

The Hsp70 system recruits ClpB/Hsp104 to the surface of stress-induced protein aggregates and prion fibrils.
Abstract
Hsp100 and Hsp70 chaperones in bacteria, yeast, and plants cooperate to reactivate aggregated proteins. Disaggregation relies on Hsp70 function and on ATP-dependent threading of aggregated polypeptides through the pore of the Hsp100 AAA+ hexamer. In yeast, both chaperones also promote propagation of prions by fibril fragmentation, but their functional interplay is controversial. Here, we demonstrate that Hsp70 chaperones were essential for species-specific targeting of their Hsp100 partner chaperones ClpB and Hsp104, respectively, to heat-induced protein aggregates in vivo. Hsp70 inactivation in yeast also abrogated Hsp104 targeting to almost all prions tested and reduced fibril mobility, which indicates that fibril fragmentation by Hsp104 requires Hsp70. The Sup35 prion was unique in allowing Hsp70-independent association of Hsp104 via its N-terminal domain, which, however, was nonproductive. Hsp104 overproduction even outcompeted Hsp70 for Sup35 prion binding, which explains why this condition prevented Sup35 fragmentation and caused prion curing. Our findings indicate a conserved mechanism of Hsp70–Hsp100 cooperation at the surface of protein aggregates and prion fibrils.
Introduction
Stress conditions perturb cellular protein homeostasis, inducing misfolding of proteins into amorphous aggregates. In bacteria, yeast, and plant cells, reactivation of aggregated proteins is mediated by the hexameric AAA+ chaperone Hsp100, termed ClpB in bacteria and Hsp104 in yeast, which is essential for thermotolerance development (Sanchez and Lindquist, 1990; Squires et al., 1991). The protein disaggregation activity of ClpB/Hsp104 requires cooperation with a cognate Hsp70 chaperone system, i.e., the bacterial DnaK system (consisting of DnaK and its cochaperones DnaJ and GrpE) or the yeast Ssa system (consisting of Ssa and its cochaperones Ydj1 or Sis1 and Sse or Fes1). The Hsp70 system has an essential function at the initial stage of the disaggregation process, where it acts to permit the subsequent ATP-driven substrate-threading activity of ClpB/Hsp104. This results in extraction of single unfolded polypeptides from aggregates through the central pore of the AAA+ hexamer (Lum et al., 2004; Schlieker et al., 2004; Weibezahn et al., 2004; Zietkiewicz et al., 2004; Tessarz et al., 2008). Why the Hsp70 system is required at the initial phase of disaggregation is still unclear and a matter of controversy. On the one hand, the binding of DnaK and DnaJ to aggregates allows for the targeting of misfolded polypeptides to the substrate-processing central pore of ClpB (Haslberger et al., 2007), implying that ClpB might require DnaK/DnaJ for aggregate binding. Such a dependency was demonstrated in vitro for an aggregated model protein (Acebrón et al., 2009). On the other hand, Hsp70-independent association of ClpB with protein aggregates in vitro was also observed (Schlieker et al., 2004; Barnett et al., 2005). Further, the presence of DnaK/DnaJ even reduces the binding of ClpB to aggregated model substrates in vitro, implying competition instead of cooperation between the two chaperone systems during substrate interaction (Nagy et al., 2010). Finally, ClpB and Hsp104 also exert an Hsp70-independent disaggregation activity on a subset of aggregated substrates in the presence of ATP/ATPγS mixtures (Doyle et al., 2007a,b); the physiological relevance of such activity is, however, still unclear. Together, these disparate results do not provide a unifying concept concerning the role for the Hsp70 system in the ClpB/Hsp104-dependent disaggregation of stress-induced protein aggregates.
In addition to its role in solubilizing stress-induced protein aggregates, Saccharomyces cerevisiae Hsp104 is also required for the inheritance of several protein-based genetic elements (prions), including [PSI+] and [PIN+]/[RNQ+], that result from conversion of soluble Sup35 and Rnq1 proteins, respectively, into ordered amyloid-like aggregates (Chernoff et al., 1995; Sondheimer and Lindquist, 2000). Prion propagation involves several steps, starting from fibril growth via incorporation of soluble monomers, followed by the generation of seed templates (propagons) by fibril severing and their delivery to daughter cells during cell division (Shorter and Lindquist, 2005; Pezza and Serio, 2007). Hsp104 has been proposed to act primarily during fibril fragmentation in vivo (Satpute-Krishnan et al., 2007). Ssa1 also plays a crucial role in yeast prion propagation, although interactions with Hsp104 in this process are not well defined and, again, are controversial (Masison et al., 2009; Romanova and Chernoff, 2009; Desantis and Shorter, 2012). An Ssa1-independent severing activity of Hsp104 toward Sup35 and Ure2 prion fibers was demonstrated in a purified system (Shorter and Lindquist, 2004, 2006). In other studies, however, Hsp104 on its own either does not exhibit such activity (Krzewska and Melki, 2006) or requires additional, unknown factors for Sup35 fragmentation (Inoue et al., 2004). In yeast, the Ssa1 co-chaperone Sis1 is required for the propagation of various prions including [RNQ+] and [PSI+], and depletion of Sis1 causes a transient increase in fibril size, which suggests a role for the Ssa1/Sis1 system in fibril severing (Sondheimer et al., 2001; Aron et al., 2007; Higurashi et al., 2008). Accordingly, the processing of Sup35 prions by Hsp104-mediated threading in vivo requires Sis1 (Tipton et al., 2008), which suggests that Ssa1/Sis1 is acting at some stage of Hsp104-mediated fiber severing, similar to its role in the solubilization of stress-induced aggregates. These studies, however, rely on Sis1 depletion over several cell generations, and the possibility that the observed effects on Sup35 prion processing result from indirect effects on cell physiology (Masison et al., 2009) cannot be excluded.
Current knowledge has thus not yet generated a generally accepted definition of the roles of the Hsp70 system in ClpB/Hsp104-dependent protein disaggregation and prion fibril fragmentation, or resolved whether these roles are identical or different activities. This led us to perform a global analysis of the Hsp70/J protein and ClpB/Hsp104 chaperone cooperation in Escherichia coli and yeast. We have analyzed the interaction of DnaK/Ssa1 and ClpB/Hsp104 with stress-induced protein aggregates and prion fibrils. We have further dissected the interdependence of these chaperones with respect to substrate binding and determined the immediate consequences of Hsp70 inactivation on prion fibril severing. Together our data reveal a conserved mechanistic principle of protein disaggregation and prion fibril fragmentation, with Hsp70 chaperones acting as essential substrate-targeting factors for ClpB/Hsp104. Our data therefore resolve hitherto disparate results into a unifying explanation.
Results
Hierarchical chaperone binding to heat-induced protein aggregates in E. coli
We have recently established an experimental setup based on fluorescent monitors, enabling us to study protein aggregation and chaperone–substrate interaction in E. coli cells. Exposure of E. coli to 45°C causes the formation of protein aggregates at cell poles, followed by the localization of cellular DnaK, DnaJ, and ClpB to these sites (Winkler et al., 2010). Here, we use functional YFP fusion versions of the chaperones involved (Winkler et al., 2010) to unravel the functional relationship between these chaperones with respect to aggregate binding.
To demonstrate that the occurrence of heat-induced foci of respective chaperone fusion proteins can be used as a sensitive readout for aggregate binding, we first compared the efficiency of two major cytosolic J proteins, DnaJ and CbpA, in relocalizing DnaK-YFP. J proteins act as DnaK co-chaperones and mediate the binding of DnaK to soluble substrates and protein aggregates in vitro (Laufen et al., 1999; Acebrón et al., 2008). Polar localization of DnaK-YFP after heat stress was dependent on DnaJ (Fig. S1 A). CbpA was dispensable for DnaK-YFP targeting to aggregates, but replaced DnaJ function in ΔdnaJ mutants if produced up to higher levels (Fig. S1 A). In contrast to the behavior of DnaK-YFP, the binding of DnaJ to protein aggregates was independent of DnaK. Heat-induced DnaJ-YFP foci formation was unaltered in dnaK103(am) cells, which produce a truncated DnaK variant that cannot bind substrates (Fig. S1 A; Spence et al., 1990). Notably, in dnaK103(am) cells, polar foci of DnaJ-YFP were already detectable at 30°C, which can be explained by increased protein misfolding and aggregation in the absence of DnaK. These findings establish the formation of stress-induced chaperone foci as a valid measure to study chaperone–substrate interaction in vivo.
Next, we analyzed the interdependence of DnaK and ClpB with respect to aggregate binding. DnaK-YFP still exhibited heat-induced relocalization in ΔclpB cells, whereas the relocalization of ClpB-YFP was strongly affected in dnaK103(am) cells (Fig. 1 A). Here, ClpB-YFP fluorescence largely stayed cytosolic, and an inefficient polar localization was observed in only a few cases. The absence of ClpB-YFP foci after heat stress is not caused by differences in protein aggregation, as thermolabile MetA-YFP (Gur et al., 2002; Winkler et al., 2010) still formed heat-induced foci in dnaK103(am) cells at 45°C (Fig. S1 B). A quantitative polar localization of ClpB-YFP was restored by the expression of plasmid-encoded dnaK, demonstrating that the inefficiency of ClpB in aggregate binding in dnaK103(am) mutants is directly caused by the absence of functional DnaK. Furthermore, the expression of the DnaK-V436F protein, which is defective in substrate interaction (Mayer et al., 2000), could not complement the aggregate binding deficiency of ClpB in dnaK103(am) cells. These findings demonstrate that initial association of DnaK via DnaJ is a prerequisite for ClpB targeting to aggregates.
Figure 1.

Hsp70 chaperones target ClpB/Hsp104 to heat-induced protein aggregates. (A) E. coli wild-type (dnaK+), dnaK103, and ΔclpB cells expressing ClpB-YFP or DnaK-YFP were grown at 30°C and shifted to 45°C. DnaK-V436F is deficient in substrate interaction. The occurrence of foci indicate chaperone binding to protein aggregates. The percentage of 200 cells harboring foci and the percentage of polar fluorescence intensity was quantified (n = 20). Bars, 1 µm. (B) S. cerevisiae cells harboring a single wild-type ssa1 copy or the ssa1-45(ts) allele and expressing mCitrine-Luciferase and Hsp104-CFP were grown at 25°C and shifted to 37°C. Next, cells were shifted to 45°C to induce protein aggregation. The percentage of colocalization of mCitrine-Luciferase and Hsp104-CFP foci at 45°C was quantified in both strains (n = 100). The broken lines indicate the borders of respective yeast cells. Bars, 2 µm.
