

Inactivating ADP ribosylation of the ER chaperone BiP is a rapidly reversible mechanism for buffering acute changes in unfolded protein load.
Abstract
Gene expression programs that regulate the abundance of the chaperone BiP adapt the endoplasmic reticulum (ER) to unfolded protein load. However, such programs are slow compared with physiological fluctuations in secreted protein synthesis. While searching for mechanisms that fill this temporal gap in coping with ER stress, we found elevated levels of adenosine diphosphate (ADP)–ribosylated BiP in the inactive pancreas of fasted mice and a rapid decline in this modification in the active fed state. ADP ribosylation mapped to Arg470 and Arg492 in the substrate-binding domain of hamster BiP. Mutations that mimic the negative charge of ADP-ribose destabilized substrate binding and interfered with interdomain allosteric coupling, marking ADP ribosylation as a rapid posttranslational mechanism for reversible inactivation of BiP. A kinetic model showed that buffering fluctuations in unfolded protein load with a recruitable pool of inactive chaperone is an efficient strategy to minimize both aggregation and costly degradation of unfolded proteins.
Introduction
Biogenesis of secreted proteins initiates in the lumen of the ER. There, a host of enzymes and chaperones assist in the maturation and folding of client polypeptides into functional proteins (Sitia and Braakman, 2003). Adaptive mechanisms referred to collectively as the ER unfolded protein response (UPRer) match the ER capacity to the load of unfolded proteins. Increased unfolded protein load is met by attenuated protein synthesis and enhanced production of ER chaperones (Walter and Ron, 2011).
Chaperones of the DnaK class are among the oldest and most ubiquitous elements used by cells to cope with unfolded proteins. These abundant, bipartite proteins reversibly bind extended hydrophobic segments of unfolded polypeptide chains, shielding them from aggregation. Cycles of ATP binding, hydrolysis, and nucleotide exchange are coupled to substrate binding and release (Bukau et al., 2006). By exploiting differences in the concentration dependence of the protein folding and protein aggregation processes, chaperones of the DnaK class can promote protein folding despite the fact that they stabilize the unfolded state. Because their utility is critically dependent on the balance of chaperone and client polypeptide concentrations, the expression of DnaK class chaperones is tightly regulated at the transcriptional level by compartment-specific UPRs (Balch et al., 2008).
BiP is the DnaK class chaperone of the ER lumen (Munro and Pelham, 1986), and its expression is transcriptionally up-regulated by the UPRer (Kozutsumi et al., 1988). As this process has a latency of several hours, it has yet to be determined how cells respond to physiological fluctuations in the rate of secretory protein translation, which often occur over a shorter timescale (Itoh and Okamoto, 1980; Logothetopoulos and Jain, 1980). Furthermore, BiP has a long half-life, up to 48 h (Hendershot et al., 1988), posing the question how the ER copes with physiological declines in unfolded protein load. The significance of this problem is highlighted by experimental evidence that excess BiP or other DnaK-type chaperones are deleterious to protein secretion (Dorner et al., 1992) and cell fitness (Feder et al., 1992). Such studies suggest that excess chaperone may stabilize the unfolded state to a degree that becomes limiting to protein maturation, attaching a price tag to protection from aggregation.
A posttranslational mechanism regulating the activity of BiP could help solve this problem. An initial clue was provided by the observation that BiP undergoes ADP ribosylation (Carlsson and Lazarides, 1983; Hendershot et al., 1988; Leno and Ledford, 1989) and that amino acid starvation and protein synthesis inhibitors, which lower the flux of unfolded proteins into the ER, increase ADP ribosylation of BiP (Ledford and Jacobs, 1986), whereas manipulations that enhance the burden of unfolded proteins in the ER result in lower levels of ADP-ribosylated BiP (Hendershot et al., 1988; Leno and Ledford, 1989; Laitusis et al., 1999). These observations have led to the suggestion that ADP ribosylation may play an important role in the short-term regulation of the activity of BiP (Freiden et al., 1992). Despite the appeal of such an idea, its molecular basis has remained obscure.
Here, we provide evidence for ADP ribosylation on highly conserved residues in the substrate-binding domain of BiP. Functional analysis of the modified residues reveals that ADP ribosylation attenuates substrate binding and interferes with allosteric coupling of BiP’s two domains in the setting of declining unfolded protein load. A kinetic model reveals the potential utility of this posttranslational fast-response mechanism for regulating the activity of BiP, which serves alongside the well-studied gene expression programs of the UPRer to maintain protein-folding homeostasis.
Results
Physiological regulation of levels of ADP-ribosylated BiP
It is well established that pharmacological manipulation of unfolded protein load on the ER profoundly affects levels of ADP-ribosylated BiP (Hendershot et al., 1988; Leno and Ledford, 1989; Laitusis et al., 1999), but the extent to which this modification occurs under normal physiological conditions has so far remained unknown in detail. To address this issue, we exploited the fact that the ribosylated and nonribosylated forms of BiP can be resolved by isoelectric focusing (IEF) followed by immunoblotting (Laitusis et al., 1999). Exposure of GH3 cells (a secretory pituitary line) to the protein synthesis inhibitor cycloheximide resulted in accumulation of acidic BiP, characteristic of the ADP ribosylated form (Carlsson and Lazarides, 1983), whereas thapsigargin, an agent that promotes unfolded protein stress in the ER, led to disappearance of this species (Fig. 1 A), as previously noted (Laitusis et al., 1999).
Figure 1.
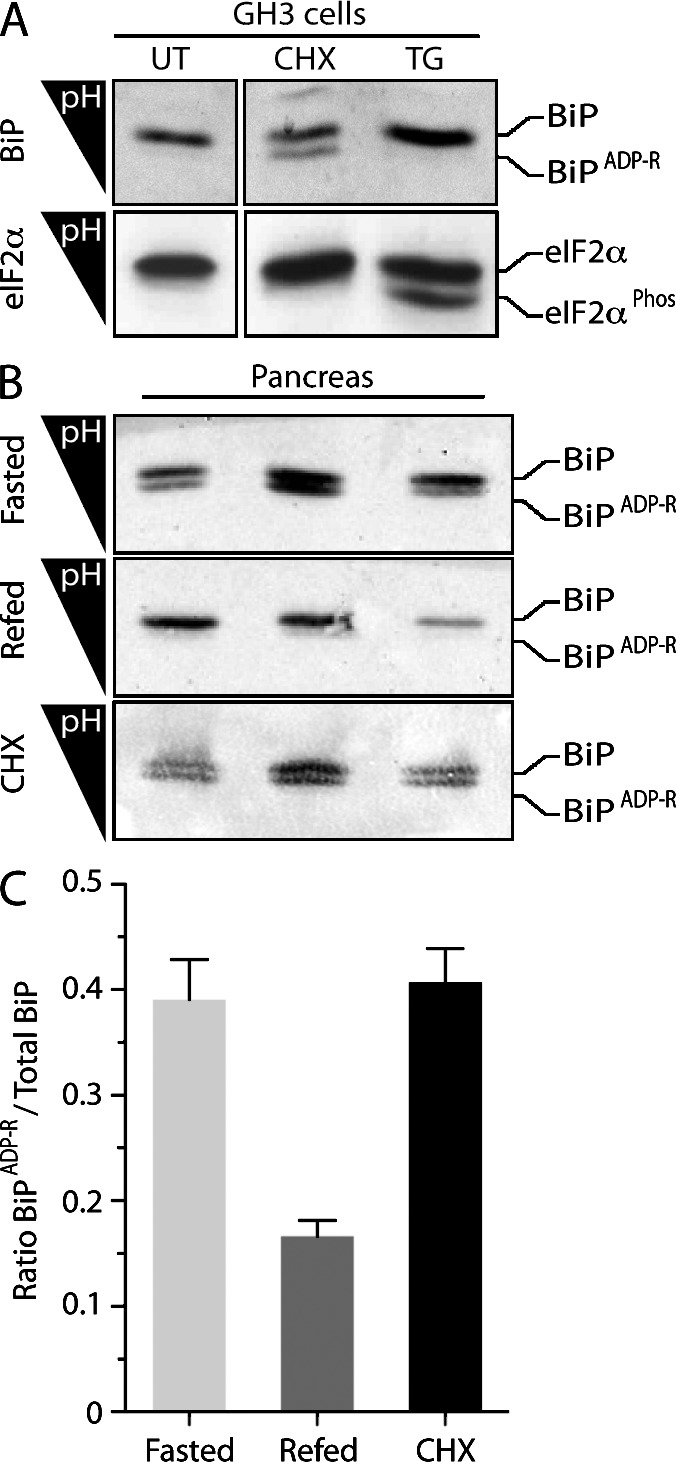
ADP ribosylation of BiP correlates with physiological activity of the ER. (A) Immunoblot of lysates from untreated GH3 cells (UT) and cells exposed to the protein synthesis inhibitor cycloheximide (CHX; 50 µg/ml) or the ER stress–causing agent thapsigargin (TG; 300 nM). The acidic, ADP-ribosylated form of BiP is resolved from the unmodified form by IEF (top). Phosphorylated eIF2α (eIF2αPhos), resolved on the same gel, serves as a marker for ER stress (bottom). The experiment shown is representative of three repeats. (B) Immunoblot of IEF gels of BiP solubilized from pancreatic microsomes of individual fasted mice (top), fasted mice 1 h after refeeding (middle), and fed mice 2 h after injection with cycloheximide (100 mg/kg; bottom). (C) Quantitation of the ratio of modified to total BiP separated by IEF in the triplicate samples displayed in B. Error bars show SEM.
Secreted protein synthesis in the pancreas is low in fasted animals and is stimulated by refeeding (Morisset and Webster, 1972; Itoh and Okamoto, 1980). IEF of protein extracts from detergent-solubilized pancreatic membranes showed the presence of acidic, ADP-ribosylated BiP in samples obtained from fasted animals and its rapid disappearance upon refeeding. Injection of cycloheximide led to reemergence of the acid form in fed animals, recapitulating the process observed in cultured cells (Fig. 1, B and C). In addition to ADP ribosylation, BiP has been reported to also undergo phosphorylation (Hendershot et al., 1988); however, prolonged exposure of BiP isolated from mouse pancreas to λ-phosphatase had no effect on its mobility on IEF, arguing against a role for phosphorylation in regulating the activity of BiP (Fig. S1).
BiP is believed to function as part of a high–molecular weight multichaperone complex (Meunier et al., 2002). Size-exclusion chromatography of proteins in detergent lysates of pancreatic membranes showed that BiP is distributed in many fractions (Fig. 2 A). Modified (acidic) BiP was enriched in the lower–molecular weight fractions, whereas BiP from the high–molecular weight, active multichaperone complex (Meunier et al., 2002) was relatively depleted of the modified form (Fig. 2 B). These observations suggested a correlation between physiological fluctuations in the functional state of BiP and its modification, prompting us to identify the modified residues.
Figure 2.

ADP-ribosylated BiP is enriched in high–molecular weight fractions of the ER extract. (A) Coomassie-stained SDS-PAGE of pancreatic microsomal proteins from fasted and fed mice resolved by gel filtration. BiP is distributed bimodally between a high–molecular weight peak containing the cochaperone ORP150 and GRP94 and lower–molecular weight peak. A representative experiment reproduced three times is shown. Abs., absorbance; mAU, milli–absorbance unit. (B) BiP immunoblot of IEF gels of the input (GF input) and fractions 9 and 14 of the fasted and 1-h refed samples above. The ratio of ADP ribosylated to total BiP in each lane is indicated (R). Black lines indicate that intervening lanes have been spliced out.
