

Abstract
Motivation: The human microbiome plays an important role in human disease and health. Identification of factors that affect the microbiome composition can provide insights into disease mechanism as well as suggest ways to modulate the microbiome composition for therapeutical purposes. Distance-based statistical tests have been applied to test the association of microbiome composition with environmental or biological covariates. The unweighted and weighted UniFrac distances are the most widely used distance measures. However, these two measures assign too much weight either to rare lineages or to most abundant lineages, which can lead to loss of power when the important composition change occurs in moderately abundant lineages.
Results: We develop generalized UniFrac distances that extend the weighted and unweighted UniFrac distances for detecting a much wider range of biologically relevant changes. We evaluate the use of generalized UniFrac distances in associating microbiome composition with environmental covariates using extensive Monte Carlo simulations. Our results show that tests using the unweighted and weighted UniFrac distances are less powerful in detecting abundance change in moderately abundant lineages. In contrast, the generalized UniFrac distance is most powerful in detecting such changes, yet it retains nearly all its power for detecting rare and highly abundant lineages. The generalized UniFrac distance also has an overall better power than the joint use of unweighted/weighted UniFrac distances. Application to two real microbiome datasets has demonstrated gains in power in testing the associations between human microbiome and diet intakes and habitual smoking.
Availability: http://cran.r-project.org/web/packages/GUniFrac
Contact: hongzhe@upenn.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Understanding the compositional differences of microbial communities (microbiomes) is essential in microbial ecology. With the development of next-generation sequencing technologies, microbiome composition can now be determined by direct DNA sequencing without the need for laborious cultivation (Tringe et al., 2005; Gill et al., 2006; von Mering et al., 2007; Dinsdale et al., 2008; Grice et al., 2009; Turnbaugh et al., 2009; Qin et al., 2010). There has been a great interest in human microbiome studies in different body sites, ranging from skin (Grice et al., 2009) to gut (Qin et al., 2010; Arumugam et al., 2011; Muegge et al., 2011; Wu et al., 2011) and respiratory tract (Charlson et al., 2010). Important insights have been gained from analysis of large-scale human microbiome data, including the discovery of enterotypes (Arumugam et al., 2011) and discovery of the link between diet and these enterotypes (Wu et al., 2011). Although the metagenomic shotgun approach is potentially more powerful and unbiased, 16S rRNA gene targeted sequencing is routinely performed to determine the taxonomic composition. The generated 16S rRNA sequence tags are usually clustered into operational taxonomic units (OTUs) with a specified amount of variability allowed within each OTU (Schloss et al., 2009; Caporaso et al., 2010). At 97% similarity, these OTUs represent ‘species’. Downstream analysis is then performed on the OTU abundance data.
Two central themes in human microbiome studies are to identify potential biological and environmental factors that are associated with microbiome composition, and to define the relationship between microbiome features and biological or clinical outcomes. The goal is to provide a better understanding of the factors that shape our microbiome and, potentially, contribute to the development of new therapeutic strategies to modulate the microbiome composition and affect human health (Spor et al., 2011; Virgin and Todd, 2011). Testing the association of microbiome composition with potential environmental factors using OTU abundances directly is difficult due to high dimensionality, non-normality and phylogenetic structure of the OTU data. Instead, distance-based non-parametric testing, in which a distance measure is defined-between any two microbiome samples, is usually used to achieve this goal (Fukuyama et al., 2012; Kuczynski et al., 2010a; Wu et al., 2011, 2010; Charlson et al., 2010). The power of the distance-based test depends on a proper choice of the distance measure. Numerous distance measures have been proposed to compare microbial communities (Kuczynski et al., 2010b; Swenson, 2011). Phylogenetic distance measures, which account for the phylogenetic relationship among the species, provide far more power because they exploit the degree of divergence between different sequences. Among these, the UniFrac distances are the most popular ones (Lozupone and Knight, 2005; Lozupone et al., 2007). There are two versions of UniFrac distances: an unweighted UniFrac distance that considers only species presence and absence information and counts the fraction of branch length unique to either community, and a weighted UniFrac distance that uses species abundance information and weights the branch length with abundance difference. Unweighted UniFrac distance is most efficient in detecting abundance change in rare lineages. When the abundance of a rare lineage falls below a certain threshold, the sequencing machine may not be able to pick it up and it will appear absent in the final dataset. On the other hand, weighted UniFrac distance is most sensitive to detect change in abundant lineages since it uses absolute abundance difference in its definition. However, unweighted/weighted UniFrac distances may not be very powerful in detecting change in moderately abundant lineages. Recently, a variance-adjusted weighted UniFrac distance (VAW-UniFrac), which moderates the branch proportion difference by its variance, was developed to account for the fact that weighted UniFrac distance does not consider the variation of the weights under random sampling (Chang et al., 2011). VAW-UniFrac was shown to increase the power over weighted UniFrac distance for detecting the difference between two microbial communities.