We also considered an alternative explanation for these data. DnaK and DnaJ may not act by initial targeting of ClpB to protein aggregates but by stabilizing intrinsically unstable interactions of ClpB with aggregated proteins. We tested this possibility by using a YFP fusion to the ClpB-E279A/E678A protein (referred to as ClpB-trap-YFP), which is deficient in ATP hydrolysis and therefore exhibits stabilized interaction with substrates in vitro (Weibezahn et al., 2003). Although ClpB-trap-YFP efficiently localized to polar foci after heat shock in ΔclpB cells, its diffuse cytosolic distribution largely remained in heat-stressed ΔclpB dnak103(am) cells (Fig. S1, C and D), which indicates that in the absence of DnaK, even the ClpB-trap protein recognizes protein aggregates only inefficiently. We also analyzed the role of ClpB N domains in aggregate interaction by monitoring the localization of ΔN-ClpB-YFP before and after heat shock. ΔN-ClpB-YFP was efficiently targeted to polar aggregates in a DnaK-dependent manner, excluding a major role of N domains in aggregate binding in vivo (Fig. S1, C and D).
Together these findings indicate a binding hierarchy for disaggregating chaperones to protein aggregates in E. coli: DnaJ mediates the association of DnaK with aggregated proteins, which then is a prerequisite for subsequent recruitment of ClpB.
The role of Hsp70—targeting ClpB/Hsp104 to heat-induced protein aggregates—is conserved in yeast
Next, we analyzed whether the hierarchical association of Hsp70 and ClpB with protein aggregates is evolutionary conserved by monitoring aggregate interaction of functional Hsp104-CFP in S. cerevisiae (Specht et al., 2011). The canonical cytosolic Hsp70 proteins of S. cerevisiae are encoded by four genes, ssa1 to ssa4, the simultaneous deletion of which is lethal. We made use of an ssa2Δ ssa3Δ ssa4Δ ssa1-45(ts) strain (referred to as ssa1-45(ts) strain), which lacks three out of the four Ssa proteins (Ssa2p, Ssa3p, and Ssa4p) and harbors the temperature-sensitive Ssa1-45 allele (Becker et al., 1996). In these cells, the single remaining Ssa1 protein harbors a mutation of the conserved proline 417 residue (P417L) located in the substrate-binding domain. Ssa1-45 can be inactivated directly by temperature upshift to 37°C, presumably destabilizing the substrate-binding domain and leading to decreased substrate-binding ability.
Hsp104-CFP was expressed in ssa1-45(ts) cells and isogenic SSA1 cells, which harbor a single SSA1 gene as a wild-type copy and served as reference. Levels of Hsp104-CFP and Ssa1 in SSA1 and ssa1-45(ts) cells were identical at all applied temperatures (Fig. S2 C). To correlate the localizations of Hsp104-CFP and protein aggregates, we coexpressed mCitrine-Luciferase. Upon incubation of the cells at 25°C, mCitrine-Luciferase exhibited diffuse cytosolic staining, whereas Hsp104-CFP was predominantly localized to the nucleus (unpublished data). Temperature upshift to 37°C did not significantly affect the localization of either fusion protein, whereas shift to 45°C caused aggregation of mCitrine-Luciferase, which formed multiple foci distributed throughout the cytosol (Fig. 1 B). In SSA1 cells, Hsp104-CFP colocalized with mCitrine-Luciferase foci, whereas in ssa1-45(ts) cells Hsp104-CFP did not relocalize. The heat shock did not reduce the viability of SSA1 and ssa1-45(ts) cells (Fig. S2 D), which indicates that inactivation of Ssa1-45 directly affects Hsp104 localization. In conclusion, binding of Hsp104 to heat-induced protein aggregates in yeast cells relies on functional Ssa1, demonstrating that the role of Hsp70 chaperones in recruiting ClpB/Hsp104 to protein aggregates is conserved in evolution.
Targeting function of Hsp70 exhibits species specificity and involves the M domain of ClpB/Hsp104
Hsp70 and Hsp100 cooperate in a species-specific manner in protein disaggregation (Glover and Lindquist, 1998; Krzewska et al., 2001; Schlee et al., 2004). Hsp104 cannot replace ClpB function in E. coli ΔclpB cells and, vice versa, bacterial ClpB does not complement the defects of S. cerevisiae hsp104Δ cells (Parsell et al., 1993; Glover and Lindquist, 1998). We hypothesized that the targeting function of Hsp70 might be responsible for the observed species specificity of the bi-chaperone system. To test this hypothesis, we expressed Hsp104-YFP in E. coli ΔclpB cells (at ClpB wild-type levels) and monitored its localization at 30°C and 45°C. Heat shock did only cause relocalization to polar sites in a minority of cells, in contrast to ClpB-YFP, which demonstrates that Hsp104 does not efficiently associate with protein aggregates in E. coli (Fig. 2 A).
Figure 2.

The ClpB/Hsp104 M domain mediates Hsp70-dependent targeting to protein aggregates. (A) E. coli ΔclpB cells expressing either ClpB-, Hsp104-, or hybrid HBH-YFP, harboring the ClpB M domain, were grown at 30°C and shifted to 45°C. The percentage of cells showing foci was determined (n = 200). The degree of polar fluorescence intensity was quantified (percentage of total fluorescence, n = 20). Bars, 1 µm. (B) E. coli ΔclpB cells expressing Luciferase and the indicated YFP fusion proteins were grown at 30°C and shifted to 45°C. The recovery of Luciferase activity after heat shock was determined. Standard deviations are given (error bars). wt, ClpB.
To investigate whether inefficient binding of Hsp104-YFP to heat-induced aggregates in E. coli is caused by the absence of the correct Hsp70 partner, we exchanged the ClpB/Hsp104-specific middle (M) domain, which was recently demonstrated in vitro to mediate species-specific cooperation between Hsp70 and ClpB/Hsp104 (Sielaff et al., 2010; Miot et al., 2011). The Hsp104 variant HBH-YFP harbors the E. coli ClpB M domain and, in contrast to Hsp104-YFP, localized to polar foci in all ΔclpB cells after heat treatment (Fig. 2 A). Also, the fraction of HBH-YFP that was recruited to polar sites upon heat stress was significantly increased when compared with cells harboring Hsp104-YFP foci. The increase in heat-induced targeting of HBH-YFP to polar sites was lost in dnaK103(am) cells, demonstrating that the cooperation with DnaK is responsible for binding of HBH-YFP to protein aggregates in E. coli (Fig. S1 E). Targeting of HBH-YFP to protein aggregates in ΔclpB cells also allowed for partial recovery of aggregated Luciferase, whereas expression of Hsp104-YFP did not allow for Luciferase reactivation (Fig. 2 B). The recovery of Luciferase activity by HBH-YFP was minor as compared with ClpB-YFP. This finding may be either due to the reduced targeting to protein aggregates or to a reduced disaggregation activity of the chimera due to less efficient cooperation between the ClpB M domain and the Hsp104 AAA domains.
We conclude that the species specificity of the ClpB/Hsp104 and Hsp70 cooperation relies at least in part upon the specific targeting function of Hsp70, and can be overcome in vivo by swapping ClpB/Hsp104 M domains, allowing for the recruitment of the AAA+ chaperone to its site of action.
Hsp104 binding to Rnq1 prion fibrils is dependent on Ssa
Next, we analyzed whether the interaction of Hsp104 with its second physiological substrate, prion fibrils, is also affected by the Hsp70 chaperone system. We first focused on the well-characterized [PIN+]/[RNQ+] prion, which is generated by the Rnq1 protein. Rnq1-GFP or Rnq1-mCherry fusion proteins have been used before to faithfully monitor the prion state of yeast cells (Aron et al., 2007). Expression of Rnq1-mCherry or Rnq1-GFP in SSA1 or ssa1-45(ts) cells led to the formation of foci, which disappeared after prolonged incubation of the cells with guanidine hydrochloride (GdnHCl). GdnHCl is known to cause prion curing by inhibiting Hsp104 activity (Ferreira et al., 2001; Jung and Masison, 2001), thus demonstrating that the observed foci represent prion fibrils (Fig. S2 A). Furthermore, Rnq1-GFP formed SDS-resistant, amyloid-like aggregates in SSA1 and ssa1-45(ts) cells at permissive (25°C) and nonpermissive (37°C) temperatures that were converted to soluble Rnq1-GFP upon GdnHCl treatment (Fig. S2 B).
To test for an interaction of either Hsp104 or Ssa1 with prion fibrils, we performed colocalization studies in yeast cells and confirmed them by line intensity plot profiles (Fig. S3, A–P). For monitoring the binding of Hsp104 to Rnq1 prions and the role of Ssa1 in this process, we coexpressed Hsp104-CFP and Rnq1-mCherry in ssa1-45(ts) and isogenic SSA1 yeast cells. At 25°C, Hsp104-CFP colocalizes with Rnq1-mCherry foci in SSA1 and ssa1-45(ts) cells (Figs. 3 A and S3 A). Colocalization was additionally confirmed by confocal microscopy (unpublished data). Upon temperature upshift to 37°C, the colocalization remained in SSA1 cells, but not in ssa1-45(ts) cells, in which Hsp104-CFP showed a diffuse cytosolic staining despite the presence of Rnq1-mCherry foci. The loss of Hsp104-CFP colocalization with Rnq1-mCherry foci indicates that the binding of Hsp104-CFP to the Rnq1-mCherry prion fibril relies on functional Ssa1. Consistently, when ssa1-45(ts) cells were shifted back from 37°C to 25°C, binding of Hsp104-CFP to Rnq1-mCherry was restored, demonstrating that Ssa1-45 inactivation is reversible (Figs. 3 B and Fig. S3 B).
Figure 3.