Mapping ADP-ribosylated residues in BiP
Mature hamster BiP was modified to contain a signal sequence, which upon cleavage exposes an N-terminal FLAG-M1 epitope tag (Petrova et al., 2008). FLAG-tagged BiP was immunopurified from 293T cells metabolically labeled with 32P-orthophosphate or 3H-adenosine; the latter selectively labels adenylated and ADP-ribosylated species (Fig. 3, A and B). Incorporation of label was increased by cycloheximide and decreased by novobiocin, a nonselective inhibitor of ADP ribosylation. MG132, a proteasome inhibitor, had no major effect on the modification (Fig. 3 A), suggesting that modification does not mark BiP for degradation. These observations on recombinant BiP mirrored those previously made on the endogenous protein (Carlsson and Lazarides, 1983; Hendershot et al., 1988; Leno and Ledford, 1989). Persistence of the labeled species upon treatment with λ-phosphatase or calf intestinal phosphatase (CIP) further implicated ADP ribosylation over phosphorylation (Fig. S2 A, second and fourth lanes); however, our attempts to remove the phosphoadenosine of the modification with snake venom phosphodiesterase were thwarted by a protease contamination of all commercial preparations of the enzyme we could find (Fig. S2 A, third, fifth, and sixth lanes). Hydroxylamine resistance of labeling (Fig. S2 A, seventh lane) suggests modification of Arg or Cys residues rather than glutamate, as the latter forms a labile carboxylate ester bond to the ADP-ribose moiety (Hsia et al., 1985).
Figure 3.

ADP ribosylation of the substrate-binding domain of BiP. (A, top) Autoradiograph (autorad) of 32P-labeled FLAG-tagged BiP immunopurified from untreated 32P-orthophosphate–labeled 293T cells (UT), cells exposed to the protein synthesis inhibitor cycloheximide (CHX; 50 µg/ml, 30 min) in the absence or presence of the proteasome inhibitor MG132 (CHX MG; 10 µM), or novobiocin, an inhibitor of ADP ribosylation (NB; 0.5 mM). (bottom) Coomassie stain of the same gel. 32P-radioactive counts (after background subtraction) and counts normalized to BiP protein content in each sample (R) are indicated. (B) Coomassie-stained gel of 3H-labeled FLAG-tagged BiP immunopurified from untreated 3H-adenosine–labeled 293T cells (UT) and novobiocin-treated cells. 3H counts from the excised BiP bands and values normalized to protein content (R) are indicated. (C) Coomassie stain (lanes 1, 2, 5, and 6) and autoradiography (lanes 3 and 4) of a tris-tricine SDS-PAGE with cyanogen bromide cleaved FLAG-tagged BiP immunopurified from metabolically labeled 293T cells (as in A and B). The bar diagram on the right depicts the 3H signal from the corresponding gel slice. The cyanogen bromide bands are numbered (I–VI), and their identity is indicated. H6-BiP denotes bacterially expressed hamster BiP provided as a standard. The 32P- and 3H-adenosine label is incorporated into two cyanogen bromide fragments (I and III) converging on the peptide sequence T434-M541 of hamster BiP. A representative experiment reproduced twice is shown. (D) The location of the labeled cyanogen bromide fragment T434-M541 is indicated (in gold) on a ribbon diagram of BiP’s substrate-binding domain modeled on E. coli DnaK (Protein Data Bank accession no. 1DKZ; rendered in MACPyMOL). Two of the six Arg residues on that fragment are conserved in all DnaK family members, and their side chains have been modeled. Note the incompatibility of the closed lid with the modification of these two residues with a bulky ADP-ribose. N, N terminus.
As we could not detect modified BiP peptides using mass spectrometry, an alternative course to identifying the modified residues was taken. Purified, metabolically labeled BiP was subjected to cyanogen bromide cleavage, and the resulting fragments were resolved on a tris-tricine gel. Autoradiography of the gel derived from the 32P-labeled sample revealed two labeled bands (I and III in Fig. 3 C). Importantly, the same two bands were labeled by 3H-adenosine (Fig. 3 C, rightmost graph). Matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry identified the bands as reflecting cleavage after Met339, Met433, and Met541. Band III corresponded to the completely cleaved Thr434-Met541 peptide, and band I corresponded to an incompletely cleaved fragment generated by skipped cleavage at the Met433-Thr434 bond. Thus, all detectable 32P- and 3H-adenosine labels in BiP were found on a single fragment emanating from the substrate-binding lobe of the chaperone (Fig. 3 D).
Whereas bacterial ADP ribosyltransferases (ARTs) are able to modify diverse substrates, the known mammalian mono-ARTs are believed to be selective for Arg residues (Koch-Nolte et al., 2008). The labeled Thr434-Met541 cyanogen bromide fragment is devoid of potentially modifiable Cys and contains two Arg residues that are conserved in all known members of the DnaK family. Mutation of Arg470 to Lys led to a nearly complete loss of labeling, whereas mutation of Arg492 to Lys led to a highly reproducible ∼50% reduction in labeling. The double mutant and the single Arg470 to Lys mutant were similarly affected (Fig. 4, A and B). Mutation of the remaining four Arg residues within this fragment had little or no effect on label incorporation when compared with wild-type BiP (Fig. S2 B). Furthermore, tryptic digestion followed by reverse-phase HPLC of labeled BiP yielded two 32P- and two 3H-adenosine–labeled fractions of indistinguishable retention time (Fig. 4, C–E). Together, these observations are consistent with the presence of labeling on both Arg470 and Arg492 and suggest that modification of Arg470 may be a prerequisite for modification of Arg492.
Figure 4.

Mutations in Arg470 and Arg492 inhibit ADP ribosylation of BiP. (A, top) Autoradiograph (autorad) of 32P-labeled wild type (WT) and the indicated FLAG-tagged mutant BiP immunopurified from 32P-orthophosphate–labeled 293T cells. 32P-radioactive counts (after background subtraction) and counts normalized to BiP protein content in each sample (R) are indicated. (bottom) Coomassie stain of the same gel. (B) Coomassie-stained gel of 3H-labeled FLAG-tagged wild type and the indicated mutant BiP immunopurified from 3H-adenosine–labeled 293T cells. 3H counts from the excised BiP bands and values normalized to protein content (R) are indicated. (C) Absorbance (Abs.) of peptides eluted from a tryptic in-gel digest of purified FLAG-tagged BiP from transfected 293T cells and resolved by reverse-phase HPLC on a C18 column. (D) 32P label profile of material eluted from a tryptic in-gel digest of purified FLAG-tagged BiP from transfected 32P-orthophosphate–labeled 293T cells resolved as in C. A typical profile reproduced three times is shown. (E) 3H label profile of material eluted from a tryptic in-gel digest of purified FLAG-tagged BiP from transfected 3H-adenosine–labeled 293T cells resolved as in C.
ADP ribosylation–mimetic charge substitutions destabilize substrate binding by BiP
Attempts to ADP ribosylate recombinant BiP in vitro either with crude ER extracts or purified cholera toxin were unsuccessful. As ADP ribosylation is predicted to modify the electrostatic properties of the substrate-binding domain of BiP, we substituted the relevant Arg residues with Glu. Although this mutation does not mimic the spatial or structural effects of ADP ribosylation, it introduces an acidic residue in place of the basic Arg and, like the acidic pyrophosphate of ADP-ribose, alters the electrostatics. Thus, it likely serves as a minimal estimate of the perturbation to BiP function imposed by ADP ribosylation. In the presence of ADP, wild-type BiP, purified from Escherichia coli, bound a fluorescently labeled substrate peptide with a measured affinity in the high micromolar range (kd = 13 µM), as previously described (Knarr et al., 2002; Marcinowski et al., 2011). Similarly, robust steady-state binding, reflected in a strong fluorescent polarization signal, was also observed for the BiPR470E mutant (kd = 9 µM; Fig. 5 A). BiPR492E and the BiPR470E;R492E double mutant, despite being properly folded, gave rise to very weak fluorescent polarization signals (Fig. 5 B).
Figure 5.

Charge-substitution mutations that mimic ADP ribosylation destabilize substrate binding by BiP. (A) Relative fluorescent anisotropy of Lucifer yellow–labeled BiP substrate peptide (NH2-HTFPAVLGSC-COOH) bound at steady state by the indicated concentration of wild type (WT) and mutant BiP in the presence of 1 mM ADP. The mean ± SEM of a measurement conducted in triplicate is shown. The highest anisotropy value for each protein (among the triplicates) was set at 100. (B) A comparison of the fluorescent anisotropy signal of the probe in A bound at steady state by the indicated concentration of wild type and mutant BiPs in the presence of 1 mM ADP (in arbitrary units [au]). Note the dramatically lower fluorescent anisotropy of the BiPR492E and BiPR470E;R492E double mutant (2E), which is similar to the signal obtained by the known substrate-binding mutant BiPV461F (Petrova et al., 2008). The mean ± SEM of a measurement conducted in triplicate is shown. (C) Fluorescent anisotropy signal of the BiP-bound–labeled peptide (as in A) after introduction of a 500-fold excess of unlabeled peptide at t = 0. The inset is a time zoom on the first 30 min of the competition. Lidless BiP lacks the C-terminal 100 residues (Δ554-654). koff: wild type = 0.012 min−1, Lidless = 0.102 min−1, R470E = 0.444 min−1, R470E Lidless = 0.197 min−1. A representative experiment reproduced three times is shown. (D) Single turnover ATP hydrolysis rates of the indicated BiP proteins in the absence (BiP alone) and the presence of substrate peptide (Pep; 30 µM) and J-domain (2 µM). BiPE201G has an ATPase mutation and is provided as a reference. The mean ± SEM of a measurement conducted in duplicate is shown. (E) Fluorescent anisotropy signal of the BiP-bound–labeled peptide (in the presence of 1 mM ADP, as in A) after introduction of an excess of ATP (4 mM) at t = 0. A representative experiment reproduced three times is shown.
Measureable steady-state substrate binding by BiPR470E enabled us to characterize further the impact of this ADP ribosylation charge–mimetic modification. Challenge of the BiP–fluorescently labeled peptide complex with excess unlabeled peptide provided an estimate of complex stability, when BiP is in its ADP-bound, high-affinity state. The rate of complex disassembly measured with the BiPR470E mutant was higher than that of wild-type BiP by a factor of 40 (Fig. 5 C), indicating that a charge substitution that mimics ADP ribosylation markedly destabilizes the chaperone–client complex.
Arg470 is conserved in all known DnaK proteins. In the crystal structure of the substrate-binding domain of E. coli DnaK, the corresponding residue, Arg445, is seen to make ionic contacts with Asp526 (Zhu et al., 1996), conserved as Asp552 in the lid domain of BiP (Fig. 3 D). Disruption of these contacts by charge-neutralizing mutations of the lid residue, DnaKD526A or DnaKD526C, markedly enhanced the dissociation of substrates from DnaK in vitro (Fernández-Sáiz et al., 2006; Schlecht et al., 2011), mirroring the effects of the BiPR470E ADP ribosylation–mimetic mutation here. Interestingly, the destabilizing effect of the BiPR470E mutation on substrate binding by BiP was more conspicuous than loss of the entire lid, whereas introduction of the BiPR470E mutation into the lidless background (BiPΔ(554–654);R470E) had only a modest further effect on binding (Fig. 5, B and C). In comparison, the lidless BiPΔ(554–654);R492E, like its full-length counterpart, had lost most detectable substrate binding (Fig. 5 B). Together, these findings suggest that the BiPR470E mutation affects substrate binding primarily by altering the disposition of the lid, whereas BiPR492E affects substrate binding by a different mechanism.