In this article, we introduce generalized UniFrac distances that unify weighted UniFrac and unweighted UniFrac distances. The new generalized UniFrac distances cover a series of distances ranging from weighted to unweighted UniFrac by adjusting the weight on the branches. The generalized UniFrac distances are designed to provide a robust and powerful tool for detecting a wider range of biologically relevant changes in microbiome composition. We conduct extensive Monte Carlo simulation studies under various conditions to evaluate their power in detecting environmental influence on microbiome composition using PERMANOVA (McArdle, 2001), a distance-based non-parametric test. Although each distance in the series can perform the best in certain scenarios, none has the optimal performance under all conditions considered. However, analyses based on the generalized UniFrac distances are shown to be more robust and has overall the best performances across a range of possible scenarios. We demonstrate the power gain of using this distance in detecting the microbiome differences by analysis of two real human gut microbiome datasets related to linking human gut microbiome composition to long-term diet (Wu et al., 2011) and testing upper respiratory tract microbiome difference between smokers and non-smokers (Charlson et al., 2010).
2 METHODS
2.1 Generalized UniFrac distances between two microbial communities
Consider two microbiome communities A and B and suppose that we have a rooted phylogenetic tree with n branches. Let bi be the length of the branch i and piA and piB are the taxa proportions descending from the branch i for community A and B, respectively. The unique fraction metric, or UniFrac, measures the phylogenetic distance between sets of taxa in a phylogenetic tree as the fraction of the branch length of the tree that leads to descendants from either one environment or the other, but not both. The original definition refers to unweighted UniFrac (Lozupone and Knight, 2005), which is mathematically defined as
where I(.) is the indicator function and only presence/absence of species of branch i, I(piA > 0) and I(piB > 0), are used in the definition. The distance definition dU completely ignores the taxa abundance information. In contrast, the (normalized) weighted UniFrac distance (Lozupone et al., 2007) weights the branch length with abundance difference and is defined as
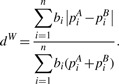 |
Note that dW cannot be reduced to dU even if we convert abundance data into presence/absence data. Also note that dW uses the absolute proportion difference |piA − piB| in its formulation. The consequence of using the absolute difference is that the value of dW is determined mainly by branches with large proportions and is less sensitive to the abundance changes on the branches with small proportions. To attenuate the weight on branches with large proportions, we may instead use the relative difference |piA − piB|/(piA + piB) (∈ [0,1]) in the formulation. We denote this distance measure as
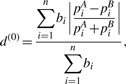 |
where 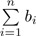 in the denominator is the normalizing factor so that d(0) ∈ [0,1]. Now if we dichotomize the abundance data using the indication function I(.), d(0) is reduced to dU. So d(0) can be seen as the ‘weighted version’ of dU. Using the relative differences, we place equal emphasis on every branch and the distance is not dominated by the branches with large proportions, since the relative difference does not depend on the magnitude of piA,piB. However, the low-abundance branches may be more noisy and the relative difference may amplify such noises. To strike a balance between relative difference and absolute difference, we weight the branch length both by the relative difference and its importance indicated by the branch proportion. We propose the following generalized UniFrac distances
in the denominator is the normalizing factor so that d(0) ∈ [0,1]. Now if we dichotomize the abundance data using the indication function I(.), d(0) is reduced to dU. So d(0) can be seen as the ‘weighted version’ of dU. Using the relative differences, we place equal emphasis on every branch and the distance is not dominated by the branches with large proportions, since the relative difference does not depend on the magnitude of piA,piB. However, the low-abundance branches may be more noisy and the relative difference may amplify such noises. To strike a balance between relative difference and absolute difference, we weight the branch length both by the relative difference and its importance indicated by the branch proportion. We propose the following generalized UniFrac distances
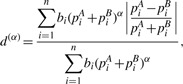 |
where α ∈ [0,1] controls the contribution from high-abundance branches, and 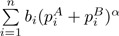 is the normalizing factor so that d(α) ∈ [0,1]. Branches with zero proportions for both communities will not be included in the calculation. As α changes from 0 to 1, more emphasis is placed on high-abundance branches. When α = 1, d(α) is reduced to dW. When α = 0, we get d(0) defined above.
is the normalizing factor so that d(α) ∈ [0,1]. Branches with zero proportions for both communities will not be included in the calculation. As α changes from 0 to 1, more emphasis is placed on high-abundance branches. When α = 1, d(α) is reduced to dW. When α = 0, we get d(0) defined above.