Ssa1 targets Hsp104 to Rnq1 prion fibrils. (A) S. cerevisiae SSA1 and ssa1-45(ts) cells expressing RNQ-mCherry and Hsp104-CFP were grown at 25°C and shifted to 37°C. Binding of Hsp104 to Rnq1 prion fibrils was solely observed at 25°C and 37°C in the SSA1 strain and at 25°C in the ssa1-45(ts) strain. The frequency of colocalization (%) is given (n = 100). (B) ssa1-45(ts) cells were grown at 25°C, and cycloheximide (CHX) was added to stop protein synthesis. Next, cells were shifted to 37°C and incubated further for 60 min at either 25°C or 37°C. At each step, the localizations of Rnq1-mCherry and Hsp104-CFP were analyzed. The reestablishment of Rnq1-mCherry and Hsp104-CFP colocalization during a recovery phase at 25°C indicates that Ssa1-45 activity is restored, enabling it to target Hsp104-CFP to Rnq1-mCherry foci. The frequency of colocalization (%) is given (n = 100). (C) Association of Ssa1 and Ssa1-45 with Rnq1 fibers was monitored by immunofluorescence in SSA1 and ssa1-45(ts) cells at 25°C and 37°C. In SSA1 cells, Ssa1 colocalizes with Rnq1-GFP foci at 25°C and at 37°C. In ssa1-45(ts) cells, efficient binding of Ssa1-45 to Rnq1-GFP foci was only observed at 25°C. The frequency of colocalization (%) is given (n = 100). The broken lines indicate the borders of respective yeast cells. Bars, 2 µm.
The similarity in Ssa1 dependence on Hsp104 binding to both heat-induced protein aggregates and prion fibrils predicts an interaction between Ssa1 and Rnq1-mCherry aggregates, which should be temperature-sensitive in the case of ssa1-45(ts) cells. We therefore tested for Ssa1 binding to Rnq1-GFP fibrils in SSA1 and ssa1-45(ts) cells by immunofluorescence. Ssa1 foci were observed at 25°C upon expression of Rnq1-GFP, and both Ssa1 and Rnq1-GFP foci colocalized in SSA1 and ssa1-45(ts) cells, demonstrating efficient interaction of Ssa1 with prion fibrils (Figs. 3 C and S3 C). Inactivation of Ssa1 in ssa1-45(ts) cells abrogated Ssa1 binding to Rnq1-GFP foci, providing an explanation for the observed loss of Hsp104-CFP binding (Figs. 3 C and S3 C).
Next, we monitored the consequences of the loss of Ssa1 function in ssa1-45(ts) cells at 37°C on the physical state of the Rnq1 prion. We determined the dynamics of Rnq1-GFP foci in the presence and absence of functional Ssa1 by fluorescence loss in photobleaching (FLIP) experiments. Here, a small cytosolic area within SSA1 or ssa1-45(ts) cells harboring Rnq1-GFP foci outside of the bleach area was repeatedly bleached with a laser pulse, and the fluorescence intensity of Rnq1-GFP foci was determined. At 25°C, a rapid drop of Rnq1-GFP foci fluorescence was observed in both cells, indicating a dynamic exchange of Rnq1-GFP molecules between cytosol and foci (Fig. 4 A). To exclude photobleaching effects as a source of fluorescence decay, we bleached an area in a neighboring cell located in close proximity to the Rnq1-GFP foci. Here, no decrease in Rnq1-GFP foci intensity was observed (Fig. 4 A). We also determined the mobility of diffuse Rnq1-GFP fluorescence in [pin−] cells, revealing a significantly higher mobility (Fig. 4 A). Together, these data show that FLIP experiments specifically report on the dynamics of Rnq1-GFP fibrils.
Figure 4.

Inactivation of Ssa1 inhibits Rnq1p fiber fragmentation by Hsp104. (A) Rnq1-GFP foci become immobile upon Ssa1 inactivation. FLIP measurements of Rnq1-GFP were performed in SSA1 and ssa1-45(ts) cells at 25°C and after temperature upshift to 37°C. For comparison, the mobility of diffuse Rnq1-GFP was determined in [pin−] cells. Mobility of Rnq1-GFP was also monitored in SSA1 cells at 37°C after addition of GdnHCl, causing Hsp104 inactivation. n = 25 cells were analyzed for each condition. Standard errors (error bars) and a photo bleaching control are given. (B) Inactivation of Ssa1 or addition of GdnHCl causes an increase in Rnq1-GFP foci size. SSA1 and ssa1-45(ts) cells were shifted from 25°C to 37°C, and fluorescence intensity of Rnq1-GFP foci was determined before (0 min) and after temperature upshift (90 min). Standard deviations are given (error bars).
The inactivation of Ssa1 in ssa1-45(ts) cells resulted in stabilization of Rnq1-GFP and was not observed in SSA1 cells (Fig. 4 A). The loss of Rnq1-GFP dynamics in the absence of functional Ssa1 suggests that severing of Rnq1-GFP fibrils is compromised. In agreement with this possibility, we observed an identical stabilization of Rnq1-GFP foci in SSA1 cells upon inhibition of Hsp104 by addition of GdnHCl (Fig. 4 A). We ensured Hsp104 inactivation by determining a strongly decreased reactivation of heat-aggregated mCitrine-Luciferase in SSA1 cells in the presence of GdnHCl (unpublished data). Stabilization of Rnq1-GFP fibrils in ssa1-45(ts) cells at 37°C was accompanied by a specific increase in foci fluorescence intensity, which suggests an increase in Rnq1-GFP fibril size (Fig. 4 B). A comparable increase in Rnq1-GFP foci intensity was observed in SSA1 cells treated with GdnHCl (Fig. 4 B). These findings demonstrate that the stabilized Rnq1-GFP foci in ssa1-45(ts) cells still act as a seed for soluble Rnq1-GFP molecules, thereby excluding a scenario in which a loss of Hsp104 interaction is caused by a change in Rnq1 aggregate structure.
We also monitored whether inhibition of Hsp104 by GdnHCl affects Hsp104 binding to Rnq1-GFP foci. The colocalization frequency of Hsp104-CFP and Rnq1-mCherry was strongly reduced upon treatment with GdnHCl, whereas Ssa1 binding to Rnq1-GFP remained unaffected (Figs. 5 and S3 D). These findings shed new light on the mechanism of Hsp104 inhibition by GdnHCl, which has been previously only demonstrated to reduce Hsp104 ATPase activity (Grimminger et al., 2004; Nowicki et al., 2012). Our data suggest that GdnHCl additionally affects Ssa1-Hsp104 cooperation, leading to reduced targeting of Hsp104 to fibrils.
Figure 5.
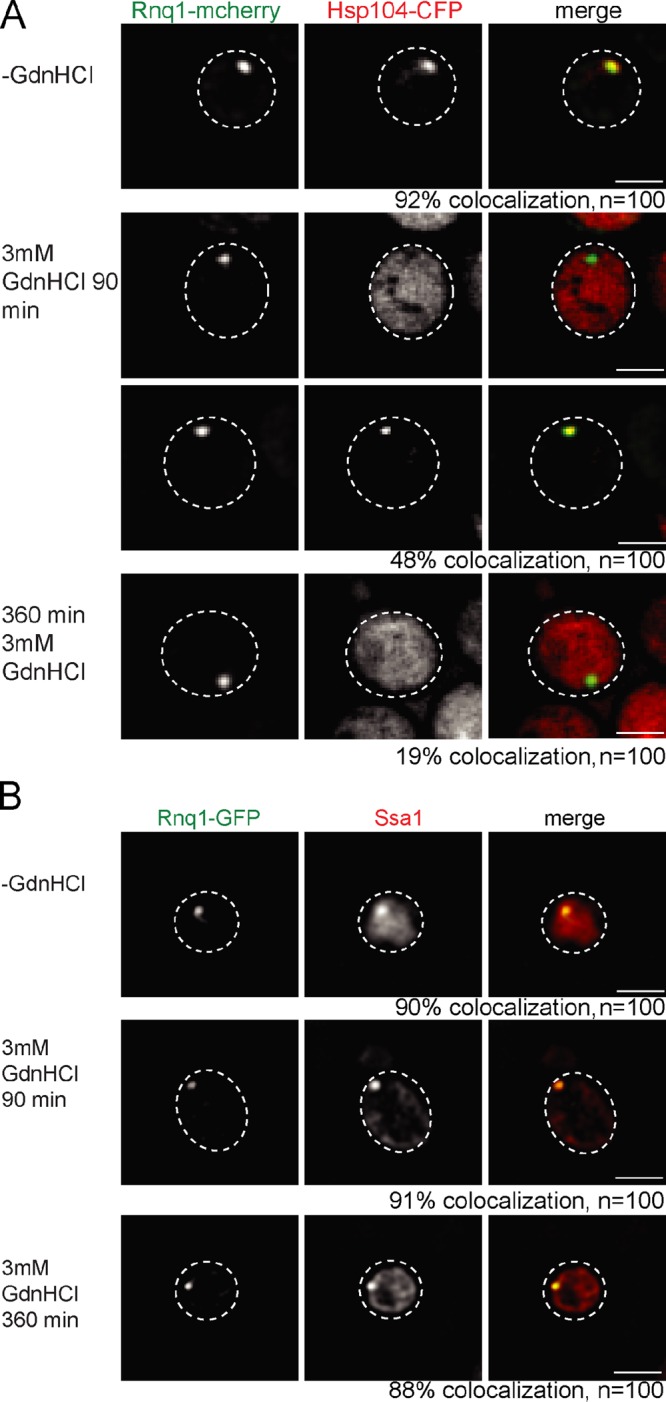
GdnHCl inhibits Hsp104 binding to Rnq1 fibrils. S. cerevisiae SSA1 cells expressing Rnq1-mCherry or Rnq1-GFP were treated for 0, 90, and 360 min with 3 mM GdnHCl. Colocalization of Rnq1 foci with Hsp104-CFP (A) or SSA1 (B) was analyzed. SSA1 was detected by immunofluorescence. Colocalization of Hsp104-CFP and Rnq1-mCherry is strongly reduced upon prolonged GdnHCl treatment. Addition of GdnHCl does not affect Ssa1 association with Rnq1-GFP fibrils. The frequency (%) of colocalization is given (n = 100). The broken lines indicate the borders of respective yeast cells. Bars, 2 µm.
