010;12:5330–5333. doi: 10.1021/ol102371x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.For a recent example of a gold-catalyzed Ritter reaction albeit one not involving aziridinium ions, see: Ibrahim N, Hashmi ASK, Rominger F. Adv Synth Catal. 2011;353:461–468.
- 252.Li H, Widenhoefer RA. Org Lett. 2009;11:2671–2674. doi: 10.1021/ol900730w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Donohoe TJ, Callens CKA, Flores A, Lacy AR, Rathi AH. Chem Eur J. 2011;17:58–76. doi: 10.1002/chem.201002323. [DOI] [PubMed] [Google Scholar]
- 254.Michaelis DJ, Shaffer CJ, Yoon TP. J Am Chem Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. [DOI] [PubMed] [Google Scholar]
- 255.Despite its exotic appearance, compound 402 is readily available through the oxidation of N,N′-di-tert-butylurea: Du H, Zhao B, Shi Y. Org Synth. 2009;86:8392. doi: 10.1021/jo901685c.
- 256.Du H, Zhao B, Shi Y. J Am Chem Soc. 2007;129:762–763. doi: 10.1021/ja0680562. [DOI] [PubMed] [Google Scholar]
- 257.Du H, Yuan W, Zhao BG, Shi YA. J Am Chem Soc. 2007;129:11688–11689. doi: 10.1021/ja074698t. [DOI] [PubMed] [Google Scholar]
- 258.(a) Xu L, Shi YA. J Org Chem. 2008;73:749–751. doi: 10.1021/jo702167u. [DOI] [PubMed] [Google Scholar]; (b) Xu L, Du H, Shi Y. J Org Chem. 2007;72:7038–7041. doi: 10.1021/jo0709394. [DOI] [PubMed] [Google Scholar]
- 259.Ramirez TA, Zhao B, Shi Y. Tetrahedron Lett. 2010;51:1822–1825. doi: 10.1016/j.tetlet.2010.01.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Du H, Zhao BG, Shi YA. J Am Chem Soc. 2008;130:8590–8591. doi: 10.1021/ja8027394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Fu R, Zhao BG, Shi YA. J Org Chem. 2009;74:7577–7580. doi: 10.1021/jo9015584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Timberlake JW, Alender J, Garner AW, Hodges ML, Özmeral C, Szilagyi S, Jacobus JO. J Org Chem. 1981;46:2082–2089. [Google Scholar]
- 263.Wang B, Du H, Shi Y. Angew Chem Int Ed. 2008;47:8224–8227. doi: 10.1002/anie.200803184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yuan W, Du H, Zhao B, Shi Y. Org Lett. 2007;9:2589–2591. doi: 10.1021/ol071105a. [DOI] [PubMed] [Google Scholar]
- 265.Shimizu H, Nagasaki I, Matsumura K, Sayo N, Saito T. Acc Chem Res. 2007;40:1385–1393. doi: 10.1021/ar700101x. [DOI] [PubMed] [Google Scholar]
- 266.Du H, Zhao B, Yuan W, Shi Y. Org Lett. 2008;10:4231–4234. doi: 10.1021/ol801605w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhao BG, Du HF, Shi YA. J Org Chem. 2009;74:8392–8395. doi: 10.1021/jo901685c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zhao BG, Peng XG, Cui SL, Shi YA. J Am Chem Soc. 2010;132:11009–11011. doi: 10.1021/ja103838d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Wen Y, Zhao BG, Shi YA. Org Lett. 2009;11:2365–2368. doi: 10.1021/ol900808z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zhao B, Yuan W, Du H, Shi Y. Org Lett. 2007;9:4943–4945. doi: 10.1021/ol702061s. [DOI] [PubMed] [Google Scholar]
- 271.Cornwall R, Zhao GB, Shi Y. Org Lett. 2011;13:434–437. doi: 10.1021/ol102767j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.(a) Hoffmann H, Lindel T. Synthesis. 2003:1753–1783. [Google Scholar]; (b) Al-Mourabit A, Zancanella MA, Tilvi S, Romo D. Nat Prod Rep. 2011;28:1229–1260. doi: 10.1039/c0np00013b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhao BG, Du H, Shi YA. Org Lett. 2008;10:1087–1090. doi: 10.1021/ol702974s. [DOI] [PubMed] [Google Scholar]
- 274.Sharpless KB, Singer SP. J Org Chem. 1976;41:2504–2506. [Google Scholar]
- 275.Bruncko M, Khuong TAV, Sharpless KB. Angew Chem Int Ed. 1996;35:454–456. [Google Scholar]
- 276.Fukuyama T, Jow CK, Cheung M. Tetrahedron Lett. 1995;36:6373–6374. [Google Scholar]
- 277.Natsugari H, Whittle RR, Weinreb SM. J Am Chem Soc. 1984;106:7867–7872. [Google Scholar]
- 278.Jiang H, Nielsen JB, Nielsen M, Jørgensen KA. Chem Eur J. 2007;13:9068–9075. doi: 10.1002/chem.200700696. [DOI] [PubMed] [Google Scholar]
- 279.Simmons B, Walji AM, MacMillan DWC. Angew Chem Int Ed. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]