Therefore, by varying α from 1 to 0, we achieve a series of distances ranging from dW to d(0). Note that dU is obtained by dichotomizing the abundance in d(0), but is different from d(0). We are particularly interested in d(0.5), the distance in the middle of the distance series
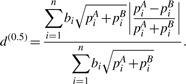 |
We also compare dW,d(0.5),d(0) and dU with VAW-UniFrac distance dVAW (Chang et al., 2011), which is defined as:
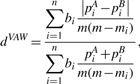 |
where mi is the total number of individuals/reads from both communities on the i th branch and m is total number of individuals/reads.
2.2 Statistical test based on UniFrac distances
We study the power of generalized UniFrac distances using the distance-based non-parametric test for association of microbiome composition with environmental covariates. Suppose we have a set of m environmental covariates. We assume that we have collected microbiome data and the m-dimensional covariates data X on n samples. We apply the PERMANOVA procedure (McArdle, 2001) [Permutational Multivariate Analysis of Variance Using Distance Matrices, ‘adonis’ function from R package ‘vegan’ (Oksanen et al., 2011)], which partitions the distance matrix among sources of variation, fits linear models to distance matrices and uses a permutation test with pseudo-F ratios to obtain the P-values. The pseudo-F statistic is defined as
where tr(.) is the trace function of a matrix, H = X(XTX)−1XT is the hat (projection) matrix of the design matrix X, G is Gower's centered matrix and n and m is the number of samples and the number of predictors, respectively. Let dij be the generalized UniFrac distance between community i and j and denote 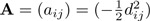 . The Gower's matrix is defined as
. The Gower's matrix is defined as
where 1 is a vector of 1's.
Since dU and dW reflect the abundance change in either rare lineages or abundant lineages, combining dU and dW may potentially increase the overall power. Instead of applying Bonferroni correction to the P-values from separate PERMANOVA tests using dU or dW to control the family-wise Type I error rate, a more powerful approach is to take the maximum of pseudo-F statistics for dU and dW as a new test statistic. The significance of the pseudo-F statistics is assessed based on permutations.
2.3 Simulation strategies
We use two simulation strategies to evaluate the power of the generalized UniFrac distances under various conditions. The first strategy is a modification of the simulation method proposed by Schloss (2008), where we draw points (16S rRNA sequences) from a 2D circle with known densities (Fig. 1A). This strategy facilitates simulations of different community characteristics such as species evenness and species richness. The Euclidean distance between points is analogous of the genetic distance between the sequences. The diameter of the circle represents the maximum genetic divergence between any pair of sequences within a sample. The area of the circle is proportional to the richness and the density distribution of the circle is proportional to the evenness. By varying the centroid positions (o) and their radius (r), it is possible to vary the fraction of shared membership and species richness within each sample (Fig. 1B and D). By varying the point distribution on the circle (density proportional to rα, where α controls the degree of evenness and α = 0.5 for uniformly distribution), it is possible to change the species evenness (Fig. 1C). We also simulate scenarios where lineages of different abundance levels change by a k fold (Fig. 1E–G). These are achieved by simulating the community with point mass concentrated at the circle center (r1.0) and varying the point density in different regions of the 2D circle corresponding to abundant lineages (0–0.2r from the center; Fig. 1E), moderately abundant lineages (0.4r–0.8r from the center; Fig. 1F) and rare lineages (0.8r–1.0r from the center; Fig. 1G). We further bin the sampled points into small hexagons as ‘OTU’s before calculating the UniFrac distance [‘hexbin’ function from the R package ‘hexbin’ (Carr et al., 2011)]. The phylogenetic tree of these ‘OTU’s is built using NJ algorithm (Neighbor Joining, ‘nj’ function in R) and rooted by midpoint rooting method. Generalized UniFrac distances are then calculated based on the NJ tree and ‘OTU’ abundances. Each replication consists of drawing 400 points from each community, a bin size of 0.015 units to form ‘OTUs’ (∼ 300 OTUs per sample), and the maximum distance between any two points is 0.3 units (r = 0.15), corresponding to typical phylum level divergence of 30% for 16S rRNA gene. These conditions allow us to simulate the sampling intensity and biodiversity found within a typical 16S rRNA gene targeted sequencing experiment (Schloss, 2008).
Fig. 1.