Ssa-independent binding of Hsp104 to NM-YFP prion fibrils can occur but is insufficient for prion fragmentation
To investigate whether our results obtained for [RNQ+] are also valid for other prions, we analyzed the role of Ssa1 for the interaction of Hsp104 with the [PSI+] prion. We coexpressed Hsp104-CFP and NM-YFP in ssa1-45(ts) and SSA1 cells. NM-YFP contains the prion-forming NM domain of Sup35 and has been successfully used previously to monitor prion propagation in yeast (Patino et al., 1996; Ganusova et al., 2006; Mathur et al., 2010; Tyedmers et al., 2010). Overproduction of NM-YFP caused formation of foci in both yeast strains at 25°C, which were lost after prolonged incubation with GdnHCl (Fig. S2 E). NM-YFP formed SDS-resistant aggregates in SSA1 and ssa1-45(ts) cells at permissive (25°C) and nonpermissive (37°C) temperatures that were converted to monomeric NM-YFP upon GdnHCl treatment (Fig. S2 F), further confirming that the NM-YFP foci represent the prion form.
At 25°C, Hsp104-CFP binds to NM-YFP foci in SSA1 and ssa1-45(ts) cells (Figs. 6 A and S3 E). Colocalization was additionally confirmed by confocal microscopy (unpublished data). Interestingly, colocalization was not altered upon inactivation of Ssa1. This contrasts the observed Ssa dependence of Hsp104 binding on either heat-induced aggregates (Fig. 1 B) or Rnq1-mCherry prion fibrils (Fig. 3 A).
Figure 6.

Ssa1 modulates the interaction of Hsp104 with NM-YFP fibers. (A) S. cerevisiae SSA1 and ssa1-45(ts) cells expressing NM-YFP and Hsp104-CFP were grown at 25°C and shifted to 37°C. NM-YFP foci colocalize with Hsp104-CFP in SSA1 cells and in ssa1-45(ts) cells at 25°C and 27°C. The frequency of colocalization (%) is given (n = 100). The broken lines indicate the borders of respective yeast cells. Bars, 2 µm. (B and C) Hsp104-CFP and NM-YFP become immobilized upon Ssa1-45 inactivation. FLIP experiments of Hsp104-CFP (B) and NM-YFP (C) were performed in SSA1 and ssa1-45(ts) cells at 25°C and 37°C. For comparison, the motilities of diffuse Hsp104-CFP located outside the foci (cytosol) or of diffuse NM-YFP in [psi−] cells were determined. The mobility of NM-YFP was also monitored in SSA1 cells at 37°C after addition of GdnHCl. n = 25 cells analyzed for each condition. Standard errors and photo bleaching controls are given (error bars). (D) Inactivation of Ssa1 or addition of GdnHCl causes an increase in NM-YFP foci size. SSA1 and ssa1-45(ts) cells were shifted from 25°C to 37°C, and the fluorescence intensities of NM-YFP foci were determined before (0 min) and after temperature upshift (90 min). Standard deviations are given (error bars).
To test whether this discrepancy is caused by differences in Ssa1 binding to NM-YFP fibrils as compared with Rnq1-GFP fibrils, we monitored Ssa1 localization in SSA1 and ssa1-45(ts) cells harboring NM-YFP foci by immunofluorescence (Figs. S3 H and S4 A). Colocalization between Ssa1 and NM-YFP foci was observed at 25°C in both yeast strains and was confirmed by confocal microscopy (unpublished data), demonstrating efficient interaction of Ssa1 with NM-YFP fibrils. Inactivation of Ssa1 in ssa1-45(ts) cells caused a specific loss of Ssa1 interaction with NM-YFP (Figs. S3 H and S4 A), similar to the situation found for Rnq1-GFP. We conclude that Hsp104-CFP still binds to NM-YFP fibrils in the absence of Ssa1.
To elucidate potential differences in the mode of Hsp104 association with NM-YFP fibrils (as compared with Rnq1-mCherry fibrils), we investigated the dynamics of this interaction in the presence and absence of functional Ssa1 by FLIP experiments. A rapid drop of Hsp104-CFP fluorescence colocalizing with NM-YFP foci was observed at 25°C in SSA1 and ssa1-45(ts) cells, indicating a dynamic exchange of Hsp104-CFP with prion fibrils (Fig. 6 B). We excluded photobleaching as a potential source of fluorescence decrease and determined a higher mobility of cytosolic Hsp104-CFP located outside the foci, demonstrating that FLIP experiments allow one to specifically monitor the dynamics of Hsp104-CFP engaged in prion interaction (Fig. 6 B).
When shifting ssa1-45(ts) cells from 25°C to 37°C, Hsp104-CFP binding to NM-YFP was stabilized when compared with Ssa1 cells (Fig. 6 B). Thus, although Ssa1 is not required for binding of Hsp104 to NM-YFP fibrils, its inactivation renders the association of Hsp104 largely immobile.
The static binding of Hsp104 to prion fibrils in the absence of functional Ssa1 suggests a deficiency of Hsp104 chaperone function, which should result in a reduction of prion fibril mobility. We therefore probed the physical state of NM-YFP foci in SSA1 and ssa1-45(ts) cells at 25°C and 37°C by FLIP experiments (Fig. 6 C). NM-YFP foci were mobile in both cells at 25°C, which indicates a dynamic exchange of NM-YFP molecules with the cytosol, in agreement with previous findings (Kawai-Noma et al., 2006; Satpute-Krishnan et al., 2007; Kawai-Noma et al., 2009). We again excluded photobleaching effects and determined a higher mobility of diffuse NM-YFP fluorescence in [psi−] cells, demonstrating that FLIP experiments faithfully report on the mobility of NM-YFP prion fibrils (Fig. 6 C).
Inactivation of Ssa1 in ssa1-45(ts) significantly reduced the mobility of NM-YFP when compared with SSA1 cells (Fig. 6 C). Thus, inactivation of Ssa1 affects the dynamics of NM-YFP fibrils and Hsp104 in the same way.
Our findings indicate that Ssa1 is required for the release of NM-YFP molecules from prion fibrils. To further substantiate the role of Ssa1 in NM-YFP prion severing, we compared the consequences of Ssa1 and Hsp104 inactivation by GdnHCl addition on prion mobility. We found a reduction of NM-YFP dynamics in the presence of GdnHCl in SSA1 cells that was almost identical to the one observed upon Ssa1 inactivation in ssa1-45(ts) cells, providing solid evidence for a loss of NM-YFP prion fragmentation in the absence of functional Ssa1 (Fig. 6 C). Along the same line, stabilization of NM-YFP foci in ssa1-45(ts) cells at 37°C, or in SSA1 cells upon GdnHCl treatment, caused a similar increase in NM-YFP foci fluorescence, which suggests an increase in NM-YFP fibril size (Fig. 6 D).
To demonstrate that key findings obtained for NM-YFP also hold true for full-length Sup35 in a [PSI+] background, we analyzed the binding of red fluorescent Hsp104-eqFP611 to Sup35-GFP, which had been characterized previously (Wu et al., 2006; Satpute-Krishnan et al., 2007). Hsp104-eqFP611 still colocalized with Sup35 in ssa1-45(ts) cells at 37°C; however, the interaction became immobile in the absence of functional Ssa1 (Figs. S2 G and S3, F and G). Accordingly, the mobility of Sup35-GFP foci was especially reduced in ssa1-45(ts) cells at 37°C, matching the stabilization determined in SSA1 cells upon Hsp104 inhibition by GdnHCl treatment (Fig. S2 H).
Collectively, Hsp104 can bind independently from Ssa1 to NM domain or full-length Sup35 fibers. However, this interaction is nonproductive and does not result in prion fragmentation.
Overproduction of Hsp104 outcompetes Ssa1 for binding to NM-YFP fibrils
To gain mechanistic insights into the Ssa1-independent but nonproductive association of Hsp104 with NM-YFP fibrils, we considered the possibility that association is mediated by the N domain of Hsp104. This hypothesis is driven by the observation that although the N domain is not required for [PSI+] propagation, it is essential for the curing of yeast cells from Sup35 prions by high Hsp104 levels, which implies an interaction between the Hsp104 N domain and Sup35 fibrils (Hung and Masison, 2006). We therefore produced NM-YFP in SSA1 or ssa1-45(ts) cells expressing ΔN-Hsp104-CFP and performed immunofluorescence against Hsp104 (Figs. 7 A and S3 L). At 25°C, ΔN-Hsp104 colocalized with NM-YFP foci in both strains, whereas at 37°C ΔN-Hsp104 colocalized with NM-YFP foci only in SSA1 cells but not in ssa1-45(ts) cells. As a control, we also performed immunofluorescence with full-length Hsp104 in SSA1 or ssa1-45(ts) cells expressing NM-YFP. Here, binding of Hsp104 to NM-YFP foci was still observed in both strains at 37°C (Figs. S3 J and S5 A). These findings confirm the key role for Ssa1 in Hsp104 targeting to prion fibrils. It further indicates that NM-YFP fibrils are distinct from Rnq1-mCherry fibrils, as they harbor a binding site for the Hsp104 N domain that can target Hsp104 to NM fibrils in the absence of Ssa1. This special feature of NM-YFP is supported by the observation that high levels of full-length Hsp104 cure [PSI+] but no other yeast prions (Chernoff et al., 1995; Derkatch et al., 1997; Moriyama et al., 2000).
Figure 7.

The N domain of Hsp104 mediates binding to NM-YFP fibers and enables high Hsp104 levels to outcompete Ssa1 for NM-YFP association. (A) ΔN-Hsp104-CFP binding to NM-YFP fibers is dependent on SSA1. S. cerevisiae SSA1 and ssa1-45(ts) cells expressing ΔN-Hsp104-CFP and NM-YFP were grown at 25°C and shifted to 37°C. Binding of ΔN-Hsp104-CFP to NM-YFP foci was analyzed by immunofluorescence. Binding of ΔN-Hsp104 with NM-YFP foci was only observed at 25°C and 37°C in SSA1 cells and at 25°C in ssa1-45(ts) cells. The frequency of colocalization (%) is given (n = 100). (B) Hsp104 but not ΔN-Hsp104 competes with Ssa1 for binding to NM-YFP fibers. Ssa localization was determined by immunofluorescence in SSA1 cells overexpressing Hsp104/ΔN-Hsp104 and NM-YFP. The colocalization frequency (%) of Ssa1 and NM-YFP foci was determined (n = 200). The broken lines indicate the borders of respective yeast cells. vc, vector control. (C) FLIP experiments of NM-YFP were performed at 25°C in SSA1 cells overexpressing ΔN-Hsp104 or Hsp104. NM-YFP foci showed reduced mobility in the presence of high levels of Hsp104. n = 25 cells were analyzed for each condition. Standard errors are given (error bars). vc, vector control.