The structural integrity of the mutant BiPs is attested to by their normal circular dichroism (CD) spectra (Fig. S3 A). Furthermore, like other DnaK chaperones, BiP is an ATPase (Wei et al., 1995; Mayer et al., 2003). Single-turnover hydrolysis of prebound ATP and multiple-turnover hydrolysis were stimulated by substrate and by a DnaJ cofactor, as expected (Petrova et al., 2008), but were similar in the wild-type, BiPR470E, and BiPR492E mutants (Figs. 5 D and S3 B). In keeping with these observations, a mutation that disrupts the corresponding ionic interaction in DnaK (Asp526 to Ala) is also without effect on ATP hydrolysis (Fernández-Sáiz et al., 2006). Furthermore, addition of ATP to preformed wild-type or BiPR470E mutant chaperone–substrate complexes led to their rapid dissociation followed by gradual complex reformation at similar rates, as ATP was hydrolyzed (Fig. 5 E). Thus, the ADP ribosylation–mimetic mutations destabilized substrate binding without measurably affecting nucleotide hydrolysis or exchange, attesting to their functional integrity.
Upon exposure to peptide, BiPR470K and BiPR492K, with charge-conserving substitutions, gave anisotropy signals that were similar to the wild type and resulted in modest threefold-higher peptide offrates (Fig. S3, C and D). Thus, these in vitro peptide-binding experiments support the conclusion that the Arg to Glu substitutions exerted their effects on peptide binding by disrupting ionic interactions. Furthermore, these in vitro observations argue that the effect of the BiPR470K and BiPR492K mutations on ADP ribosylation in vivo reflected loss of ribosylation sites and not a structural perturbation that interferes with ADP ribosylation indirectly.
We were unable to purify sufficient quantities of modified BiP (away from the unmodified species) from mouse pancreas to measure the effect of the modification on substrate binding in vitro. Therefore, an alternative approach was taken to gauge the functional similarity of modified pancreatic BiP and bacterially expressed BiP bearing mutations that mimic ADP ribosylation. SubA is the effector of an enterotoxigenic E. coli that cleaves the interdomain linker of mouse BiP at L417-L418 with exquisite specificity (Paton et al., 2006). In DnaK-type chaperones, the disposition of the linker is affected by nucleotide- and substrate-mediated allosteric transitions (Liu and Hendrickson, 2007; Swain et al., 2007; Bertelsen et al., 2009), which correlate with the linker’s sensitivity to proteases (Buchberger et al., 1995). In the ATP-bound state, the linker was substantially protected from SubA, as predicted by structural studies (Liu and Hendrickson, 2007; Swain et al., 2007; Bertelsen et al., 2009); however, addition of peptide substrate to wild-type ATP-bound BiP markedly sensitized the linker to cleavage by purified SubA in vitro (Fig. 6 A). In contrast, the interdomain linker of BiPR470E and BiPR470; R492 mutant BiP remained substantially protected from the protease (Fig. 6, B and C), suggesting that the ADP ribosylation–mimetic mutations interfered with substrate-mediated allosteric coupling that alters linker accessibility.
Figure 6.

Both ADP ribosylation of the endogenous protein and its mimetic mutations interfere with interdomain allosteric coupling in BiP. (A) Coomassie-stained SDS-PAGE of bacterially expressed wild-type (WT) BiP (7.5 µM) exposed for the indicated time to the SubA protease (SubA Inc.; 0.6 µM; which cleaves the interdomain linker) in the presence of 2 mM ATP in the absence or presence of 0.5 mM substrate peptide. The nucleotide-binding (46 kD) and substrate-binding (27 kD) fragments of BiP produced by proteolysis are resolved. The experiments shown are representative of two repeats. (B) Same as A, with bacterially expressed R470E mutant BiP. (C) Same as A, with bacterially expressed R470E:R492E double mutant BiP. (D) Coomassie-stained gel with BiP purified from mouse pancreas or E. coli. (E) Coomassie-stained IEF gel of endogenous BiP purified from pancreas of cycloheximide-treated mice and exposed to SubA in the presence of ATP but in the absence of substrate peptide. The unmodified and ADP-ribosylated forms of BiP and the more basic nucleotide-binding fragment (BiP NBF) are indicated. The more acidic, substrate-binding fragment is not resolved on this gel. (F) Same as D but in the presence of 0.5 mM substrate peptide. (G) Immunoblot of an IEF gel of the samples shown in the first five lanes of E, probed sequentially with a rabbit serum raised to the N terminus (BiP; left) and a mouse monoclonal antibody to the C terminus (KDEL; right) of mouse BiP. Note the selective reactivity of the cleavage product (BiP NBF; at the top of the gel) with the N-terminal rabbit serum.
BiP was purified to homogeneity from the pancreas of cycloheximide-treated mice (Fig. 6 D) and was roughly equally distributed between the basic, unmodified, and acidic modified forms. A time course of exposure to SubA, followed by IEF, revealed that in the presence of ATP, the linker of both the modified and unmodified BiP resisted digestion (Fig. 6 E). Addition of peptide selectively sensitized the unmodified BiP to SubA, whereas the modified form, like the bacterially expressed BiPR470E and BiPR470;R492 mutant, retained its resistance (Fig. 6 F). The identity of the BiP species is revealed by the immunoblot of the IEF gel (Fig. 6 G). The molecular basis for the effect of the modifications and mutations on the allosteric transitions reflected in sensitivity to SubA remains to be explained. Nonetheless, the concordant behavior of the endogenous modified BiP and the bacterially expressed mutant BiP in this assay indicates that the mutations capture an important aspect of the functional consequences of the naturally occurring modifications and suggests that ADP ribosylation substantially inactivates the chaperone.
Reversible inactivation of BiP enhances the ability of cells to cope with fluctuations in unfolded protein load
To gain insight into the significance of ADP ribosylation of BiP, a kinetic model was used to contrast a cell that responds to fluctuations in unfolded protein load solely by changes in the rate of BiP synthesis (ADPr−) with a cell that can also draw on a pool of inactive, ADP-ribosylated BiP to buffer changes in load (ADPr+; Fig. 7 A and Table 1). The protein-folding environment in the ER of a hypothetical secretory cell that cycles diurnally between low- and high-synthesis periods (exemplified by the pancreas of a fasted and fed animal) was simulated using each model (translational control is not relevant to this comparison, as it acts upstream of the fluctuation in unfolded protein load). In both models, the concentration of ER constituents (BiP, ADP-ribosylated BiP, unfolded protein, and complexes thereof) and outputs (folded, misfolded, and degraded protein) converged on a periodic steady state that was independent of the initial conditions (Fig. 7 [B–D] and Table 2) and required BiP to defend against misfolding (Fig. S4). The success of the ADPr− model is consistent with the ability of simple organisms, like yeast, that are not known to implement ADP ribosylation, to maintain protein-folding homeostasis in their ER.
Figure 7.

Kinetic models of the ER without (ADPr−) and with (ADPr+) ADP ribosylation arrive at a periodic steady state. (A) Kinetic model of the ER. Unfolded proteins (U) are introduced into the ER at variable rates (ks) from a source (S). In the ER, they can fold (to F, with a rate constant kf), misfold, and aggregate (to A, with a rate constant ka) or bind with BiP (B), forming a reversible complex (UB, with a forward rate constant kon and a reverse rate constant koff). The unfolded protein in the UB complex can be degraded (to Du, with rate constant kdub), releasing BiP. The production of BiP is proportional to the burden of unfolded protein in the ER and set by the rate constant ksb. The delay factor t0 models the latency of the transcriptionally based UPR. BiP is turned over by degradation to DB, with rate constant kdb. Degradation of BiP from complex with substrate liberates free U. In the ADPr+ model, B is in a dynamic equilibrium with its inert ADP-ribosylated form Br (governed by the rate constants krf and krb). (B and C) Time evolution of the concentration of U, UB, B, and Br in the ADPr− and ADPr+ models. At the 0 time point, the ER was assumed to have a minimal volume of 10−14 L and to have no content of U or B. (D) Time evolution of the diurnal variation in the synthesis of U in a hypothetical pancreatic cell (with a volume of 3×10−12 L) of a mouse that fasts during the day and feeds at night. Note that after a transient phase characterized by high concentration of U, a functional ER that attains a periodic steady state is constituted in both models.
Table 1.
The rate constants used in the model
| Constant | Definition | Value | Unit |
| ks | Synthesis of U | 0.2–1.2 | mol−min−1 |
| kf | Folding of U | 2 | min−1 |
| ka | Aggregation of U | 5 | m3(n−1)−mol−(n−1)−min−1 |
| n | Power dependence of aggregation | 3 | Dimensionless |
| kon | Formation of UB from U and B | 10 | m3−mol−1−min−1 |
| koff | Dissociation of UB | 0.01 | min−1 |
| kdub | Degradation of U (from UB with release of B) | 0.02 | min−1 |
| kdb | Degradation of B | 0.0005 | min−1 |
| ksb | Synthesis of B | 0.012 | mol−1−min−1 |
| t0 | Time delay in B synthesis dependence on U | 120 | min |
| krb | Conversion of Br to B | 0.1 | min−1 |
| krf | Conversion of B to Br | 0.4 | min−1 |
| α | Dependence of V on B and UB and Br | 1 | m3−mol−1 |
| V0 | Initial volume of ER | 0.1 | m3 |
Table 2.
Correlating changes in the parameters kf, ka, and n with outputs in the ADPr− and ADPr+ models
| kf(min−1) | ka(m3(n−1)−mole−(n−1)−min−1) | n | Aggregation | Degradation | ||
| ADPr− | ADPr+ | ADPr− | ADPr+ | |||
| mol/24 h | mol/24 h | |||||
| 0.2 | 5 | 3 | 136.6 | 107.7 | 314.6 | 300.7 |
| 2 | 5 | 3 | 16.4 | 14.8 | 108.0 | 80.2 |
| 20 | 5 | 3 | 1.1 | 1.1 | 13.2 | 9.0 |
| 2 | 38.2 | 4 | 16.4 | 14.0 | 108.0 | 80.2 |
| 2 | 290 | 5 | 16.3 | 13.3 | 108.0 | 80.3 |
The effects of changing the rate of folding (kf) or the terms governing the concentration dependence of aggregation (ka and n) on rates of aggregation and degradation integrated over a 24-h cycle after the ER had reached a periodic steady state in the two models are shown.
Protein folding is a first-order process that is largely concentration independent, whereas the higher-order process of aggregation is strongly concentration dependent. Formation of a complex with chaperones lowers the concentration of unfolded proteins and preferentially disfavors aggregation (Mayer and Bukau, 2005), but a quality control strategy based on this principle requires an adequate pool of chaperone for the unfolded protein load. As a consequence, the burden of aggregation was especially conspicuous after the transition from the low- to high-synthesis state, as the relatively slow transcriptional UPRer struggled to match the rapidly rising unfolded protein concentration in the ADPr− model (Fig. 8 A). These problems are marginally less severe in the ADPr+ model because the availability of a pool of inactive, ADP-ribosylated BiP that is rapidly converted to active chaperone can assist in limiting aggregation (9.5% less aggregation in the ADPr+ model integrated over a 24-h period).
Figure 8.