Two simulation strategies to evaluate the generalized UniFrac distances. (A–G), 2D circle-based simulation of microbial communities with different characteristics. (A) The microbial community is represented by a 2D circle. Points are drawn from the circle to simulate the 16S-based sampling process. These points are further binned into small hexagons as OTUs. UPGMA or NJ method is used to build the OTU phylogenetic tree. Six scenarios are investigated, where the difference occurs in: community membership (B), evenness (C), richness (D), most abundant lineages (E), moderately abundant lineages (F) and rare lineages (G). The affected lineages are indicated by a red circle or ring. H, tree-based simulation of microbial communities based on the phylogenetic tree and DM model. A real OTU phylogenetic tree from a throat microbial community dataset is used. These OTUs are roughly divided into 20 clusters (lineages) by performing PAM method using the OTU patristic distance matrix. Each cluster is subjected to abundance change in response to the environment. Counts are generated from a DM model.
The second set of simulations utilize a real upper respiratory tract microbiome dataset consisting of 60 samples and 856 OTUs from Charlson et al. (2010) (Fig. 1H). A common way of modeling multivariate count data is to use the multinomial model. However, the multinomial model assumes fixed underlying proportions for each sample, which do not hold for real microbiome data due to high degree of heterogeneity among the samples. The real OTU count distribution (Supplementary Fig. S1A) exhibits more variance than expected from a multinomial model (Supplementary Fig. S1B). To realistically simulate the data, it is important to model extra-variation or overdispersion of the OTU counts. This can be achieved by using the Dirichlet-multinomial (DM) model (Mosimann, 1962), which assumes the underlying proportions of the multinomial model come from a Dirichlet distribution. The density function of a DM random variable N is given as
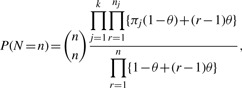 |
where 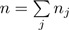 is total count, k is the OTU number and proportion mean π = (π1,π2,⋯,πk) and dispersion θ are parameters. When θ = 0, it is reduced to multinomial model. We estimate the DM parameters π,θ using maximum likelihood method (‘dirmult’ function from R package ‘dirmult’). We then generate OTU counts using the DM model with the estimated parameters and 1000 counts per sample. Supplementary Figure S1C shows an OTU heatmap generated by the DM model, in which the overdispersion is similar to that of the real data. To study the power of UniFrac variants for identifying potential environmental factors, we let the abundance of a certain OTU cluster change in response to environment. We use the UPGMA tree of the OTUs based on the OTU distance matrix calculated under the K80 nucleotide substitution model (Felsenstein, 2004), QIIME (FastTree algorithm (?)) and partition the 856 OTUs into 20 clusters using Partitioning Around Medoids (PAM) (‘pam’ function from R package ‘cluster’) based on patristic distances (the length of the shortest path linking two OTUs on the tree). These OTU clusters are highlighted in different colors in Figure 1H.
is total count, k is the OTU number and proportion mean π = (π1,π2,⋯,πk) and dispersion θ are parameters. When θ = 0, it is reduced to multinomial model. We estimate the DM parameters π,θ using maximum likelihood method (‘dirmult’ function from R package ‘dirmult’). We then generate OTU counts using the DM model with the estimated parameters and 1000 counts per sample. Supplementary Figure S1C shows an OTU heatmap generated by the DM model, in which the overdispersion is similar to that of the real data. To study the power of UniFrac variants for identifying potential environmental factors, we let the abundance of a certain OTU cluster change in response to environment. We use the UPGMA tree of the OTUs based on the OTU distance matrix calculated under the K80 nucleotide substitution model (Felsenstein, 2004), QIIME (FastTree algorithm (?)) and partition the 856 OTUs into 20 clusters using Partitioning Around Medoids (PAM) (‘pam’ function from R package ‘cluster’) based on patristic distances (the length of the shortest path linking two OTUs on the tree). These OTU clusters are highlighted in different colors in Figure 1H.
We call the first strategy 2D circle-based simulation and the second tree-based simulation. For power calculation, we use 2000 replications.