The reason for prion curing by Hsp104 overproduction is unknown. High levels of Hsp104 have been suggested to either affect the partitioning of propagons during cell division or to either increase or decrease the efficiency of prion propagation (Kryndushkin et al., 2003; Tuite and Cox, 2003; Masison et al., 2009). The data presented here tempted us to speculate that the ability of Hsp104 to bind to NM-YFP fibrils via its N domain enables Hsp104 to specifically outcompete Ssa1 for fibril binding if present at high levels. To test this scenario, we first expressed NM-YFP in SSA1 cells, followed by copper-controlled overproduction of either Hsp104 or ΔN-Hsp104 (Fig. S5 B). In agreement with earlier findings (Zhou et al., 2001), high Hsp104 levels specifically led to the appearance of ribbon-like NM-YFP structures in some cells, probably representing an intermediate prion state (Alberti et al., 2009; Tyedmers et al., 2010), and also an increased cytosolic fluorescence of NM-YFP, which indicates prion curing (Fig. S5 C). Importantly, overproduction of Hsp104 strongly reduced Ssa1 binding to NM-YFP foci and ribbons, as determined by immunofluorescence, whereas overproduction of ΔN-Hsp104 had no effect in comparison to the vector-only control (Figs. 7 B and S3 K). A still-detectable Ssa1/NM-YFP colocalization in a few cells is likely explained by cell-to-cell differences in the Hsp104 expression levels. As a control, we monitored the consequences of Hsp104 overproduction on Ssa1 binding to Rnq1-GFP foci because increased Hsp104 levels do not lead to [RNQ+] curing (Fig. S5 D). Ssa1 association with Rnq1-GFP foci was not affected, demonstrating that increased Hsp104 levels specifically outcompete Ssa1 for NM-YFP binding. We also tested the consequences of Hsp104 and ΔN-Hsp104 overproduction on NM-YFP foci mobility by FLIP experiments. High Hsp104 levels reduced NM-YFP mobility, whereas ΔN-Hsp104 had no significant effect (Fig. 7 C). This finding confirms that an Ssa1-independent binding of Hsp104 prevents NM-YFP fragmentation and underlines the key role of Ssa1 in prion severing.
The deletion of the Hsp90 co-chaperone Sti1 and inhibition of Hsp90 activity by Radicicol treatment have been shown to largely diminish [PSI+] curing by Hsp104 overproduction (Moosavi et al., 2010; Reidy and Masison, 2010). We therefore tested whether a reduction of Hsp90 activity has an impact on Ssa1 binding to NM-Sup35 fibers upon Hsp104 overproduction. In the presence of Radicicol, we observed a strong increase in the fraction of SSA1 cells showing colocalization of Ssa1 and NM-YFP foci in the presence of high Hsp104 levels (Fig. 8 A). Similar findings were obtained when colocalization of Ssa1 and NM-YFP foci in SSA1 sti1Δ cells upon Hsp104 overproduction was determined (Fig. 8 B). Thus cellular conditions that prevent [PSI+] curing upon Hsp104 overproduction reestablish Ssa1 binding to NM-YFP fibrils, supporting our model in which [PSI+] curing by high Hsp104 levels is caused by preventing Ssa1 association with prion fibrils.
Figure 8.

Inhibition of Hsp90 activity and deletion of sti1Δ prevents outcompetition of Ssa1 by high Hsp104 levels. (A and B) Ssa localization was determined by immunofluorescence in SSA1 pdr5Δ cells or SSA1 sti1Δ overexpressing Hsp104. SSA1 pdr5Δ cells were analyzed in the absence or presence of the Hsp90 inhibitor Radicicol or DMSO (control). Colocalization frequencies with NM-YFP foci were determined (n = 100). The broken lines indicate the borders of respective yeast cells. vc, vector control. Bars, 2 µm.
Ssa1 is a general targeting factor of Hsp104 to prion fibrils
To further generalize our observation that Ssa1 recruits Hsp104 to prion fibrils, we analyzed the binding of Hsp104 to the recently identified prion protein Mot3 and the potential prion proteins Lsm4 and Nrp1 (Alberti et al., 2009). To visualize the formation of respective prion fibrils in yeast cells, YFP fusion proteins of the respective prion domains were expressed at 25°C. Upon overexpression, Mot3-YFP and Lsm4-YFP formed foci. Nrp1-YFP also produced foci in most cells, whereas few cells (2%) showed ribbon-like structures (Figs. 9 and S3, N–P).
Figure 9.

Ssa1 has a general role in the targeting of Hsp104 to prion fibrils. S. cerevisiae SSA1 and ssa1-45(ts) cells expressing Hsp104-CFP and YFP fusion to the prion domains of Mot3 (A), Lsm4 (B), or Nrp1 (C) were grown at 25°C and shifted to 37°C. Binding of Hsp104 to the individual prion domains was observed at 25°C and 37°C for the SSA1 cells and solely at 25°C for ssa1-45(ts) cells. The frequency of colocalization (%) is given (n = 60). The broken lines indicate the borders of respective yeast cells. Bars, 2 µm.
The localization pattern of the prion domain–YFP fusion proteins was reverted by GdnHCl to a diffuse cytosolic distribution, demonstrating that the respective foci and ribbon structures represent proteins in the prion state (Fig. S2, I–K). Next, Hsp104-CFP was coexpressed with the individual prion–YFP fusion proteins in SSA1 and ssa1-45(ts) cells. At 25°C, Hsp104-CFP revealed colocalization with all tested prion fibrils in both cell types. Temperature upshift to 37°C specifically abrogated Hsp104-CFP binding to Mot3-YFP, Lsm4-YFP, and Nrp1-YFP foci and Nrp1-YFP ribbons in ssa1-45(ts) cells but not in SSA1 cells (Figs. 9 and S3, N–P). This binding deficiency correlated with a loss of Ssa1 prion interaction in ssa1-45(ts) cells at elevated temperatures (Figs. S4, B–D), demonstrating that the role of Ssa1 as a recruitment factor for Hsp104 binding to prion fibrils is conserved.
Discussion
In this paper, we have established the fact that bacterial ClpB and yeast Hsp104 require their cooperating Hsp70 chaperones, DnaK and Ssa1, as targeting factors for the association with two different categories of physiological substrates: aggregates of heat-denatured proteins and prion fibrils (Fig. 10).
Figure 10.

Conserved cooperation between Hsp70 and ClpB/Hsp104 in protein disaggregation and prion fragmentation. Hsp70 chaperones recruit ClpB/Hsp104 to both stress-induced amorphous protein aggregates and prion fibrils. Hsp70-mediated targeting involves the ClpB/Hsp104 M domain. ClpB/Hsp104 threading activity extracts misfolded polypeptides from protein aggregates for subsequent refolding. The same activity leads to prion fiber severing and the generation of oligomeric propagons. High Hsp104 levels bind via N domains to Sup35 fibrils, thereby preventing Hsp70 association and fiber fragmentation.
The function of Hsp70 chaperones as targeting factors for Hsp100s is in principle sufficient to explain their essential role at the initial stages of protein disaggregation. Such a role could be indirect, relying on Hsp70-driven conformational changes of the aggregates to expose segments of aggregated polypeptides that can be recognized by ClpB/Hsp104 for substrate threading. An indirect role of Hsp70, however, is difficult to reconcile with the previously noticed species specificity in the Hsp70–Hsp100 cooperation in vitro (Glover and Lindquist, 1998; Krzewska et al., 2001; Schlee et al., 2004). Indeed, we observed that yeast Hsp104 is unable to efficiently interact with protein aggregates in E. coli ΔclpB cells, which harbor an active DnaK chaperone system (Fig. 2 A). We therefore favor a direct role for Hsp70 proteins in targeting ClpB/Hsp104 to aggregates that is based on species-specific physical interactions between the cooperating chaperones. In vitro experiments revealed that species specificity is mediated by the ClpB/Hsp104 M domain (Sielaff and Tsai, 2010; Miot et al., 2011). Here, we provide the rationale for this observation, as a hybrid Hsp104 harboring the M domain of ClpB (HBH) efficiently and productively binds to protein aggregates in E. coli cells, thereby overcoming the species barrier (Fig. 2). How the M domains of ClpB and Hsp104 exert this function remains unclear, but it must involve a mechanism that allows targeting of the chaperone to Hsp70 aggregate complexes (Cashikar et al., 2002; Schirmer et al., 2004; Haslberger et al., 2007).
Our finding that the hierarchy of chaperone binding to stress-induced protein aggregates holds true for prion fibrils is of particular interest because Hsp104 has been found to have prion-severing activity on its own (Shorter and Lindquist, 2004, 2006), which suggests that the need for and mode of Hsp70-Hsp104 cooperation in this process is different. We used the temperature-sensitive Ssa1-45 allele, which enabled us to monitor in vivo the immediate consequences of Ssa1 inactivation at 37°C on Hsp104–prion interactions and fibril dynamics. We demonstrate that a productive interaction of Hsp104 with all tested prion substrates, including Sup35-NM and Rnq1, requires prior association of Ssa1 with the fibrils. We consider this effect to be direct because Ssa1-45 inactivation was entirely reversible, restoring Hsp104 targeting upon return to the permissive temperature. The role of Ssa1 as a targeting factor is in line with large quantities of Ssa1/2 co-isolated with Sup35 fibrils from yeast cell lysates (Bagriantsev et al., 2008). However, in contrast to the originally suggested inhibitory effect of Ssa1/2 on Hsp104 activity (Bagriantsev et al., 2008), we demonstrate that Ssa1 binding is a prerequisite for Hsp104 activity.