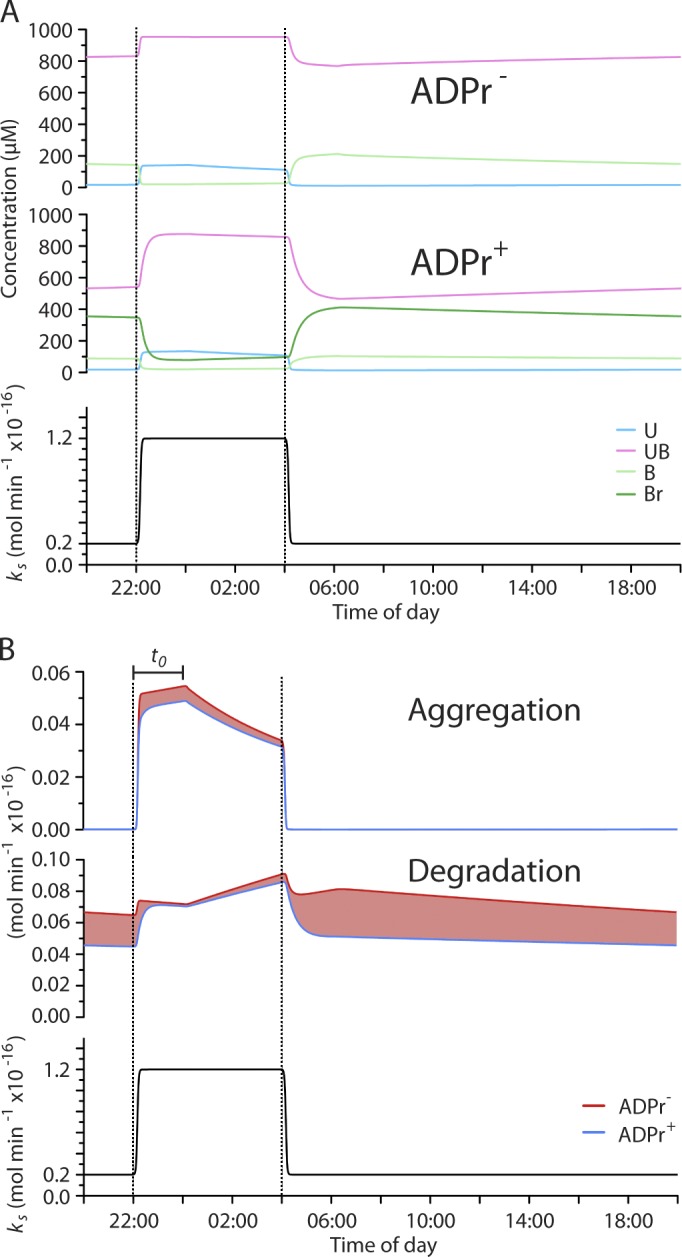
ADP ribosylation of BiP affords protection against misfolding while minimizing the cost of unfolded protein degradation in a kinetic model of the ER. (A) Time evolution of the concentration of U, UB, B, and Br over a 24-h period (at periodic steady state; see Fig 6) in a model ER that is unable (ADPr−) or able (ADPr+) to regulate BiP by ADP ribosylation. The bottom graph shows the diurnal variation in the synthesis of U in a hypothetical pancreatic cell (with a volume of 3×10−12 L) of a mouse that fasts during the day and feeds at night. (B) Comparison of the time evolution of the rates of aggregation and degradation of unfolded protein in the ADPr− and ADPr+ models. The red shading highlights surplus aggregation or degradation of unfolded proteins in the ADPr− model. Note the dominance of the red trace in aggregation during the high-synthesis phase of the diurnal variation in translation (top) and in degradation during the low-synthesis phase of the cycle. The latent phase of the response of BiP synthesis to changes in the burden of unfolded proteins is indicated by the bar (t0).
Despite its importance to preventing aggregation, chaperone binding competes with folding (Dorner et al., 1992) and channels unfolded proteins to degradation (Otero et al., 2010). Rapid inactivation of BiP during the negative phase of the diurnal excursion in protein synthesis allowed the ADPr+ model to reequilibrate the pool of BiP to the declining burden of unfolded proteins, despite the long half-life of BiP (Hendershot et al., 1988). In contrast, in the ADPr− model, the perdurance of BiP favored persistence of the chaperone-unfolded protein complex, even as the burden of unfolded proteins was decreasing (compare the UB trace in the ADPr− and ADPr+ graphs of Fig. 8 A). In the ADPr− model, excessive partitioning of unfolded proteins to a complex with BiP resulted in degradation of many unfolded proteins that folded correctly and were secreted in the ADPr+ model (25.8% less degradation in the ADPr+ model integrated over a 24-h period; Fig. 8 B, middle graph). The benefit of the ADPr+ system is manifested in the superior efficiency with which it defends against aggregation while minimizing gratuitous degradation of unfolded proteins in an ER subject to large fluctuations in load. Such benefits were observed over a range of diurnal excursions in protein synthesis rates (Fig. S4, A and B) and a range of intensities of the transcriptional UPR (Fig. S4, C and D).
The advantages of the ADPr+ model were also maintained when the ADP-ribosylated BiP was modeled, not as a completely inert agent that does not bind unfolded proteins but more conservatively as having 40-fold higher offrates for substrate binding (observed experimentally with BiPR470E in the presence of ADP; Fig. S5, A and B). Similarly, ADP ribosylation afforded an advantage to the ER that was evident if one assumed that all the BiP in the cell turned over with similar kinetics or if only free BiP was subject to turnover (Fig. S5, C and D).
Discussion
ADP ribosylation correlates with physiological fluctuations in ER client protein load and is regulated on a timescale shorter than that of the better-known transcriptional and translational strands of the UPRer. Evidence pointing to conserved Arg residues in the substrate-binding domain as major modification sites in BiP fits well with previous observations that modification compromises substrate binding. Together, these new findings support the hypothesis that ADP ribosylation has evolved to inactivate BiP when the load of unfolded protein diminishes and to provide a pool of latent BiP that can be rapidly recruited to service a surge of incoming unfolded proteins. A kinetic model provides further support for the utility of such regulation of a major ER chaperone and explains how this posttranslational strand might be integrated into the UPRer.
Though we were unable to detect ADP-ribosylated BiP peptides directly by mass spectrometry, several lines of evidence support the assignment of the modified residues. Labels incorporated from tritiated adenosine or intracellular phosphate pools were found on a single cyanogen bromide fragment from FLAG-tagged hamster BiP purified from cells. In a tryptic digest, the label was recovered in only two peaks of reverse-phase HPLC. Whereas the early eluting peak may correspond to a breakdown product of the modification, the simplicity of the pattern in both the 32P- and 3H-labeled experiments suggests that the modifications of BiP occur on no more than two major sites. The small number of modifications is also suggested by the simplicity of the IEF pattern, which shows a single basic and a single acid band in several cell types, though the latter is also consistent with a highly processive incorporation of several modifications (and their highly processive reversal).
The evidence for Arg470 as a modification site in vivo is especially compelling, as the conservative Arg470 to Lys substitution eliminates all labeling, with modest affects on the function of BiP in vitro. The Arg492 to Lys mutation also reproducibly lowered label incorporation into BiP in vivo but only by ∼50%. Therefore, it is impossible to distinguish between processive modification of Arg470 followed by Arg492 and a scenario whereby the (conservative) Arg492 to Lys mutation affected the structure of the substrate-binding domain to preclude modification at Arg470. However, preserved substrate binding by BiPR492K in vitro argues against a major structural modification of the substrate-binding domain.
As we are unable to produce ADP-ribosylated BiP in vitro, the analysis that we presented was based on the idea that the functional consequences of the modification are caused, in part, by the electrostatic effects associated with this modification. The Arg to Glu substitutions that we used are likely to mimic the replacement of the positive charge of the guanidino group of Arg by the negative charge of the pyrophosphates of the ADP-ribose. However, as this substitution does not fully reproduce the spatial and structural consequences of the addition of a bulky ADP-ribose, the functional perturbation to BiP elicited by R470E and R492E mutations is likely a conservative estimate of the consequences of ADP ribosylation at the same sites.
The structure of the substrate-binding domains of DnaK family members is a good model for BiP and readily predicts a perturbation in client binding by ADP ribosylation of the two highly conserved Arg residues. Merely replacing the positive by a negative charge in BiPR492E eliminates detectable binding of a fluorescent peptide in vitro. This may be a result of the disruption of a predicted conserved hydrogen bond between the η2-nitrogen of Arg492 (DnaK Arg467) and the backbone carbonyl of Ala454 (DnaK Ala429), destabilizing the L5,6 loop, which forms the wall of the substrate-binding groove (Zhu et al., 1996). Arg470 (DnaK Arg445) is predicted to engage lid residue Asp552 (DnaK Asp526) in a conserved salt bridge that would be disrupted by ADP ribosylation. Lid–substrate-binding domain interactions are known to stabilize the bound substrate in the high-affinity ADP state of DnaK (Mayer et al., 2000; Fernández-Sáiz et al., 2006). As predicted by these earlier studies, BiPR470E exhibits high offrates for substrate binding. Interestingly, disruption of the Arg470-Asp552 salt bridge is even more perturbing than removal of the lid. And, in the absence of a lid, the R470E mutation has no further destabilizing effect on substrate binding by BiP. Together, these observations are consistent with a regulatory role for lid interactions outside the substrate-binding domain on substrate binding (Schlecht et al., 2011). Thus, we speculate that alternative interactions of the lid (perhaps with the nucleotide-binding domain) might be favored by the destabilizing effect of ADP ribosylation on competing lid–substrate domain interactions. Such interactions may also contribute to the marked effect of ADP ribosylation on interdomain allosteric coupling revealed by the persistent protection of the interdomain linker in modified BiP exposed to both ATP and substrate.
No gross effect of the mutations on ATP hydrolysis was noted. At first glance, this might suggest a measure of inefficiency, as the ADP-ribosylated, inactive, latent chaperone could hydrolyze ATP in futile cycles. However, it is important to note that the in vitro measurements reporting on very slow ATP hydrolysis do not take into account nucleotide exchange factors that accelerate the process in vivo (Mayer and Bukau, 2005) and whose interaction with BiP could be subject to regulation by ADP ribosylation. Furthermore, the assays we performed do not take into account the full catalytic cycle of the chaperone, with its many allosteric couplings. Thus, this is an area that needs to be examined in further detail.
The rapid ADP ribosylation and deribosylation of BiP in response to changes in unfolded protein load suggest that these modifications are enzyme mediated. There are no known ER-localized ARTs, but mammals are endowed with several genes encoding secreted and membrane-bound, Arg-specific ARTs (Koch-Nolte et al., 2008). Thus, a relevant enzymatic activity is present, at least transiently in the ER of mammalian cells. Furthermore, an experiment in which PARP1, a poly–ADP-ribose polymerase, was mislocalized to the ER lumen revealed that the substrate NAD+ too is present in the ER lumen at concentrations adequate to support high levels of poly–ADP-ribose production (Dölle et al., 2010).
Candidates for a glycohydrolase that removes the modification are less forthcoming. A mitochondrial activity that removes ADP-ribose from Cys119 of glutamate dehydrogenase has been described, but it appears to be specific for modified Cys (Herrero-Yraola et al., 2001; Choi et al., 2005). A mammalian ADP-ribosyl Arg hydrolase has been characterized and its gene cloned (Moss et al., 1992), but the encoded protein is cytosolic and thus unlikely to contribute the regulation of BiP activity.