3 RESULTS
3.1 Comparison of the power of different UniFrac variants using 2D circle-based simulations
We use PERMANOVA to test for environmental effect and compare the power of dW,d(0.5),d(0), dU and dVAW. Specifically, we simulate two environmental conditions (e.g. smoking versus non-smoking) under which we draw 10 samples each. We then vary the degree of community difference under these two conditions and produce the power curve over a grid of 10 for each UniFrac distance. We investigate six scenarios, where the environmental factor affects the community membership, species evenness, species richness, most abundant lineages moderately abundant lineages and rare lineages (Fig. 1B–G). For each scenario, we vary one community characteristic (Supplementary Table S1). Suppose x1 and x2 are the mean values of the community characteristic for Conditions 1 and 2. We simulate 10 communities for each condition with community characteristic value xij ∼ Uniform(xj − s, xj + s) for i = 1…10 and j = 1,2, where s controls the variation within each condition and takes different values for different scenarios (see Supplementary Table S1). Each community is sampled once. Initially, we let x1 = x2 (no difference) and then increase the difference between x1 and x2 to simulate stronger environmental effect. PERMANOVA is then performed on the distance matrices and the power curve is created over a grid of 10 using Type I error α = 0.05. Figure 2 shows the power curves for different UniFrac distances under the six scenarios considered. When the environmental factor has no effect (x1 = x2), PERMANOVA controls the Type I error at the nominal level of 0.05 for all five UniFrac distances. As the environmental effect becomes stronger, all the distances have better power. When the environmental factor affects the community membership or richness (Panels 1 and 3), all the distances give a similar power and their power curves are nearly identical. For the evenness change scenario (Panel 2), the power of dW and d(0.5) is very close and is more powerful than d(0) and dU. dW is the most powerful for detecting change in most abundant lineages (Panel 4) but is much less powerful for change in rare lineages (Panel 6). dU shows an opposite trend: it is the most powerful for detecting change in rare lineages (Panel 6) but has almost no power for change in most abundant lineages (Panel 4). In contrast, d(0.5) is the most powerful for detecting change in moderately abundant lineages (Panel 5). They are also the most robust among the distances investigated: its power is close to the best UniFrac distance under all scenarios. The performance of d(0) lies between d(0.5) and dU and is also very robust. Finally, under the 2D circle simulations, the performance of dVAW is almost identical to that of d(0.5).
Fig. 2.

Power comparison of different UniFrac variants for detecting environmental effect using 2D circle-based simulation. PERMANOVA is used for testing hypotheses. The specific community difference caused by different environmental conditions is indicated in the panel title. The power curves are created by varying the degree of environmental effect. The initial point of the power curve is the power when there is no environmental effect.
In the above simulations, we use a bin size of 0.015 to form ‘OTU’s (∼300 OTUs per sample). To study the effect of bin size, we compare the power curves of the generalized UniFrac distances using a smaller bin size of 0.01 (∼700 OTUs per sample) or a larger bin size of 0.03 (∼80 OTUs per sample). The bin size does not change the general conclusion (Supplementary Fig. S2). To study the effect of tree construction methods, we also construct the phylogenetic tree using UPGMA. The general conclusions still hold (Supplementary Fig. S3).
3.2 Comparison of the power of different UniFrac variants using tree-based simulations
We also compare the power of different UniFrac distances for detecting environmental effect using tree-based simulations that mimic the throat microbiome data (see Section 3.4 for details). A recently proposed variance adjusted UniFrac distance (dVAW) (Chang et al., 2011) is also compared in this setting. dVAW was developed to moderate the branch proportion difference by its variance and was shown to increase the power of detecting the difference between two microbial communities. We use the phylogenetic tree of the 856 OTUs from the throat microbiome dataset and divide them into 20 clusters (Fig. 1H). The mean OTU proportions and the dispersion parameter are estimated from the real data by fitting a DM model. We assume that the environmental factor causes an increase of the abundance of a particular OTU cluster. Specifically, suppose that the proportion of the i th OTU cluster under Condition 1 is pi. For Condition 2, the proportion of i th OTU cluster is increased by k fold where k varies from 1 (no difference) to 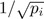 (strong effect) on a grid of 10. The proportion vector is re-normalized to sum to 1. Next, 10 samples are simulated for each condition with their OTU counts generated by the DM model with the corresponding proportion vector and the common dispersion parameter. As expected, the five UniFrac distances differ in their power for detecting environmental effect for the 20 OTU clusters tested. Except for d(0), all the UniFrac distances have their best-performance scenarios. dW, d(0.5), dU and dVAW achieve the highest power in seven, six, three and one cases, respectively. For the remaining three cases, dW and d(0.5) are equally the most powerful (Supplementary Fig. S4). The results are consistent with the 2D circle-based simulation: dW is most powerful in detecting the environmental effect on most abundant lineages, d(0.5) is most powerful for moderately abundant lineages and dU is most powerful for rare lineages. In contrast, performance of the test with d(0) and dVAW is generally between dU and d(0.5). The power of dW and dU has a reciprocal relationship and neither of them is as robust as d(0.5). Figure 3A shows the power curves of four representative cases. As the proportion of the affected cluster decreases from 19.7% to 0.9%, dW becomes less powerful and the power of dU has the opposite trend.