These findings support and extend previous studies, demonstrating that depletion of the yeast Hsp40, Sis1, affects prion propagation and prion protein threading by Hsp104 in vivo (Higurashi et al., 2008; Tipton et al., 2008). These studies relied on the continuous depletion of Sis1 levels by cell division and only allowed monitoring of the consequences of Hsp70 inactivation after several generations. Here, we unambiguously demonstrate that loss of Hsp70 activity has an instant effect on Hsp104 binding and prion severing, leading to strongly reduced prion fibril dynamics and, consequently, an increase in prion fibril size.
Our findings contradict reports demonstrating an Hsp104-only mediated severing of [PSI+] prion fibrils of Sup35 proteins in vitro (Shorter and Lindquist, 2004, 2006). Although we cannot fully explain this discrepancy, we describe here the finding that [PSI+] prion fibrils harbor a binding site for the Hsp104 N domain, a unique feature not observed for other prion proteins analyzed. This feature allows for Ssa1-independent association of Hsp104 with NM-YFP fibrils in vivo. However, we found this interaction to be nonproductive, as Hsp104-NM-YFP complexes are immobile and do not result in prion severing (Fig. 5). We expect that such N domain–mediated binding of Hsp104 to Sup35 NM fibrils only occurs in the absence of functional Hsp70 or upon Hsp104 overproduction. Our observation is consistent with previous in vitro findings, which show that Hsp104 can bind to Sup35NM fibrils but requires additional factors for fragmentation (Inoue et al., 2004).
We further demonstrate that this unique feature of NM-YFP explains the puzzling observation that Hsp104 overproduction abrogates Sup35 prion severing and cures [PSI+] (Chernoff et al., 1995; Kryndushkin et al., 2003). We found that the Ssa1-independent, direct association of Hsp104 (but not of ΔN-Hsp104; Fig. 6) with NM-YFP fibrils outcompetes Ssa1 for aggregate binding. Ssa1 displacement by Hsp104 overproduction thus prevents the normal hierarchy of chaperone association required for formation of productive Ssa1–Hsp104 interactions on the NM-YFP fibril surface. Our findings furthermore provide an explanation for several enigmatic observations, including that (a) Sup35NM fibril sizes increase upon Hsp104 overproduction (Kryndushkin et al., 2003); (b) simultaneous Ssa1 and Hsp104 co-overproduction does not result in [PSI+] curing, as such conditions probably prevent the displacement of Ssa1 from Sup35 fibrils (Newnam et al., 1999); and (c) Hsp104 overproduction inhibits propagation of [PSI+] but not of other prions (Moriyama et al., 2000). Interestingly, an Hsp104-binding site was recently identified in the M domain of Sup35 (Helsen and Glover, 2012). The observation that this binding site is not required for [PSI+] propagation but is essential for curing by Hsp104 overproduction (Helsen and Glover, 2012) is in perfect agreement with our findings.
Summing up, we have identified Hsp70 chaperones as crucial for the targeting of ClpB/Hsp104 to stress-induced protein aggregates and prion fibrils. The previously controversial mechanism of chaperone cooperation is conserved for both types of aggregates. Despite a shared mechanistic principle, the efficiencies of the protein disaggregation reactions are different because Hsp70/Hsp104 are capable of completely solubilizing heat-induced protein aggregates, whereas prion fibrils largely persist in presence of the bi-chaperone system.
The key role identified for Hsp70 also has a broad-spectrum impact on prion propagation in metazoa, which lack an Hsp104 homologue. We suggest that Hsp70 chaperones play a key role in prion diseases in metazoa and may cooperate with additional chaperones to increase disaggregation activity and replace Hsp104 function. In line with such a model, the Sup35NM domains can propagate as a prion in mammalian cells, which suggests the existence of cellular components ensuring prion propagation in higher eukaryotes (Krammer et al., 2009).
Materials and methods
Plasmids and strains
All plasmids, E. coli K12, and yeast strains used in this study are listed in Tables S1–S4. Fluorescent fusion proteins for expression in plasmids were constructed by fusion PCR. Yeast chromosomal Hsp104 cerulean fluorescent protein (CFP) or eqFP611 (red fluorescent protein) C-terminal tagging was performed by chromosomal integration of a PCR-amplified cassette according to Janke et al. (2004) and Wach et al. (1997). The pBS10 (for CFP labeling) and the pYM51 plasmid (for eqFP611 labeling) were used as templates for PCR. ΔN-Hsp104-CFP was generated by replacement of the chromosomal copy of Hsp104 by ΔN-hsp104-cfp-hph. sti1 and pdr5 deletion strains were constructed by one-step PCR-mediated gene replacement with the pFA6-natNT2 module. Genomic gene modifications were confirmed by PCR (pdr5Δ, sti1Δ, n-hsp104-cfp, hsp104-cfp, and hsp104-eqfp611) and Western blotting (sti1Δ, n-hsp104-cfp, and hsp104-cfp). Isolated prion domain–YFP fusions were constructed according to Alberti et al. (2009). Sup35-GFP was provided by T.R. Serio (Brown University, Providence, RI).
E. coli and S. cerevisiae growth conditions
E. coli cells were grown if not indicated otherwise in lysogeny broth (LB) media to an OD600 of 0.6–0.8. For heat treatment, cells were shifted to 45°C for 20 min in a water bath shaker Gyrotory G76. When appropriate, antibiotics were added to standard concentrations. Gene expression was induced by the addition of indicated concentrations of IPTG or arabinose (Table S2). To stop protein translation, tetracycline (25 µg/ml) was added 10 min before the start of the experiment.
Yeast cells were grown to mid-exponential growth phase in synthetic media without the respective amino acids and with or without antibiotics. Expression of Gal promoter constructs was induced by diluting the cells in synthetic media with 2% galactose. Strains derived from YWO0625 and YWO0622 were grown at 25°C. Ssa1 was inactivated in ssa1-45(ts) cells (provided by D. Wolf, Institute of Biochemistry, University of Stuttgart, Stuttgart, Germany) by incubating the cells to 37°C for 1.5 h. For the coexpression experiment of mCitrine-Luciferase/prion domains and Hsp104-CFP, 25 µg/ml cycloheximide was added 10 min before transferring the cells to 37°C.
Simultaneous overexpression of Hsp104 and NM-YFP or Rnq1-GFP was performed with Hsp104 controlled by a copper-inducible promoter and prion-fluorophore fusions controlled by a galactose-inducible one. Yeast cells were shifted from Raffinose- to Galactose-containing media for 2 h before inducing Hsp104 by addition of 100 µM copper sulphate. Inhibition of Hsp90 activity was achieved by addition of 68 µM Radicicol for 8 h. 3 mM GdnHCl was used in liquid media or on plates to cure prion aggregates.
Western Blotting, SDS-PAGE, and semi-denaturating detergent agarose gel electrophoresis (SDD-AGE)
SDS-PAGE and Western blotting were performed according to standard protocols. SDD-AGE was performed as described previously (Tessarz et al., 2008). Hsp104 and Ssa antibodies were used at 1:10,000 dilutions, and YFP antibodies were used at 1:20,000 dilution.
Determination of Luciferase activity
E. coli ΔclpB cells coexpressing Luciferase and ClpB/Hsp104 variants were grown at 30°C to logarithmic growth phase and shifted to 45°C for 20 min. Similarly, S. cerevisiae SSA1 cells expressing mCitrine-Luciferase were grown at 30°C, shifted to 37°C for 60 min, and transferred to 45°C for 20 min. Luciferase activities and cell culture densities (OD600) were determined before and after 45°C heat shock and during a subsequent recovery phase at 30°C as described previously (Schröder et al., 1993). Luciferase activities determined before heat shock were normalized to the respective cell culture densities and set to 100%. Data represent the mean ± SD of three independent experiments.
Plating assay
For viability analysis, overnight cultures of yeast cells were diluted into fresh YPD medium and grown to mid-log phase at 25°C. Cells were first shifted to 37°C for 60 min and then incubated at 45°C for the indicated period of time. Samples were taken, serially diluted fivefold, and spotted onto YPD plates. Survival of cells was determined by calculating the plating efficiency after 2 d of growth at 25°C.
Fluorescence microscopy
For snapshot imaging, cells were pelleted for 5 min at 5,000 rpm, resuspended in TEB (E. coli) or PBS buffer (yeast), and immobilized on 1% agarose pads. Agarose pads were sealed with grease (Apiezon) and covered with coverslips. Imaging was performed using a wide-field system (xcellence IX81; Olympus) equipped with a Plan-Apochromat 100×/1.45 NA oil objective lens, a camera (Orca-R2; Hamamatsu), and appropriate filter settings (dual-band CFP/YFP sbx HC and GFP/mCherry sbx ET filter sets). For yeast imaging, z series were taken with step sizes of 0.25–0.35 µm. Images were restored by Wiener filtering (except images in Fig. S4, C and D) using the Olympus deconvolution software.
Confocal images for selected samples were acquired using a confocal microscope system (LSM 780; Carl Zeiss) equipped with a Plan-Apochromat 100×/1.40 NA oil objective lens. YFP, Alexa Fluor 568/mCherry, and CFP were excited with 514 nm, 561 nm, and 405 nm laser lines, respectively. Two-color 3D imaging was performed with 0.3 µm step sizes along the z axis, and corresponding planes for both channels and the merged images are shown. Confocal settings were chosen to allow for clear spectral separation of the simultaneously visualized fluorophore pairs (YFP/CFP, CFP/mCherry, and YFP/Alexa Fluor 568 nm).
For time-lapse experiments, yeast cells were immobilized on Concanavalin A–coated glass-bottom culture dishes (MatTek Corporation) filled with PBS. Temperature was kept at 37°C by an incubation chamber attached to a temperature control unit (PeCon). For image analysis, ImageJ software was used. Quantification of polar and cytosolic fluorescence intensities for E. coli were calculated from 20 individual cells using the following formula: percentage polar fluorescence = (fluorescence of the ROI − background)/(fluorescence of the whole cell background). For yeast time-lapse experiments, foci intensities (maximum projections) and corresponding background intensities of 35–45 cells were measured over a time period of 90 min and analyzed with ImageJ. Colocalization of protein aggregates and chaperones was confirmed by line intensity plots using the ImageJ macro RGB profile tool. No manipulations other than contrast and brightness adjustments for images were applied.