A kinetic model reveals at least two conspicuous benefits to rapidly regulating BiP’s activity posttranslationally, most obviously as a rapidly deployable defense against protein misfolding to maintain homeostasis during the latency period of the conventional transcriptionally mediated UPRer. Less obvious, but of potentially equal importance, is the role of ADP ribosylation in down-regulating BiP activity when the concentration of unfolded proteins declines. Folding and aggregation are concentration-dependent processes affecting unfolded proteins. Chaperones bind to unfolded proteins and lower their effective concentration. This negatively impacts both rates of folding and aggregation. Aggregation is a higher-order process compared with folding and is therefore sensitive to concentration. Thus, at high-substrate (unfolded protein) concentrations, chaperones preferentially inhibit aggregation and favor folding by default. However, at low-substrate concentration, chaperones, like BiP, have the potential to inhibit protein folding by channeling unfolded proteins to degradation pathways (Otero et al., 2010). This explains the negative impact of BiP overexpression on secretion of folded protein (Dorner et al., 1992). Rapid inactivation of BiP by ADP ribosylation allows cells to benefit from the protection against aggregation, afforded by ample chaperones, without paying the cost of enhanced degradation of unfolded protein that are engaged in a fruitless complex with BiP. This represents a significant advantage over a UPRer that is exclusively transcriptionally mediated.
It is interesting to reflect on the fact that in simple eukaryotes, the UPRer is largely transcriptionally mediated. As the complexity of secretion imposed by multicellularity increased, this simple coping mechanism proved inadequate. Protein kinase RNA–like ER kinase (PERK)–mediated translational control over rates of unfolded protein synthesis, a metazoan invention (Harding et al., 1999), affords cells the ability to cope with fluctuations in the demand for secretory proteins with a lower steady-state pool of chaperones (Trusina et al., 2008). But translational control caps the gain on rates of secretion, and its excess reduces fitness (Lin et al., 2009). ADP ribosylation of BiP appears to have evolved as a further refinement of the UPRer to help resolve the tension between the benefit of protecting the long-lived cells of complex organisms against protein aggregation and the cost of slowing down protein maturation by excess chaperoning and degradation. The magnitude of the activity-dependent changes in BiP modification suggest that in some tissues (like the pancreas), reversible ADP ribosylation of BiP may be the first line of defense in coping with physiological fluctuations in unfolded protein load.
Materials and methods
Mammalian cell culture
293T and GH3 cells were cultured in DME and 25 mM low-glucose DME, respectively (Sigma-Aldrich), supplemented with 10% FetalClone II calf serum (Hyclone; Thermo Fisher Scientific) and 2 mM l-Glutamine (Sigma-Aldrich). Cells were maintained at 37°C in an atmosphere containing 5% CO2.
Mammalian expression plasmids
N-terminally FLAG-tagged hamster BiP (27–654) was expressed from the pFLAG-CMV-1 vector (Sigma-Aldrich; Petrova et al., 2008). Point mutations were introduced by PCR and confirmed by sequencing. 293T cells were transfected by calcium phosphate precipitation.
Bacterial expression and purification of proteins
Site-directed mutagenesis and deletion of the lid domain of BiP (554–654) were performed by PCR and confirmed by sequencing. H6-tagged hamster BiP was cloned into a pQE10 plasmid and expressed in M15 Escherichia coli. Cultures were induced with 1 mM IPTG at an optical density of 0.6 at 600 nm and were grown for a further 6 h before harvesting by centrifugation. Cell pellets were lysed in nitrilotriacetic acid (NTA) lysis buffer (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 0.2% Triton X-100, 10% glycerol, 20 mM imidazole supplemented with 0.2 mM PMSF, 2 µM leupeptin, 4 µg.ml−1 aprotinin, and 1 µg.ml−1 pepstatin) using a high-pressure homogenizer (Nano DeBEE; BEE International). Lysate was cleared by centrifugation at 20,000 g for 30 min before incubation with 2 ml Ni-NTA beads (QIAGEN) for 2 h at 4°C. Beads were washed five times with 20 ml NTA lysis buffer supplemented with either 30 mM imidazole, 1% Triton X-100, 1 M NaCl, 5 mM ATP, or 0.5 M Tris-HCl, pH 7.5, sequentially. BiP was eluted in NTA lysis buffer lacking Triton X-100 and supplemented with 500 mM imidazole and dialyzed into HKM buffer (150 mM KCl, 50 mM Hepes, pH 7.5, and 10 mM MgCl2).
Mouse H6-Smt3-fusion P58 J-domain was expressed in Rosetta(DE3) E. coli by the aforementioned protocol with the following amendments: IPTG induction was performed at 18°C for 6 h. Protein-bound Ni-NTA beads were washed with 20 ml NTA wash buffer before incubation with yeast Ulp1 protease at 4°C overnight to release untagged P58 protein. Further purification and buffer exchange into HKM buffer were performed by gel filtration using a Superdex 200 column (GE Healthcare).
H6-tagged SubA expression was performed in Origami E. coli and followed the protocol described for H6-BiP; however, Ni-NTA beads were washed only with 20 ml NTA lysis buffer and 20 ml NTA lysis buffer supplemented with 500 mM Tris-HCl, pH 7.5. H6-tagged eIF2α was expressed in DH5α E. coli and was purified as described for H6-BiP, with the following amendments: IPTG induction was performed at 30°C for 6 h. Protein-bound Ni-NTA beads were washed with 20 ml NTA lysis buffer before protein elution. All proteins were stored at −80°C.
Assessment of H6-BiP folding by CD
H6-BiP was buffer exchanged into 10 mM NaH2PO4/Na2HPO4, pH 7.5, by PD10 column (GE Healthcare) size-exclusion chromatograph. Measurements were obtained with a spectropolarimeter (J-810; Jasco) using a path length of 0.2 mm at 25°C. A wavelength range of 180–260 nm was recorded, and data from seven acquisitions were averaged. Data were smoothed by a Savitzky–Golay smoothing filter.
Preparation of mouse pancreas microsomal extracts
All experiments performed on mice were approved by the ethical committee of the University of Cambridge and by the UK home office. Mice were either subjected to an overnight fasting period, a fasting followed by a 1-h feeding, or injected with a 10-µl.g−1 body weight of a 10-µg.µl−1 solution of cycloheximide in PBS in the fed state. Pancreatic tissue was Dounce homogenized in homogenization buffer (250 mM sucrose, 25 mM Hepes, pH 7.2, and 25 mM KCl) before adjusting to 140 mM KCl and 5 mM MgCl2. Homogenate was centrifuged at 800 g for 5 min to remove nuclei and whole cells, and ER membranes were isolated from the supernatant fraction by OptiPrep (Sigma-Aldrich) density gradient centrifugation for 3 h at 200,000 g. ER membranes isolated from the 15–25% OptiPrep step gradient interface were diluted in homogenization buffer adjusted to 140 mM KCl and 5 mM MgCl2 and pelleted by centrifugation at 100,000 g for 30 min. The pelleted membrane fractions were solubilized by sonication in 50 mM KCl, 20 mM Hepes, pH 7.5, and 1% vol/vol Triton X-100 and cleared by ultracentrifugation at 100,000 g, and the protein-rich supernatant was snap frozen and stored at −80°C. All buffers were supplemented with 0.2 mM PMSF, 2 µM leupeptin, 4 µg.ml−1 aprotinin, and 1 µg.ml−1 pepstatin.
For SubA digestion, BiP was purified from microsomal extracts of cycloheximide-treated mice following a published protocol (Tokunaga et al., 1992), modified to eliminate the second hydroxyapatite column purification.
Analysis of endogenous ADP-ribosylated BiP from cell lysate or pancreatic microsome extracts
Where indicated, pancreatic microsome extracts were separated by a Superdex 200 10/300 gel filtration column equilibrated with 50 mM KCl, 50 mM NaCl, and 20 mM Hepes, pH 7.2, and elution fractions were analyzed by SDS-PAGE. ADP-ribosylated BiP was separated from its unmodified form by IEF, following certain modifications of the published procedure (Laitusis et al., 1999): a 3.75% acrylamide slab gel containing 8.8 M urea, 1.25% CHAPS, and 5% Pharmalytes, pH 4.5–5.4 (GE Healthcare), was used. Samples were prepared in IEF loading buffer (8 M urea, 5% CHAPS, 50 mM dithiothreitol, and 2% Pharmalytes, pH 4.5–5.4) and heated at 30°C before gel loading. A sample overlay of 0.5 M urea and 2% Pharmalytes was added before separation. 0.01 M Glu was used as the anode buffer and 0.05 M His as the cathode buffer. Proteins were transferred to nitrocellulose membranes under basic conditions and immunoblotted with polyclonal antibody raised against mouse BiP (a gift from R. Zimmermann, Saarland University, Saarbrücken, Germany) or monoclonal antibody raised against mouse eIF2α. Immunoblots were visualized using a fluorescent scanner (Odyssey; LI-COR Biosciences).
λ-phosphatase treatment of pancreatic microsome extract
Pancreatic microsome extract was incubated for 1 and 16 h in phosphatase buffer (50 mM Hepes, pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 0.01% Brij-35, 2 mM DTT, and 2 mM MnCl2) in the presence or absence of λ-phosphatase at 30°C. Proteins were buffer exchanged into IEF loading buffer by G-50 column (GE Healthcare) size-exclusion chromatography and separated by IEF before transfer to nitrocellulose membrane for immunoblotting. To assess λ-phosphatase activity, purified eIF2α was incubated with GST-Sepharose–immobilized recombinant PERK in phosphatase buffer for 30 min at 37°C. PERK-bound Sepharose beads were removed by centrifugation, and the supernatant was subsequently incubated in the presence or absence of λ-phosphatase for 1 and 16 h. Proteins were prepared for SDS-PAGE and separated on a 15% acrylamide gel containing 50 µM Phos-tag (NARD Institute, Ltd.) and 100 µM MnCl2. Proteins were transferred to nitrocellulose membrane and probed with a polyclonal antibody raised to eIF2α.
Cell transfection, metabolic labeling, and immunoprecipitation
Cells were transfected with FLAG-BiP plasmid and 36 h later were labeled overnight with 32P-orthophosphate (MP Biomedicals). Metabolic labeling with two 3H-adenosines (GE Healthcare) was performed 48 h after transfection of cells for a period of 6 h. Where indicated, 0.5 mM novobiocin (LC Laboratories) was added for the duration of labeling, or 100 µg.ml−1 of cycloheximide with or without 10 µM MG132 (Sigma-Aldrich) was added during the last hour of labeling. Cell lysates were prepared in a Ca2+-containing buffer (1% Triton X-100, 150 mM NaCl, 20 mM Hepes, pH 7.4, 10% glycerol, 10 mM CaCl2, 1 mM DTT, 1 mM PMSF, 4 µg.ml−1 aprotinin, and 2 µg.ml−1 pepstatin A), clarified by ultracentrifugation, and treated with 1 µg.ml−1 RNase A. Radiolabeled proteins were immunopurified with α-FLAG M1 affinity gel overnight. The bound proteins were eluted from α-FLAG M1 in 10 mM EDTA, 20 mM Tris, pH 7.5, and 0.02% Tween 20, resolved by SDS-PAGE, and detected by the Storm PhosphorImager (Molecular Dynamics).