(strong effect) on a grid of 10. The proportion vector is re-normalized to sum to 1. Next, 10 samples are simulated for each condition with their OTU counts generated by the DM model with the corresponding proportion vector and the common dispersion parameter. As expected, the five UniFrac distances differ in their power for detecting environmental effect for the 20 OTU clusters tested. Except for d(0), all the UniFrac distances have their best-performance scenarios. dW, d(0.5), dU and dVAW achieve the highest power in seven, six, three and one cases, respectively. For the remaining three cases, dW and d(0.5) are equally the most powerful (Supplementary Fig. S4). The results are consistent with the 2D circle-based simulation: dW is most powerful in detecting the environmental effect on most abundant lineages, d(0.5) is most powerful for moderately abundant lineages and dU is most powerful for rare lineages. In contrast, performance of the test with d(0) and dVAW is generally between dU and d(0.5). The power of dW and dU has a reciprocal relationship and neither of them is as robust as d(0.5). Figure 3A shows the power curves of four representative cases. As the proportion of the affected cluster decreases from 19.7% to 0.9%, dW becomes less powerful and the power of dU has the opposite trend.
Fig. 3.

Power comparison of different UniFrac variants for detecting environmental effect using tree-based simulation. PERMANOVA is used for testing hypotheses. The power curves are created by varying the degree of environmental effect. (A) The environmental factor affects a particular lineage (OTU cluster). Four example lineages of different abundance levels that are affected by environment are given. The lineage abundance is given in parentheses in the panel title. (B) The environmental factor affects a random lineage (left panel) or a random subset of 40 OTUs (right panel). The initial point of the power curve is the power when there is no environmental effect.
In the simulations presented above, the power is calculated assuming the affected cluster is known. Since the affected cluster can be abundant or rare, we randomly choose an affected OTU cluster in each replication and calculate the power over 2000 replications. We also report the power for the test combining dW and dU by taking the maximum of their pseudo- F statistics. We denote this method as dMAX. Figure 3B (left plot) shows that dU has the lowest overall power and d(0.5) and dMAX have the best power, indicating combining dU and dW can lead to power gain. In contrast, the power of d(0), dVAW and dW is in between. As the environmental effect becomes stronger, d(0) becomes as powerful as d(0.5) and dMAX. Finally, we assume that the environmental factor affects a random set of 40 OTUs instead of a random OTU cluster. At this extreme where the phylogenetic relationship is no longer important, d(0.5) has even higher power than the other distances, followed by d(0), dMAX, dW, dU and dVAW (see Fig. 3B, right plot). Overall, d(0.5) has a better power than other UniFrac distances including the one that combines dW and dU.
3.3 Results from analysis of a dataset linking long-term diet to gut microbiome composition
Diet strongly affects the human health, partly by modulating gut microbiome composition. Wu et al. (2011) studied the habitual diet effect on the human gut microbiome, where the diet information was converted into a vector of micro-nutrient intakes. A total of 98 healthy volunteers were enrolled in this cross-sectional study. Habitual long-term diet information was collected using food frequency questionnaire (FFQ). The questionnaires were converted to intake amounts of 214 micro-nutrients. Nutrient intake was further normalized using the residual method to standardize for caloric intake. Stool samples were collected and DNA samples were analyzed by 454/Roche pyrosequencing of 16S rRNA gene segments of the V1-V2 region. The pyrosequences were denoised (Quince et al., 2009) prior to taxonomic assignment yielding an average of 9265 ± 3864(SD) reads per sample. The denoised sequences were then analyzed by the QIIME pipeline (Caporaso et al., 2010) with the default parameter settings in the QIIME pipeline. The OTU table contains 3068 OTUs after discarding the singleton OTUs. One objective of the study is to identify nutrients that have a significant impact on the gut microbiome composition. We use PERMANOVA to test for association of microbiome composition with nutrient intake based on different UniFrac distance matrices. We compare d(0.5) with dU, dW, their combination dMAX and dVAW. We plot the number of selected nutrients against different P-value cutoffs to create a ROC-like curve (Fig. 4). For P < 0.01, all distances except the dU identify the same number of nutrients. For P > 0.03, the curve for d(0.5) is above all the other four curves. Wilcoxon signed-rank tests show that d(0.5) results in smaller P-values than other distances (P < 0.05), indicating that d(0.5) is most powerful in selecting the relevant microbiome-associated nutrients. Using dW or dU alone could miss important associations when the nominal P-value of 0.03–0.05 is used. Although using a relatively large nominal P-value can certainly lead to inclusion of possible false-positive nutrient-microbiome associations, there are situations when one might want to include all possible associations for further validation or replications. dVAW performs the second best. Interestingly, dMAX, the joint use of dW and dU, does not increase the power over dW, indicating most associations can be recovered by dW alone.