FLIP experiments
For FLIP experiments, yeast cells were immobilized on Concanavalin A–coated glass-bottom culture dishes (MatTek Corporation) filled with PBS. The dishes were placed into an integrated temperature control chamber (HT200; BioDiagnostics, Inc.) to keep the temperature at 25°C or 37°C, respectively. A confocal microscope system (A1R; Nikon) equipped with a 60×/1.40 NA oil objective lens and 407-nm and 488-nm laser lines was used for CFP and YFP image acquisition and photobleaching. Laser and filter settings allowed for simultaneous imaging of both channels. An area of 1 µm2 was bleached at a rate of 40 iterations per 2 s over 90 s. Laser intensities for bleaching were set to 5% for the 407 nm and 8% for the 488 nm. Laser settings for imaging were set to 0.5% for the 488 nm laser and 3% for the 407 nm laser. Quantification of FLIP experiments was performed using the NIS-Elements software (Nikon).
FLIP experiments with SSA1 and ssa1-45(ts) cells expressing Sup35-GFP and Hsp104-eqFP611 were performed using a confocal microscope (LSM 780; Carl Zeiss) equipped with a 63×/1.40 NA oil objective lens and 488-nm and 561-nm laser lines for GFP and eqFP611 image acquisition and photobleaching. Samples for FLIP experiments were prepared as described. Laser and filter settings allowed for simultaneous imaging of both channels. A defined area was bleached 150 times for 230 s. Laser intensities for bleaching were set to 100% for the 488- and the 561-nm laser. Laser settings for imaging were set to 1% for the 488-nm laser and 0.5% for the 561-nm laser. Quantification of FLIP experiments was performed using ZEN 2010 software (Carl Zeiss).
The fluorescence intensity of the background (Fbg), the region of interest (ROI) at a particular time point (Ft), and the fluorescence intensity before the first bleach (F0) was determined. To calculate the loss of fluorescence at a particular time point, the formula (Ft − Fbg)/(F0 − Fbg) × 100 was used. Curves represent the mean of 15–25 cells and the corresponding standard error. In case of Sup35-GFP and Hsp104-eqFP611, curves were additionally corrected for bleaching. Therefore, for each strain and fluorophore, the fluorescence intensities of neighboring, nonbleached cells was determined and bleaching curves were normalized accordingly.
Immunofluorescence
Immunofluorescence was performed essentially as described previously (Specht et al., 2011). Spheroplasts were generated by the addition of 200 µg/ml Zymolase T100 and incubation at 30°C for 30 min. Cells were attached to poly-l-lysine–coated slides and blocked for 1 h with 1% Triton X-100 in PBS-BSA (1% BSA) in a humid chamber. Incubation with Ssa1 antibody (1:1,000 dilution) or affinity-purified Hsp104 antibody (1:2,000) and the secondary antibody Alexa Fluor 568 (anti–rabbit, 1:2,000 dilution; Molecular Probes) was performed in a humid chamber with PBS containing 0.1% Triton X-100. Incubation of yeast cells with the secondary antibody alone in YWO0625/YWO0622 or in combination with the Hsp104 antibody in BY4741 hsp104Δ cells did not result in any significant fluorescence signal. Preincubation of the Ssa1 antibody with 50 µg/ml purified Ssa1 protein for 1 h at room temperature abolished the fluorescence signal, demonstrating the specificity of the primary antibody. Samples were embedded in PBS with 50% glycerol before imaging.
Online supplemental material
Fig. S1 shows the dependencies of Hsp70 and Hsp100 chaperones during aggregate binding on Hsp40 and Hsp70 proteins, respectively. Fig. S2 (A, B, E, F, and I–K) shows that fluorescent prion fusion proteins retain crucial prion characteristics. Fig. S2 (C and D) shows that S. cerevisiae ssa1-45(ts) cells do not display altered Ssa1 and Hsp104 levels or cell viabilities under the chosen experimental conditions. Fig. S2 (G and H) shows that the dynamics of Hsp104-eqFP611 and Sup35-GFP foci are reduced in ssa1-45(ts) cells at 37°C. Fig. S3 shows and validates colocalizations of Ssa1 and Hsp104 with prion model proteins. Fig. S4 shows loss of Ssa1-45 colocalization with prion proteins at nonpermissive temperatures. Fig. S5 A shows colocalization of Hsp104 with NM-YFP by immunofluorescence. Fig. S5 (B–D) shows that overproduction of full-length Hsp104 specifically cures NM-YFP in S. cerevisiae wild-type cells. Tables S1–S4 show E. coli and S. cerevisiae plasmids and strains used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201201074/DC1.
Supplementary Material
Acknowledgments
We are grateful to D. Wolf for providing SSA1 and ssa1-45(ts) strains and T.R. Serio for providing the Sup35-GFP plasmid. We thank the Nikon Imaging Center at Bioquant Heidelberg and the Zentrum für Molekulare Biologie der Universität Heidelberg Imaging facility for use of microscopes and technical support. We are grateful to L. Guilbride, Y. Oguchi, and F. Seyffer for critical reading of the manuscript.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Bu617/17-1) to B. Bukau and A. Mogk and by grants from the Bundesministerium für Bildung und Forschung (AgeNet) to B. Bukau. J. Winkler was supported by the Network Aging Research Heidelberg.
Footnotes
Abbreviations used in this paper:
- CFP
- cerulean fluorescent protein
- FLIP
- fluorescence loss in photobleaching
- GdnHCl
- guanidine hydrochloride
- SDD-AGE
- semi-denaturating detergent agarose gel electrophoresis
References
- Acebrón S.P., Fernández-Sáiz V., Taneva S.G., Moro F., Muga A. 2008. DnaJ recruits DnaK to protein aggregates. J. Biol. Chem. 283:1381–1390 10.1074/jbc.M706189200 [DOI] [PubMed] [Google Scholar]
- Acebrón S.P., Martín I., del Castillo U., Moro F., Muga A. 2009. DnaK-mediated association of ClpB to protein aggregates. A bichaperone network at the aggregate surface. FEBS Lett. 583:2991–2996 10.1016/j.febslet.2009.08.020 [DOI] [PubMed] [Google Scholar]
- Alberti S., Halfmann R., King O., Kapila A., Lindquist S. 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 137:146–158 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron R., Higurashi T., Sahi C., Craig E.A. 2007. J-protein co-chaperone Sis1 required for generation of [RNQ+] seeds necessary for prion propagation. EMBO J. 26:3794–3803 10.1038/sj.emboj.7601811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev S.N., Gracheva E.O., Richmond J.E., Liebman S.W. 2008. Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol. Biol. Cell. 19:2433–2443 10.1091/mbc.E08-01-0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett M.E., Nagy M., Kedzierska S., Zolkiewski M. 2005. The amino-terminal domain of ClpB supports binding to strongly aggregated proteins. J. Biol. Chem. 280:34940–34945 10.1074/jbc.M505653200 [DOI] [PubMed] [Google Scholar]
- Becker J., Walter W., Yan W., Craig E.A. 1996. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol. Cell. Biol. 16:4378–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashikar A.G., Schirmer E.C., Hattendorf D.A., Glover J.R., Ramakrishnan M.S., Ware D.M., Lindquist S.L. 2002. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol. Cell. 9:751–760 10.1016/S1097-2765(02)00499-9 [DOI] [PubMed] [Google Scholar]
- Chernoff Y.O., Lindquist S.L., Ono B.-i., Inge-Vechtomov S.G., Liebman S.W. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science. 268:880–884 10.1126/science.7754373 [DOI] [PubMed] [Google Scholar]
- Derkatch I.L., Bradley M.E., Zhou P., Chernoff Y.O., Liebman S.W. 1997. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 147:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis M.E., Shorter J. 2012. The elusive middle domain of Hsp104 and ClpB: location and function. Biochim. Biophys. Acta. 1823:29–39 10.1016/j.bbamcr.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle S.M., Hoskins J.R., Wickner S. 2007a. Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proc. Natl. Acad. Sci. USA. 104:11138–11144 10.1073/pnas.0703980104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle S.M., Shorter J., Zolkiewski M., Hoskins J.R., Lindquist S., Wickner S. 2007b. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat. Struct. Mol. Biol. 14:114–122 10.1038/nsmb1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira P.C., Ness F., Edwards S.R., Cox B.S., Tuite M.F. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 40:1357–1369 10.1046/j.1365-2958.2001.02478.x [DOI] [PubMed] [Google Scholar]
- Ganusova E.E., Ozolins L.N., Bhagat S., Newnam G.P., Wegrzyn R.D., Sherman M.Y., Chernoff Y.O. 2006. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol. Cell. Biol. 26:617–629 10.1128/MCB.26.2.617-629.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.R., Lindquist S. 1998. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 94:73–82 10.1016/S0092-8674(00)81223-4 [DOI] [PubMed] [Google Scholar]
- Grimminger V., Richter K., Imhof A., Buchner J., Walter S. 2004. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J. Biol. Chem. 279:7378–7383 10.1074/jbc.M312403200 [DOI] [PubMed] [Google Scholar]
- Gur E., Biran D., Gazit E., Ron E.Z. 2002. In vivo aggregation of a single enzyme limits growth of Escherichia coli at elevated temperatures. Mol. Microbiol. 46:1391–1397 10.1046/j.1365-2958.2002.03257.x [DOI] [PubMed] [Google Scholar]
- Haslberger T., Weibezahn J., Zahn R., Lee S., Tsai F.T., Bukau B., Mogk A. 2007. M domains couple the ClpB threading motor with the DnaK chaperone activity. Mol. Cell. 25:247–260 10.1016/j.molcel.2006.11.008 [DOI] [PubMed] [Google Scholar]
- Helsen C.W., Glover J.R. 2012. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J. Biol. Chem. 287:542–556 10.1074/jbc.M111.302869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi T., Hines J.K., Sahi C., Aron R., Craig E.A. 2008. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. USA. 105:16596–16601 10.1073/pnas.0808934105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung G.C., Masison D.C. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics. 173:611–620 10.1534/genetics.106.056820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y., Taguchi H., Kishimoto A., Yoshida M. 2004. Hsp104 binds to yeast Sup35 prion fiber but needs other factor(s) to sever it. J. Biol. Chem. 279:52319–52323 10.1074/jbc.M408159200 [DOI] [PubMed] [Google Scholar]
- Janke C., Magiera M.M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., Knop M. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 21:947–962 10.1002/yea.1142 [DOI] [PubMed] [Google Scholar]