Immunoprecipitates from cells labeled with 3H-adenosine were resolved by SDS-PAGE and transferred to a PVDF membrane, and the signal was detected by the KODAK BioMax TranScreen LE system (Sigma-Aldrich). For direct quantification of the 3H signal, immunopurified FLAG-BiP was resolved by SDS-PAGE and visualized by staining with Coomassie Brilliant blue R450. The gel slice corresponding to FLAG-BiP (and an additional slice in the same lane to serve as a background control) was excised, thoroughly destained with alternating washes of 25 mM NH4HCO3 and 50:50 25 mM NH4HCO3/CH3CN (vol/vol), dried down in a vacuum concentrator (SpeedVac 5301; Eppendorf) concentrator, and hydrolyzed overnight with 50 mM NaOH at 37°C. After alkaline hydrolysis, alternating addition of CH3CN and 5% formic acid hydrates and dehydrates the gel slices to extract the fluid phase, which was subsequently concentrated and dissolved in 20 µl of H2O. After addition of 1 ml of OptiPhase SuperMix liquid scintillation cocktail (PerkinElmer), the counts were measured in the 1450 Microbeta Plus Liquid Scintillation Counter (Wallac).
To test the sensitivity of the radiolabel in BiP to phosphatases, EDTA-eluted, 32P-labeled FLAG-BiP was desalted using the ProbeQuant G-50 Micro Column (GE Healthcare) and treated with either CIP in 50 mM Tris, pH 8.0, 100 mM NaCl, 15 mM MgCl2, λ-phosphatase in 50 mM Tris, pH 7.5, 100 mM NaCl, and 2 mM MnCl2 or 0.5 M NH2OH at room temperature for 1 h.
Tryptic digest and reverse-phase HPLC separation
Metabolically labeled, immunopurified FLAG-BiP was denatured in solution with 0.5% SDS at 65°C for 30 min. Samples were then diluted to reduce SDS concentration to 0.1% and digested with 2 µg of mass spectrometry grade trypsin (Promega) in a total volume of 50 µl for 16 h at room temperature. The generated tryptic peptides were acidified by addition of trifluoroacetic acid to 0.1% and injected on a 150 × 1.0–mm i.d. Luna 5-µm C18 100 Å column (Phenomenex). Chromatography was performed at a flow rate of 0.1 ml.min−1, and a solvent system of a linear gradient elution from buffer A to B over 90 min was used. Buffer composition of H2O/CH3CN/trifluoroacetic acid for A was 99.9%:0%:0.1% and for B was 0%:99.91%:0.09% (vol/vol). Absorbance of the eluted peptides was monitored at λ = 230, and 100-µl fractions were collected. 32P-labeled fractions were analyzed by direct Cerenkov counting. 3H-labeled fractions were dried down and analyzed by liquid scintillation counting using 0.25 ml of OptiPhase SuperMix liquid scintillation cocktail in a 96-well format.
Fluorescence anisotropy measurement of BiP substrate binding
Assessment of substrate binding by BiP was performed as previously described (Marcinowski et al., 2011). In brief, the peptide sequence NH2-HTFPAVLGSC-COOH was labeled with a Lucifer yellow iodoacetamide fluorescent probe (Invitrogen) and separated from free label by HPLC using a C-18 Poroshell 120 column (Agilent Technologies). Various concentrations of H6-BiP were incubated with 1 µM fluorescent peptide in the presence of 0.005% Triton X-100 in HKM buffer for 16 h at 25°C before fluorescence anisotropy measurement in a plate reader (Infinite F500; Tecan). Lucifer yellow moiety was excited at λ = 430 nm, and emission was detected at λ = 535 nm. Substrate offrates were analyzed by challenging preformed complexes of 25 µM BiP and 1 µM Lucifer yellow peptide with 500 µM unlabeled peptide and recording change in fluorescence anisotropy over time.
BiP ATPase assay
BiP ATPase activity under single turnover conditions was assayed essentially as published (Mayer et al., 1999): 30 µM BiP was incubated with 800 µM ATP supplemented with 10 µCi.µl−1 α-[32P]ATP (PerkinElmer) in buffer A (25 mM Hepes, pH 7.5, 50 mM KCl, and 10 mM MgCl2) for 2 min on ice. BiP was separated from free ATP by NICK column (GE Healthcare) gel filtration and snap frozen for subsequent assay use. α-[32P]ATP–bound BiP was added to buffer A in the presence or absence of 2 µM P58 J-domain and 30 µM substrate peptide and incubated at 30°C. Fractions of the reaction were removed periodically and spotted onto thin-layer chromatography plates (PEI Cellulose F; Merck & Co., Inc.) prespotted with 5 mM ATP and 5 mM ADP solution to assist with plate development. Thin-layer chromatography plates were developed with 10% vol/vol HOAc and 400 mM LiCl, and signal detection was performed using the Storm PhosphorImager.
To assay ATPase activity under multiturnover conditions, 25 µM BiP was incubated with 2 mM ATP supplemented with 125 µCi.ml−1 α-[32P]ATP in the presence or absence of 30 µM substrate peptide and 2 µM mouse P58 J-domain in buffer A at 30°C. 1-µl fractions were removed periodically and spotted onto thin-layer chromatography plates and developed as described for single turnover experiments.
SubA digestion
SubA digestion was performed essentially as previously described (Paton et al., 2006). 7.5 µM BiP (H6-tagged hamster BiP purified from E. coli or pancreatic mouse BiP) was incubated with 0.5 mM peptide substrate (NH2-HTFPAVL-COOH) and 2 mM ATP before addition of 0.6 µM His-tagged SubA protease. Reactions were stopped by addition of TCA at a final concentration of 15% and incubation at 4°C for 30 min. Protein precipitates were pelleted by centrifugation, washed with −20°C acetone, and resuspended in either SDS-PAGE sample buffer or IEF sample buffer containing 8 M urea before separation by SDS-PAGE or IEF-PAGE, respectively.
Kinetic modeling of the effects of BiP ADP ribosylation on protein-folding homeostasis in the ER
Description of the kinetic models and their assumptions.
Two kinetic models were designed to compare two modes by which the ER copes with fluctuations in unfolded protein load. In the first model, the ER can recruit inactive BiP from an ADP-ribosylated pool (ADPr+), whereas in the second model, the ER lacks this mechanism (ADPr−). In both cases, the load of unfolded proteins regulates the production of BiP positively, reflecting the activity of the transcriptionally mediated UPR. Translational control is not relevant to this comparison, as it acts upstream of the fluctuation in unfolded protein load. It was not considered further.
The ER was modeled as a space in which three primary agents are operating: unfolded proteins (U), BiP (B), and complexes between the two (UB). Unfolded proteins are introduced into the space from a source (S), and the rate of their production (ks) depends on the physiological demands on the cell; ks represents a key parameter of the model. Within the ER, unfolded proteins can fold, misfold, and aggregate or associate reversibly with BiP to form complexes (UB). Both folded and aggregated proteins are assumed to exit the space and not to interact with its active agents. This simplification is justified in the case of the compensated ER, as folded proteins exit the compartment, and misfolded proteins can be rapidly degraded.
We assumed that complexes between unfolded proteins and BiP (UB) can dissociate to their constituents and that they also represent a conduit to degradation of unfolded proteins and release of free B. The last assumption follows from the observations that association with BiP can enhance the degradation of unfolded proteins and that BiP overexpression results in less secretion (Dorner et al., 1992; Otero et al., 2010). Thus, the output from the ER is measured in terms of production of folded (F), aggregated (A), and degraded (DU) proteins.
BiP synthesis was considered to be proportional to the burden of unfolded proteins in the ER; because the two are linked by a latent transcriptional program (the UPR), a delay term, t0, of 120 min was introduced to account for the time it takes to induce the UPR and translate BiP mRNA into protein. Furthermore, as activation of the UPR leads to an increase in ER volume (Stefan et al., 1987; Bernales et al., 2006), the volume of the ER was modeled as proportional to the total content of chaperones in it, that is, to the total amount of BiP.
BiP degradation follows first-order kinetics and is a relatively slow process (Hendershot et al., 1988; Hu et al., 2009). In the ADPr+ model, the ADP-ribosylated form of BiP was modeled as an inert species that does not interact with unfolded proteins and is in a dynamic equilibrium with free BiP. The interconversion between the two forms of BiP was assumed to proceed via unregulated first-order processes. Thus, in this model, ADP-ribosylated BiP represents a buffer, the recruitment of which is governed by mass action. This assumption stringently tests the benefits of the ADPr+ model, as regulating the enzymatic reactions that interconvert the two forms of BiP by subordinating them to the burden of unfolded proteins would exceed the benefits of an unregulated system. We also tested a more conservative model in which modified BiP interacts with unfolded proteins but with offrates that are 40 times greater than unmodified BiP.
Folding (F) was assumed to proceed with first-order kinetics, and the rate at which folded proteins are produced is proportional to the load of free unfolded protein in the ER and is independent of their concentration. As aggregation (A) requires the interaction of two or more free unfolded proteins, it was considered as a concentration-dependent process that proceeds at a rate proportional to a power n of the concentration of unfolded protein; n is specific for the aggregation process of the protein being modeled (Knowles et al., 2009).
In this kinetic model, BiP functions merely as a holdase, and the benefit to the system arises solely from the ability of BiP to buffer increases in free unfolded protein concentration and thus favor folding over aggregation (Mayer and Bukau, 2005). Were BiP endowed with a more deterministic role in protein folding (a foldase), the benefits of a pool of ADP-ribosylated BiP would likely increase. Thus, this assumption too provides for a stringent test for the benefits of the ADPr+ model.
The rate equations.
The time evolution in the number of protein molecules in various states is defined by a system of differential equations. We adopted the following notation: F, number of folded proteins; A, number of aggregated proteins; D, number of degraded proteins; U, number of free unfolded proteins; B, number of free BiP proteins; Br, number of ADP-ribosylated BiP proteins; UB, number of complexes of unfolded and BiP proteins; V, ER volume.
The following equation shows the time evolution of the number of folded protein molecules, F:
| (1) |
The rate of production of folded proteins depends on the product between kf and U.
The following equation shows the time evolution of the number of aggregated protein molecules, A:
| (2) |
The rate of production of aggregated protein depends on the product between ka and the concentration of unfolded proteins to the power n (Un/Vn). The value of n defines the order of the concentration dependence of the aggregation process (Knowles et al., 2009).
The following equation shows the time evolution of the number of degraded protein molecules, Du:
| (3) |
The rate of degradation of unfolded protein depends on the product between kdub and UB.
The following equation shows the time evolution of the number of unfolded protein molecules in the ER:
| (4) |
This equation has three positive contributions: the rate of U synthesis (ks), the rate of dissociation of the UB complex (koffUB), and the release of U from UB by BiP degradation (kdbUB). It has three negative contributions: the rate of folding (kfU), the rate of aggregation (or misfolding, ), and the rate of formation of the UB complex with B ().
The following equation shows the time evolution of the number of molecules of free BiP:
| (5) |
This equation has four positive contributions: the rate of B synthesis (ksbU(t−t0)), where t0 is the time delay in the activation of the transcriptional response, the dissociation of the UB complex (koffUB), the conversion of ADP-ribosylated BiP to BiP (krbBr), and the release of B by the degradation of U from the UB complex (kdubUB). It has three negative contributions: the rate of formation of the UB complex with U (), the conversion of BiP to ADP-ribosylated BiP (krfB), and the degradation of free BiP (kdbB).
The following equation shows the time evolution of the number of molecules of the UB complex:
| (6) |
In this equation, the positive term describes the formation of the complex from U and B (), and the three negative terms describe the dissociation of UB complexes to their constituents (koffUB), the degradation of U in complex with B (kdubUB), and the degradation of B in complex with U (kdbUB).
The following equation shows the time evolution of the number of molecules of ADP-ribosylated BiP:
| (7) |
The positive term (krfB) describes the ADP ribosylation of free BiP, and the two negative terms describe the deribosylation of modified BiP (krbBr) and its degradation (kdbBr).