Fig. 4.

Comparison of different UniFrac variants for detecting nutrient effects on gut microbiome composition. PERMANOVA is used for testing hypotheses. 214 nutrients are included in the testing. The curves are generated by varying the P-value cutoffs.
3.4 Results from analysis of a throat microbiome dataset of smokers and non-smokers
Cigarette smokers have an increased risk of multiple diseases, including upper respiratory tract infections. Previous studies had linked smoking to specific respiratory tract bacteria, but the consequences of smoking for global airway microbial community composition had not been fully clarified. Charlson et al. (2010), investigated the smoking effect on the oropharyngeal and nasopharyngeal bacterial communities using 454 pyrosequencing of 16S sequence tags. Specifically, a total of 291 swab samples from the right and left nasopharynx and oropharynx of 29 smoking and 33 non-smoking healthy asymptomatic adults were collected. The variable region 1-2 (V1-V2) of the bacterial 16S rRNA gene was PCR-amplified using individually barcoded primer and subject to multiplexed pryosequencing. The pyrosequences were denoised (Quince et al., 2009) prior to taxonomic assignment and yielded an average of 1335 ± 603(SD) reads per airway sample. The denoised sequences were then analyzed using the QIIME pipeline (Caporaso et al., 2010) with default parameter setting. We use the left oropharyngeal samples in this study. After removing two samples with read number < 500 and discarding singleton OTUs, i.e OTU with only one read, we finally have an OTU table of 60 samples (28 smokers versus 32 non-smokers) and 856 OTUs.
We test the smoking effect on the throat microbial community composition by applying PERMANOVA (10000 permutations). All the five UniFrac distances achieve statistical significance at α = 0.05 level, indicating smoking alters the community composition. However, test using d(0.5) produces the smallest P-value of 0.006, followed by 0.008 from d(0). The P-values based on dW, dU and dVAW are 0.012, 0.019 and 0.043, respectively. We also perform a principle coordinate analysis using the distance matrices and plot the samples on the first two principle coordinates (Fig. 5). The distance d(0.5) separates the samples better than the other three distance measures. This indicates that smoking might affect not only the predominant lineages but also these less abundant lineages in the throat microbial community. We then perform Wilcoxon rank-sum or Fisher’s exact test to select the differential OTUs. At α = 0.05 level, we identify 32 OTUs. These OTUs belong to genera Prevotella (8), Lachnospiraceae (5), Veillonella (3), Streptococcus (2), Fusobacterium (2), Treponema (2), Neisseria (1), Haemophilus (1), Megasphaera (1), Dialister (1), Moryella (1), Erysipelotrichaceae (1) and four genera from Actinobacteria. Most of the selected OTUs are moderately abundant or rare, so we expect d(0.5) and d(0) to have better power.
Fig. 5.

Comparison of different UniFrac variants for clustering samples from smokers and non-smokers. Principle coordinate analysis is performed on the distance matrices of dW, d(0.5), dU and dVAW. The samples are plotted on the first two principle coordinates. The PERMANOVA P-values are also indicated in this figure. The ellipse center indicates groups means, its main axis corresponds to the first two principle components from principle component analysis and the height and width are variances on that direction.
4 DISCUSSION
Microbiome data are multivariate count data in their original form and are statistically challenging to analyze due to their high dimensionality, phylogenetic constraints among species/OTUs, overdispersion and excessive zeros. To circumvent the difficulty, the data are often summarized in the form of distance matrix. Testing association of microbiome composition with environmental covariates is performed using the distance matrix. We have demonstrated in simulations that the weighted and unweighted UniFrac impose large weight either to abundant lineages or to rare lineages; they can be underpowered in detecting change in moderately abundant lineages. Since microbiome composition change could occur in any lineages, our generalized UniFrac distances, which unify the weighted and unweighted UniFrac in a common framework, enable us to detect a much wider range of biologically relevant changes. Our simulation studies have clearly demonstrated that the generalized UniFrac distance d(0.5) is more robust than dW or dU, and its performances are in general comparable to the best UniFrac distances among the scenarios we considered. In addition, the generalized UniFrac distances are very robust to tree constructing methods. We suggest the use of d(0.5) for testing association of microbiome composition with environmental covariates to avoid missing important findings.