- Jung G., Masison D.C. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr. Microbiol. 43:7–10 10.1007/s002840010251 [DOI] [PubMed] [Google Scholar]
- Kawai-Noma S., Ayano S., Pack C.G., Kinjo M., Yoshida M., Yasuda K., Taguchi H. 2006. Dynamics of yeast prion aggregates in single living cells. Genes Cells. 11:1085–1096 10.1111/j.1365-2443.2006.01004.x [DOI] [PubMed] [Google Scholar]
- Kawai-Noma S., Pack C.G., Tsuji T., Kinjo M., Taguchi H. 2009. Single mother-daughter pair analysis to clarify the diffusion properties of yeast prion Sup35 in guanidine-HCl-treated [PSI] cells. Genes Cells. 14:1045–1054 10.1111/j.1365-2443.2009.01333.x [DOI] [PubMed] [Google Scholar]
- Krammer C., Kryndushkin D., Suhre M.H., Kremmer E., Hofmann A., Pfeifer A., Scheibel T., Wickner R.B., Schätzl H.M., Vorberg I. 2009. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc. Natl. Acad. Sci. USA. 106:462–467 10.1073/pnas.0811571106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin D.S., Alexandrov I.M., Ter-Avanesyan M.D., Kushnirov V.V. 2003. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278:49636–49643 10.1074/jbc.M307996200 [DOI] [PubMed] [Google Scholar]
- Krzewska J., Melki R. 2006. Molecular chaperones and the assembly of the prion Sup35p, an in vitro study. EMBO J. 25:822–833 10.1038/sj.emboj.7600985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzewska J., Langer T., Liberek K. 2001. Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 489:92–96 10.1016/S0014-5793(00)02423-6 [DOI] [PubMed] [Google Scholar]
- Laufen T., Mayer M.P., Beisel C., Klostermeier D., Mogk A., Reinstein J., Bukau B. 1999. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA. 96:5452–5457 10.1073/pnas.96.10.5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum R., Tkach J.M., Vierling E., Glover J.R. 2004. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem. 279:29139–29146 10.1074/jbc.M403777200 [DOI] [PubMed] [Google Scholar]
- Masison D.C., Kirkland P.A., Sharma D. 2009. Influence of Hsp70s and their regulators on yeast prion propagation. Prion. 3:65–73 10.4161/pri.3.2.9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur V., Taneja V., Sun Y., Liebman S.W. 2010. Analyzing the birth and propagation of two distinct prions, [PSI+] and [Het-s](y), in yeast. Mol. Biol. Cell. 21:1449–1461 10.1091/mbc.E09-11-0927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.P., Schröder H., Rüdiger S., Paal K., Laufen T., Bukau B. 2000. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7:586–593 10.1038/76819 [DOI] [PubMed] [Google Scholar]
- Miot M., Reidy M., Doyle S.M., Hoskins J.R., Johnston D.M., Genest O., Vitery M.C., Masison D.C., Wickner S. 2011. Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc. Natl. Acad. Sci. USA. 108:6915–6920 10.1073/pnas.1102828108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosavi B., Wongwigkarn J., Tuite M.F. 2010. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI(+)] prion but not for prion propagation. Yeast. 27:167–179 [DOI] [PubMed] [Google Scholar]
- Moriyama H., Edskes H.K., Wickner R.B. 2000. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell. Biol. 20:8916–8922 10.1128/MCB.20.23.8916-8922.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy M., Guenther I., Akoyev V., Barnett M.E., Zavodszky M.I., Kedzierska-Mieszkowska S., Zolkiewski M. 2010. Synergistic cooperation between two ClpB isoforms in aggregate reactivation. J. Mol. Biol. 396:697–707 10.1016/j.jmb.2009.11.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newnam G.P., Wegrzyn R.D., Lindquist S.L., Chernoff Y.O. 1999. Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol. 19:1325–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicki L., Leźnicki P., Morawiec E., Litwińczuk N., Liberek K. 2012. Role of a conserved aspartic acid in nucleotide binding domain 1 (NBD1) of Hsp100 chaperones in their activities. Cell Stress Chaperones. 17:361–373 10.1007/s12192-011-0312-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell D.A., Taulien J., Lindquist S. 1993. The role of heat-shock proteins in thermotolerance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 339:279–285, discussion :285–286 10.1098/rstb.1993.0026 [DOI] [PubMed] [Google Scholar]
- Patino M.M., Liu J.-J., Glover J.R., Lindquist S. 1996. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 273:622–626 10.1126/science.273.5275.622 [DOI] [PubMed] [Google Scholar]
- Pezza J.A., Serio T.R. 2007. Prion propagation: the role of protein dynamics. Prion. 1:36–43 10.4161/pri.1.1.3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy M., Masison D.C. 2010. Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104. Mol. Cell. Biol. 30:3542–3552 10.1128/MCB.01292-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanova N.V., Chernoff Y.O. 2009. Hsp104 and prion propagation. Protein Pept. Lett. 16:598–605 10.2174/092986609788490078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y., Lindquist S.L. 1990. HSP104 required for induced thermotolerance. Science. 248:1112–1115 10.1126/science.2188365 [DOI] [PubMed] [Google Scholar]
- Satpute-Krishnan P., Langseth S.X., Serio T.R. 2007. Hsp104-dependent remodeling of prion complexes mediates protein-only inheritance. PLoS Biol. 5:e24 10.1371/journal.pbio.0050024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer E.C., Homann O.R., Kowal A.S., Lindquist S. 2004. Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol. Biol. Cell. 15:2061–2072 10.1091/mbc.E02-08-0502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee S., Beinker P., Akhrymuk A., Reinstein J. 2004. A chaperone network for the resolubilization of protein aggregates: direct interaction of ClpB and DnaK. J. Mol. Biol. 336:275–285 10.1016/j.jmb.2003.12.013 [DOI] [PubMed] [Google Scholar]
- Schlieker C., Tews I., Bukau B., Mogk A. 2004. Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett. 578:351–356 10.1016/j.febslet.2004.11.051 [DOI] [PubMed] [Google Scholar]
- Schröder H., Langer T., Hartl F.-U., Bukau B. 1993. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 12:4137–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J., Lindquist S. 2004. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 304:1793–1797 10.1126/science.1098007 [DOI] [PubMed] [Google Scholar]
- Shorter J., Lindquist S. 2005. Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 6:435–450 10.1038/nrg1616 [DOI] [PubMed] [Google Scholar]
- Shorter J., Lindquist S. 2006. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol. Cell. 23:425–438 10.1016/j.molcel.2006.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sielaff B., Tsai F.T. 2010. The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J. Mol. Biol. 402:30–37 10.1016/j.jmb.2010.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sielaff B., Lee K.S., Tsai F.T. 2010. Crystallization and preliminary X-ray crystallographic analysis of a GroEL1 fragment from Mycobacterium tuberculosis H37Rv. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66:418–420 10.1107/S1744309110004409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N., Lindquist S. 2000. Rnq1: an epigenetic modifier of protein function in yeast. Mol. Cell. 5:163–172 10.1016/S1097-2765(00)80412-8 [DOI] [PubMed] [Google Scholar]
- Sondheimer N., Lopez N., Craig E.A., Lindquist S. 2001. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 20:2435–2442 10.1093/emboj/20.10.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht S., Miller S.B., Mogk A., Bukau B. 2011. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J. Cell Biol. 195:617–629 10.1083/jcb.201106037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J., Cegielska A., Georgopoulos C. 1990. Role of Escherichia coli heat shock proteins DnaK and HtpG (C62.5) in response to nutritional deprivation. J. Bacteriol. 172:7157–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires C.L., Pedersen S., Ross B.M., Squires C. 1991. ClpB is the Escherichia coli heat shock protein F84.1. J. Bacteriol. 173:4254–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P., Mogk A., Bukau B. 2008. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol. Microbiol. 68:87–97 10.1111/j.1365-2958.2008.06135.x [DOI] [PubMed] [Google Scholar]
- Tipton K.A., Verges K.J., Weissman J.S. 2008. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol. Cell. 32:584–591 10.1016/j.molcel.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite M.F., Cox B.S. 2003. Propagation of yeast prions. Nat. Rev. Mol. Cell Biol. 4:878–890 10.1038/nrm1247 [DOI] [PubMed] [Google Scholar]
- Tyedmers J., Treusch S., Dong J., McCaffery J.M., Bevis B., Lindquist S. 2010. Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation. Proc. Natl. Acad. Sci. USA. 107:8633–8638 10.1073/pnas.1003895107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat A., Alberti-Segui C., Rebischung C., Philippsen P. 1997. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast. 13:1065–1075 [DOI] [PubMed] [Google Scholar]
- Weibezahn J., Schlieker C., Bukau B., Mogk A. 2003. Characterization of a trap mutant of the AAA+ chaperone ClpB. J. Biol. Chem. 278:32608–32617 10.1074/jbc.M303653200 [DOI] [PubMed] [Google Scholar]
- Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E.U., Dougan D.A., Tsai F.T., et al. 2004. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 119:653–665 10.1016/j.cell.2004.11.027 [DOI] [PubMed] [Google Scholar]
- Winkler J., Seybert A., König L., Pruggnaller S., Haselmann U., Sourjik V., Weiss M., Frangakis A.S., Mogk A., Bukau B. 2010. Quantitative and spatio-temporal features of protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J. 29:910–923 10.1038/emboj.2009.412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.X., Masison D.C., Eisenberg E., Greene L.E. 2006. Application of photobleaching for measuring diffusion of prion proteins in cytosol of yeast cells. Methods. 39:43–49 10.1016/j.ymeth.2006.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Derkatch I.L., Liebman S.W. 2001. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI(+)] and [PIN(+)]. Mol. Microbiol. 39:37–46 (PIN(+) 10.1046/j.1365-2958.2001.02224.x [DOI] [PubMed] [Google Scholar]
- Zietkiewicz S., Krzewska J., Liberek K. 2004. Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J. Biol. Chem. 279:44376–44383 10.1074/jbc.M402405200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.