The following equation shows the time evolution of the volume of the ER:
| (8) |
The model parameters (Table 1).
The units were chosen as follows: volume is measured in cubic meters, and quantities of the inputs, agents, and outputs are measured in moles and time in minutes. To express the inputs and outputs of the model on a biologically relevant scale, the model ER space of several cubic meters is related (scaled) to a single pancreatic cell that is assumed to be a cube of 14 µm on each side. Its volume is thus 3×10−15 m3 or 3,000 femtoliters, ∼30 times the size of a red blood cell. If we assume that the volume of the ER of the pancreatic cell fluctuates between 10 and 30% of the total cell volume (Stefan et al., 1987), it thus varies between 3 and 9×10−16 m3. In the model, the volume of the ER varies conservatively between 3 and 6 m3; therefore, the scaling factor for the relevant parameters from the model to the cell is 1016.
To simulate physiologically relevant excursions in unfolded protein load, a pancreatic secretory cell of a mouse that is subject to a diurnal cycle consisting of a period of fasting alternating with a period of feeding was chosen (Ahrén, 2000). Rates of protein synthesis increase by 5–10 fold between the fasted (low glucose) and fed (high glucose) state in pancreatic β cells (Itoh and Okamoto, 1980; Logothetopoulos and Jain, 1980; Harding et al., 2001). Thus, the model ER was confronted with an 18-h period of low unfolded protein synthesis and a 6-h period of sixfold-higher protein synthesis. The physiological transition between the two states was set to take place gradually over a 20-min period.
The values of ks were chosen in consideration of the ER volume and were based on simple considerations: the total concentration of proteins in the ER is estimated to be 100 g/L, and half are assumed to be resident proteins and the other half unfolded proteins (mostly in complex with BiP; Despa, 2010). Thus, the concentration of total U (i.e., U + UB) is 50 g/L. If the mean residence time of an unfolded protein in the ER is 30 min (Jamieson and Palade, 1971), then the ER turns over 1.5 g/L−min of U. If a mean protein has a molecular mass of 30,000 D, then ks/V is 1.5 g/L−min/30,000 g/mol = ∼0.05 mol/m3−min. ks in the resting fasted ER was set to be about twofold lower than this number, and ks in the active fed state was set to be threefold higher.
Proteins fold at vastly different rates. For the purpose of the model, a protein that folds with a half-life of 30 s was chosen (kf = 2/min). However, the benefits of the ADPr+ model were assessed over a range of kf (see Table 2).
The aggregation propensities of proteins are also heterogenous. Therefore, we considered a range of aggregation regimes by varying ka and n. To simulate realistic conditions, the parameters were chosen such that overall, the production of aggregated protein (A) would not exceed a few percent of the total unfolded protein synthesized. The benefits of the ADPr+ model were sustained over a range of values of n (see Table 2). In the main text, our discussion refers to the case in which aggregation proceeds by the third power of the concentration of U.
Similarly, the rate at which unfolded protein are degraded by the ER varies considerably. We chose kdub such that no more than 30% of the total unfolded protein synthesized would undergo ER-associated degradation.
The reported half-life of BiP varies from 6 h (Hu et al., 2009) to exceeding 48 h (Hendershot et al., 1988). Therefore, kdb was given a value of 0.0005/min to give a half-life of around 24 h.
The production of BiP was set to be regulated by the burden of unfolded proteins in the ER, which is an assumption valid also if BiP has a role in repressing the UPR transducers (Kimata and Kohno, 2011). The rate constant ksb was set to ensure that the concentration of total BiP in the ER would remain of ∼1 mM (Sidrauski et al., 2002).
In vivo, the association and dissociation of BiP from its substrates are regulated by the bound nucleotide and are rate limited by cofactor-stimulated nucleotide exchange and hydrolysis reactions. To model this complex multistate process with a single on- and offrate constant, we assumed the dissociation of the UB complex to be near the offrates measured for the ADP state (koff = 10−2/min; Fig. 5; Marcinowski et al., 2011). The on-rates were modeled as lying somewhere between the low on-rates of the ADP state and the very high on-rates of the ATP state (Liebermeister et al., 2001), kon = 104 L/mol−min (or 10 m3/mole−min in the units used in the model).
There is limited experimental information to guide the selection of rate constants for the dynamic equilibrium between the modified (Br) and unmodified BiP (B). As the modified form disappears within minutes of application of ER stress (Laitusis et al., 1999), we chose a krb value of 0.1/min for the Br→B conversion. krf, the rate constant for the reverse reaction B→Br, was set to 0.4/min, as this value was found to provide an ER in which, at low burden of U, about half of BiP is modified, which is in agreement with experimental observations (Fig. 1; Laitusis et al., 1999).
Implementation and testing of the model.
The aforementioned system of rate equations (Eqs. 1–8) was implemented in MATLAB (version 7.13; MathWorks) and numerically integrated via the dde23 solver (Shampine and Thompson, 2001), which is specifically tailored for delay differential equations with constant time lags.
The rate constants chosen for implementation are presented in Table 1. Confronted with a fixed rate of unfolded protein synthesis (ks), the agents acting in the ADPr+ model (U, B, Br, UB, and V) or in the ADPr− model reached steady-state values after several days. In either model, the values were dependent on ks but independent of the initial conditions set, attesting to the adequacy of the feedback mechanisms in maintaining protein homeostasis.
Under a physiologically relevant regime of diurnal variation in ks, both the ADPr+ and the ADPr− models attained a periodic steady state that defines a canonical 24-h cycle of variation in U, B, Br, UB, V, and the rates of folding , aggregation , and degradation (Fig. 7, B–D).
The key response to unfolded protein load in both the ADPr− and ADPr+ models is the expansion of the ER volume, which is expected as an adaptation to the strong concentration dependence of aggregation and indeed observed experimentally (Stefan et al., 1987; Bernales et al., 2006). Aggregation and degradation were found to be inversely correlated with the rate of folding, as expected. However, the beneficial effects of the ADPr+ model endured with variation in kf over two orders of magnitude. Similarly, the ADPr+ model is better at coping with a range of aggregation processes characterized by different values of ka and n (Table 2).
The ADPr+ model showed a modest advantage at coping with aggregation over a range of diurnal excursions in unfolded protein synthesis (kshigh–kslow), showing both models to be competent in defending against excessive aggregation (Fig. S4 A). Strikingly, the ADPr+ model limited levels of aggregation with much less protein degradation when compared with the ADPr− model, achieving increased levels of folding in doing so (Fig. S4 B). The ADPr+ model afforded a significant advantage across a wide range of intensities of the transcriptional UPR (Fig. S4 D).
The aforementioned models assume that the ADP-ribosylated BiP is inert. This seems a safe assumption given the poor binding of peptide to the BiPR470E;R492E double mutant (Fig. 5 B) and the finding that modified BiP is excluded from complexes with substrate (Hendershot et al., 1988). However, to test the consequences of ADP ribosylation under more stringent conditions, we made the very conservative assumption that the BiPR470E mutation fully mimics the consequences of ADP ribosylation (this is a highly conservative assumption because it ignores the inhibitory impact of the bulky modification on lid closure).
In this conservative model (depicted schematically in Fig. S5 A), the ADP ribosylated BiP forms a complex with unfolded proteins (UBr, which has the same functional properties as the UB complex), but the UBr complex dissociates with offrates (koff2) that are 40-fold higher than those measured for the UB complex of unfolded proteins with unmodified BiP. As a consequence, an additional equation describing the time evolution of the concentration of UBr complexes in the ER was added to the system, shown as
in which the offrate of the UBr complex (koff2) is set at 40 × koff of the UB complex, whereas Eqs. 3, 4, 7, and 8 were modified by including the appropriate additional terms.
We simulated the same diurnal variation in unfolded protein production described above, allowing the ADPr− and (modified) ADPr+ models to reach a periodic steady state and then compared the aggregation and degradation over a 24-h period. Fig. S5 B shows the advantage of the modified ADPr+ model over the ADPr− model in terms of limiting degradation while preventing excessive aggregation. Thus, the benefits of a recruitable pool of ADP-ribosylated BiP persist even if the modified chaperone is not completely inert but merely enfeebled in its substrate binding.
The pathways governing the degradation of BiP remain poorly understood. The model used in the main text made the plausible assumption that all forms of BiP (B, Br, and UB) have equal half-lives and that their degradation is determined by the same rate constant kdb. However, this assumption is not critical to the benefits of ADP ribosylation, as an alternative ADPr+ model, in which only the free unmodified BiP (B) was subject to degradation (obtained by setting kdb for substrate bound [UB] and modified [Br] pools to to 0), also defended against aggregation while degrading less unfolded proteins than the counterpart ADPr− model (Fig. S5, C and D).
Online supplemental material
Fig. S1 shows that the acidic form of BiP is not affected by phosphatase in vitro. Fig. S2 shows that 32P metabolic labeling of BiP survives phosphatase and hydroxylamine in vitro and is limited to two Arg residues. Fig. S3 shows structural and functional integrity of the mutant BiP proteins in vitro. Fig. S4 shows that the advantages of ADPr+ model over the ADPr− model are maintained over a range of diurnal excursions in protein synthesis rates and gains of the transcriptional UPR. Fig. S5 shows that alternative ADPr+ models for substrate binding activity of ADP-ribosylated BiP and BiP degradation also afford an advantage over an ADPr− model. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201202005/DC1.
Supplementary Material
Acknowledgments
We are grateful to Matthias Meyer (Zentrum für Molekulare Biologie der Universität Heidelberg, Heidelberg, Germany), Ursula Jakob (University of Michigan, Ann Arbor, MI) Johannes Buchner (Technische Universität München, Munich, Germany), and Friedrich Koch-Nolte (University of Hamburg, Hamburg, Germany) for scientific advice; Richard Zimmermann for the gift of antisera to BiP; Tim Newton (University of Cambridge, Cambridge, England, UK) for CD spectral analysis of BiP proteins; Steven Blais, Pawel Sadowski, and Tom Neubert (New York University, New York, NY) and Kathryn Lilley (University of Cambridge, Cambridge, England, UK) for efforts at protein identification by mass spectrometry; Arza Ron, Amiram Ron, and Amos Onn (Haifa, Israel) for early work on the kinetic model; Yun Yang (New York University, New York, NY) and Alisa Zyryanova (University of Cambridge, Cambridge, England, UK) for technical assistance; and Benedict Cross and Heather P. Harding (University of Cambridge, Cambridge, England, UK) for advice and suggestions.
This work was supported by National Institutes of Health grant DK047119 and by the Wellcome Trust. D. Ron is a Wellcome Trust Principal Research Fellow.