Both weighted and unweighted UniFrac distances are sensitive to sampling depth (Lozupone et al., 2010). Inflated distances at a low sampling depth are caused by sampling variation especially for the rare lineages. The generalized UniFrac distances are also sensitive to sampling depth (Supplementary Fig. S5). However, as the sampling depth increases, the distance stabilizes. For the gut microbiome dataset, we found a sequencing depth of ∼1000 reads is sufficient to stabilize the generalized UniFrac distances. To overcome the potential adverse effects of uneven sampling, rarefaction is usually used to subsample the samples to the same depth. However, when the sampling depth varies greatly across the samples, rarefaction throws away a significant portion of the 16S reads and increases the sampling variation. We found that rarefaction is not necessary, at least, in the context of testing the association of the microbiome composition with covariates (Supplementary Fig. S6).
The power of UniFrac variants can also be compared in the context of testing whether two microbial communities differ significantly as in (Schloss, 2008; Chang et al., 2011). Instead of comparing power for detecting the difference between two communities, we focus our evaluations on the performance of UniFrac distances for associating microbiome composition to environmental covariates by collecting multiple independent samples. The rational is that as the sequence depth increases, two sample comparison will have increased power to detect differences due to sources that we are not interested in (random noises), such as the individual-to-individual variability, day-to-day variability, sampling location variability or even technical variability (e.g. sample preparation). Multiple samples from a population coupled with multivariate statistical methods such as the distance-based PERMANOVA provide powerful design and analysis methods to overcome these potential random noises (Lozupone et al., 2010). As more and more large-scale microbiome datasets are being collected, we expect that our generalized UniFrac distances can help to identify important covariates that are associated with the microbiomes that could be missed using the commonly used UniFrac distances. In addition to identifying environmental covariates that may be determinants of microbiome composition, our approach would be equally suited to identifying microbiome features associated with biological or clinical outcomes, which is needed to begin to understand the impact of the microbiome on health.
Supplementary Material
ACKNOWLEDGEMENT
We thank the three reviewers for very helpful comments.
Funding: This research was supported by the National Institute of Health grants R01CA127334, U01HL098957 and DK083981.
Conflict of Interest: None declared.
REFERENCES
- Arumugam M., et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J., et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr D., et al. hexbin: Hexagonal Binning Routines. 2011 [Google Scholar]
- Chang Q., et al. Variance adjusted weighted UniFrac: a powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinformatics. 2011;12:118. doi: 10.1186/1471-2105-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlson E., et al. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One. 2010;5:e15216. doi: 10.1371/journal.pone.0015216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinsdale E., et al. Functional metagenomic profiling of nine biomes. Nature. 2008;452:629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Inferring Phytogenies. Sunderland, MA: Sinauer Associates; 2004. [Google Scholar]
- Fukuyama J., et al. Comparisons of distance methods for combining covariates and abundances in microbiome studies. Pac. Symp. Biocomput. 2012;12:213–224. [PMC free article] [PubMed] [Google Scholar]
- Gill S., et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice E., et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J., et al. Direct sequencing of the human microbiome readily reveals community differences. Genome Biol. 2010a;11:210–218. doi: 10.1186/gb-2010-11-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J., et al. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods. 2010b;7:813–819. doi: 10.1038/nmeth.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8835. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., et al. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., et al. Unifrac: an effective distance metric for microbial community comparison. ISME J. 2010;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle B. Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology. 2001;82:290–297. [Google Scholar]
- Mosimann J. On the compound multinomial distribution, the multivariate β-distribution, and correlations among proportions. Biometrika. 1962;49:65–82. [Google Scholar]
- Muegge B., et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J., et al. vegan: Community Ecology Package. 2011 [Google Scholar]
- Qin J., et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C., et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- Schloss P., et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P. Evaluating different approaches that test whether microbial communities have the same structure. ISME J. 2008;2:265–275. doi: 10.1038/ismej.2008.5. [DOI] [PubMed] [Google Scholar]
- Spor A., et al. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011;9:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- Swenson N. Phylogenetic beta diversity metrics, trait evolution and inferring the functional beta diversity of communities. PloS One. 2011;6:e21264. doi: 10.1371/journal.pone.0021264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe S., et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- Turnbaugh P., et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H., Todd J. Metagenomics and personalized medicine. Cell. 2011;147:44–56. doi: 10.1016/j.cell.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C., et al. Quantitative phylogenetic assessment of microbial communities in diverse environments. Science. 2007;315:1126–1130. doi: 10.1126/science.1133420. [DOI] [PubMed] [Google Scholar]
- Wu G., et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.