Footnotes
Abbreviations used in this paper:
- ART
- ADP ribosyltransferase
- CD
- circular dichroism
- CIP
- calf intestinal phosphatase
- NTA
- nitrilotriacetic acid
- PERK
- protein kinase RNA–like ER kinase
- UPRer
- ER unfolded protein response
References
- Ahrén B. 2000. Diurnal variation in circulating leptin is dependent on gender, food intake and circulating insulin in mice. Acta Physiol. Scand. 169:325–331 10.1046/j.1365-201x.2000.00746.x [DOI] [PubMed] [Google Scholar]
- Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. 2008. Adapting proteostasis for disease intervention. Science. 319:916–919 10.1126/science.1141448 [DOI] [PubMed] [Google Scholar]
- Bernales S., McDonald K.L., Walter P. 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen E.B., Chang L., Gestwicki J.E., Zuiderweg E.R. 2009. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA. 106:8471–8476 10.1073/pnas.0903503106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A., Theyssen H., Schröder H., McCarty J.S., Virgallita G., Milkereit P., Reinstein J., Bukau B. 1995. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J. Biol. Chem. 270:16903–16910 10.1074/jbc.270.28.16903 [DOI] [PubMed] [Google Scholar]
- Bukau B., Weissman J., Horwich A. 2006. Molecular chaperones and protein quality control. Cell. 125:443–451 10.1016/j.cell.2006.04.014 [DOI] [PubMed] [Google Scholar]
- Carlsson L., Lazarides E. 1983. ADP-ribosylation of the Mr 83,000 stress-inducible and glucose-regulated protein in avian and mammalian cells: Modulation by heat shock and glucose starvation. Proc. Natl. Acad. Sci. USA. 80:4664–4668 10.1073/pnas.80.15.4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M.M., Huh J.W., Yang S.J., Cho E.H., Choi S.Y., Cho S.W. 2005. Identification of ADP-ribosylation site in human glutamate dehydrogenase isozymes. FEBS Lett. 579:4125–4130 10.1016/j.febslet.2005.06.041 [DOI] [PubMed] [Google Scholar]
- Despa F. 2010. Endoplasmic reticulum overcrowding as a mechanism of beta-cell dysfunction in diabetes. Biophys. J. 98:1641–1648 10.1016/j.bpj.2009.12.4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölle C., Niere M., Lohndal E., Ziegler M. 2010. Visualization of subcellular NAD pools and intra-organellar protein localization by poly-ADP-ribose formation. Cell. Mol. Life Sci. 67:433–443 10.1007/s00018-009-0190-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner A.J., Wasley L.C., Kaufman R.J. 1992. Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J. 11:1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder J.H., Rossi J.M., Solomon J., Solomon N., Lindquist S. 1992. The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev. 6:1402–1413 10.1101/gad.6.8.1402 [DOI] [PubMed] [Google Scholar]
- Fernández-Sáiz V., Moro F., Arizmendi J.M., Acebrón S.P., Muga A. 2006. Ionic contacts at DnaK substrate binding domain involved in the allosteric regulation of lid dynamics. J. Biol. Chem. 281:7479–7488 10.1074/jbc.M512744200 [DOI] [PubMed] [Google Scholar]
- Freiden P.J., Gaut J.R., Hendershot L.M. 1992. Interconversion of three differentially modified and assembled forms of BiP. EMBO J. 11:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Ron D. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 397:271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D.D., Ron D. 2001. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell. 7:1153–1163 10.1016/S1097-2765(01)00264-7 [DOI] [PubMed] [Google Scholar]
- Hendershot L.M., Ting J., Lee A.S. 1988. Identity of the immunoglobulin heavy-chain-binding protein with the 78,000-dalton glucose-regulated protein and the role of posttranslational modifications in its binding function. Mol. Cell. Biol. 8:4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Yraola A., Bakhit S.M., Franke P., Weise C., Schweiger M., Jorcke D., Ziegler M. 2001. Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. EMBO J. 20:2404–2412 10.1093/emboj/20.10.2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia J.A., Tsai S.C., Adamik R., Yost D.A., Hewlett E.L., Moss J. 1985. Amino acid-specific ADP-ribosylation. Sensitivity to hydroxylamine of [cysteine(ADP-ribose)]protein and [arginine(ADP-ribose)]protein linkages. J. Biol. Chem. 260:16187–16191. [PubMed] [Google Scholar]
- Hu C.C., Dougan S.K., Winter S.V., Paton A.W., Paton J.C., Ploegh H.L. 2009. Subtilase cytotoxin cleaves newly synthesized BiP and blocks antibody secretion in B lymphocytes. J. Exp. Med. 206:2429–2440 10.1084/jem.20090782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N., Okamoto H. 1980. Translational control of proinsulin synthesis by glucose. Nature. 283:100–102 10.1038/283100a0 [DOI] [PubMed] [Google Scholar]
- Jamieson J.D., Palade G.E. 1971. Synthesis, intracellular transport, and discharge of secretory proteins in stimulated pancreatic exocrine cells. J. Cell Biol. 50:135–158 10.1083/jcb.50.1.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y., Kohno K. 2011. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 23:135–142 10.1016/j.ceb.2010.10.008 [DOI] [PubMed] [Google Scholar]
- Knarr G., Kies U., Bell S., Mayer M., Buchner J. 2002. Interaction of the chaperone BiP with an antibody domain: Implications for the chaperone cycle. J. Mol. Biol. 318:611–620 10.1016/S0022-2836(02)00166-3 [DOI] [PubMed] [Google Scholar]
- Knowles T.P., Waudby C.A., Devlin G.L., Cohen S.I., Aguzzi A., Vendruscolo M., Terentjev E.M., Welland M.E., Dobson C.M. 2009. An analytical solution to the kinetics of breakable filament assembly. Science. 326:1533–1537 10.1126/science.1178250 [DOI] [PubMed] [Google Scholar]
- Koch-Nolte F., Kernstock S., Mueller-Dieckmann C., Weiss M.S., Haag F. 2008. Mammalian ADP-ribosyltransferases and ADP-ribosylhydrolases. Front. Biosci. 13:6716–6729 10.2741/3184 [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y., Segal M., Normington K., Gething M.J., Sambrook J. 1988. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 332:462–464 10.1038/332462a0 [DOI] [PubMed] [Google Scholar]
- Laitusis A.L., Brostrom M.A., Brostrom C.O. 1999. The dynamic role of GRP78/BiP in the coordination of mRNA translation with protein processing. J. Biol. Chem. 274:486–493 10.1074/jbc.274.1.486 [DOI] [PubMed] [Google Scholar]
- Ledford B.E., Jacobs D.F. 1986. Translational control of ADP-ribosylation in eucaryotic cells. Eur. J. Biochem. 161:661–667 10.1111/j.1432-1033.1986.tb10491.x [DOI] [PubMed] [Google Scholar]
- Leno G.H., Ledford B.E. 1989. ADP-ribosylation of the 78-kDa glucose-regulated protein during nutritional stress. Eur. J. Biochem. 186:205–211 10.1111/j.1432-1033.1989.tb15196.x [DOI] [PubMed] [Google Scholar]
- Liebermeister W., Rapoport T.A., Heinrich R. 2001. Ratcheting in post-translational protein translocation: A mathematical model. J. Mol. Biol. 305:643–656 10.1006/jmbi.2000.4302 [DOI] [PubMed] [Google Scholar]
- Lin J.H., Li H., Zhang Y., Ron D., Walter P. 2009. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE. 4:e4170 10.1371/journal.pone.0004170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Hendrickson W.A. 2007. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 131:106–120 10.1016/j.cell.2007.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetopoulos J., Jain K. 1980. In vivo incorporation of [3H[ leucine and [3H] tryptophan into proinsulin-insulin and other islet cell proteins in normoglycemic, hyperglycemic, and hypoglycemic rats. Diabetes. 29:801–805 10.2337/diabetes.29.10.801 [DOI] [PubMed] [Google Scholar]
- Marcinowski M., Höller M., Feige M.J., Baerend D., Lamb D.C., Buchner J. 2011. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 18:150–158 10.1038/nsmb.1970 [DOI] [PubMed] [Google Scholar]
- Mayer M., Reinstein J., Buchner J. 2003. Modulation of the ATPase cycle of BiP by peptides and proteins. J. Mol. Biol. 330:137–144 10.1016/S0022-2836(03)00556-4 [DOI] [PubMed] [Google Scholar]
- Mayer M.P., Bukau B. 2005. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62:670–684 10.1007/s00018-004-4464-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.P., Laufen T., Paal K., McCarty J.S., Bukau B. 1999. Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J. Mol. Biol. 289:1131–1144 10.1006/jmbi.1999.2844 [DOI] [PubMed] [Google Scholar]
- Mayer M.P., Schröder H., Rüdiger S., Paal K., Laufen T., Bukau B. 2000. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7:586–593 10.1038/76819 [DOI] [PubMed] [Google Scholar]
- Meunier L., Usherwood Y.K., Chung K.T., Hendershot L.M. 2002. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell. 13:4456–4469 10.1091/mbc.E02-05-0311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisset J.A., Webster P.D. 1972. Effects of fasting and feeding on protein synthesis by the rat pancreas. J. Clin. Invest. 51:1–8 10.1172/JCI106779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J., Stanley S.J., Nightingale M.S., Murtagh J.J., Jr, Monaco L., Mishima K., Chen H.C., Williamson K.C., Tsai S.C. 1992. Molecular and immunological characterization of ADP-ribosylarginine hydrolases. J. Biol. Chem. 267:10481–10488. [PubMed] [Google Scholar]
- Munro S., Pelham H.R. 1986. An Hsp70-like protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 46:291–300 10.1016/0092-8674(86)90746-4 [DOI] [PubMed] [Google Scholar]
- Otero J.H., Lizák B., Hendershot L.M. 2010. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 21:472–478 10.1016/j.semcdb.2009.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton A.W., Beddoe T., Thorpe C.M., Whisstock J.C., Wilce M.C., Rossjohn J., Talbot U.M., Paton J.C. 2006. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 443:548–552 10.1038/nature05124 [DOI] [PubMed] [Google Scholar]
- Petrova K., Oyadomari S., Hendershot L.M., Ron D. 2008. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 27:2862–2872 10.1038/emboj.2008.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlecht R., Erbse A.H., Bukau B., Mayer M.P. 2011. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 18:345–351 10.1038/nsmb.2006 [DOI] [PubMed] [Google Scholar]
- Shampine L.F., Thompson S. 2001. Solving DDEs in MATLAB. Appl. Numer. Math. 37:441–458 10.1016/S0168-9274(00)00055-6 [DOI] [Google Scholar]
- Sidrauski C., Brickner J.H., Walter P. 2002. The unfolded protein response. Protein targeting transport and translocation. Dalbey R.E., von Heijne G., Elsevier Science Ltd, London: 151–179. [Google Scholar]
- Sitia R., Braakman I. 2003. Quality control in the endoplasmic reticulum protein factory. Nature. 426:891–894 10.1038/nature02262 [DOI] [PubMed] [Google Scholar]
- Stefan Y., Meda P., Neufeld M., Orci L. 1987. Stimulation of insulin secretion reveals heterogeneity of pancreatic B cells in vivo. J. Clin. Invest. 80:175–183 10.1172/JCI113045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J.F., Dinler G., Sivendran R., Montgomery D.L., Stotz M., Gierasch L.M. 2007. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell. 26:27–39 10.1016/j.molcel.2007.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M., Kawamura A., Kohno K. 1992. Purification and characterization of BiP/Kar2 protein from Saccharomyces cerevisiae. J. Biol. Chem. 267:17553–17559. [PubMed] [Google Scholar]
- Trusina A., Papa F.R., Tang C. 2008. Rationalizing translation attenuation in the network architecture of the unfolded protein response. Proc. Natl. Acad. Sci. USA. 105:20280–20285 10.1073/pnas.0803476105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 334:1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wei J., Gaut J.R., Hendershot L.M. 1995. In vitro dissociation of BiP-peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis. J. Biol. Chem. 270:26677–26682 10.1074/jbc.270.44.26677 [DOI] [PubMed] [Google Scholar]
- Zhu X., Zhao X., Burkholder W.F., Gragerov A., Ogata C.M., Gottesman M.E., Hendrickson W.A. 1996. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 272:1606–1614 10.1126/science.272.5268.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.