

Abstract
Summary: The MolClass toolkit and data portal generate computational models from user-defined small molecule datasets based on structural features identified in hit and non-hit molecules in different screens. Each new model is applied to all datasets in the database to classify compound specificity. MolClass thus defines a likelihood value for each compound entry and creates an activity fingerprint across diverse sets of screens. MolClass uses a variety of machine-learning methods to find molecular patterns and can therefore also assign a priori predictions of bioactivities for previously untested molecules. The power of the MolClass resource will grow as a function of the number of screens deposited in the database.
Availability and implementation: The MolClass webportal, software package and source code are freely available for non-commercial use at http://tyerslab.bio.ed.ac.uk/molclass. A MolClass tutorial and a guide on how to build models from datasets can also be found on the web site. MolClass uses the chemistry development kit (CDK), WEKA and MySQL for its core functionality. A REST service is available at http://tyerslab.bio.ed.ac.uk/molclass/api based on the OpenTox API 1.2.
Contact: jan.wildenhain@ed.ac.uk or md.tyers@umontreal.ca
1 INTRODUCTION
Bioactive molecules can serve as powerful tools for interrogation of biological systems and/or as precursors in drug discovery. An objective in chemical systems biology is to model biological systems in order to understand the effects of small molecules on cellular processes, and thereby explain the basis for small molecule action (Hopkins, 2008). Realization of this ambitious goal will require extensive experimental datasets. The generation of chemical datasets from biological screening assays is usually limited by cost and throughput. Pharmaceutical companies and academic groups use high-throughput screens to test large libraries of small molecules that elicit a desired biological response, typically against a single target or at most a few related targets. However, chemical space is estimated to contain on the order of 1060 molecular entities, which greatly exceeds even the multi-million compound libraries at the disposal of large pharmaceutical companies (Dobson, 2004). This vastness of chemical space requires that researchers devise rational approaches for identifying small bioactive molecules, particularly given the severe resource constraints on academic screening initiatives. The computational evaluation of potential bioactive molecules can drive down the high cost of screens and help extract potential drug-like compounds from pre-existing data in the public domain. To enable the extraction of information from existing chemical screen data, we have developed a suite of machine-based learning tools that statistically rank each compound for any given assay in a user-defined database. MolClass will thus facilitate the identification of specific bioactive molecules and allow the prediction of moieties that underpin biological activity.
2 WORKFLOW FEATURES
Existing resources for chemical screen data, notably PubChem, ChEMBL and ChemBank, are passive repositories that house an incomplete matrix of small molecule activity across submitted screens of various types, ranging from in vitro binding and enzyme assays to complex cellular and whole organism phenotypic assays (Fig. 1A). To interrogate such data, MolClass generates a complete matrix of compound activities across many screens and thereby enables functional predictions for all molecules, even if not tested in a specific screen. The user can upload input datasets of up to 20 000 molecules in SDF file format, in which tags distinguish hit from non-hit compounds in one or several screens. MolClass combines the datasets to generate a computational model for each screen submitted (Fig. 1B). These models are then applied to all molecules stored in MolClass to predict activity. MolClass currently provides either a composite of all molecular 2D chemical descriptors (2529 bit) or the user can independently choose 152 property descriptors, MACCS (166 bit), Substructure (306 bit), CDK extended (1024 bit) or PubChem (881 bit) fingerprints. As different machine-learning algorithms tend to generate slightly different likelihood values, a variety of algorithms are provided in MolClass including Random Forest, Naïve Bayes, SVM, KNN, Logistic Model Tree and J48. The user can apply one or several algorithms to any dataset of interest. Unbalanced datasets are boosted, to maximally double the size of the smaller part, using SMOTE (Nitesh et al. 2002) and further, if they exceed a ratio 1:5 of active versus inactive compounds, are adjusted using the WEKA under-sampling method. All models in MolClass are then applied to these molecules to generate activity fingerprints. For training and testing, MolClass uses 10-fold cross-validation. The user can examine the model statistics, the likelihood scores for screens of interest and, as shown in Figure 1C, single molecule likelihood fingerprints for existing models. Finally, MolClass also enables a substructure search using the JME Editor in the event a molecule of interest is not present in the database.
Fig. 1.
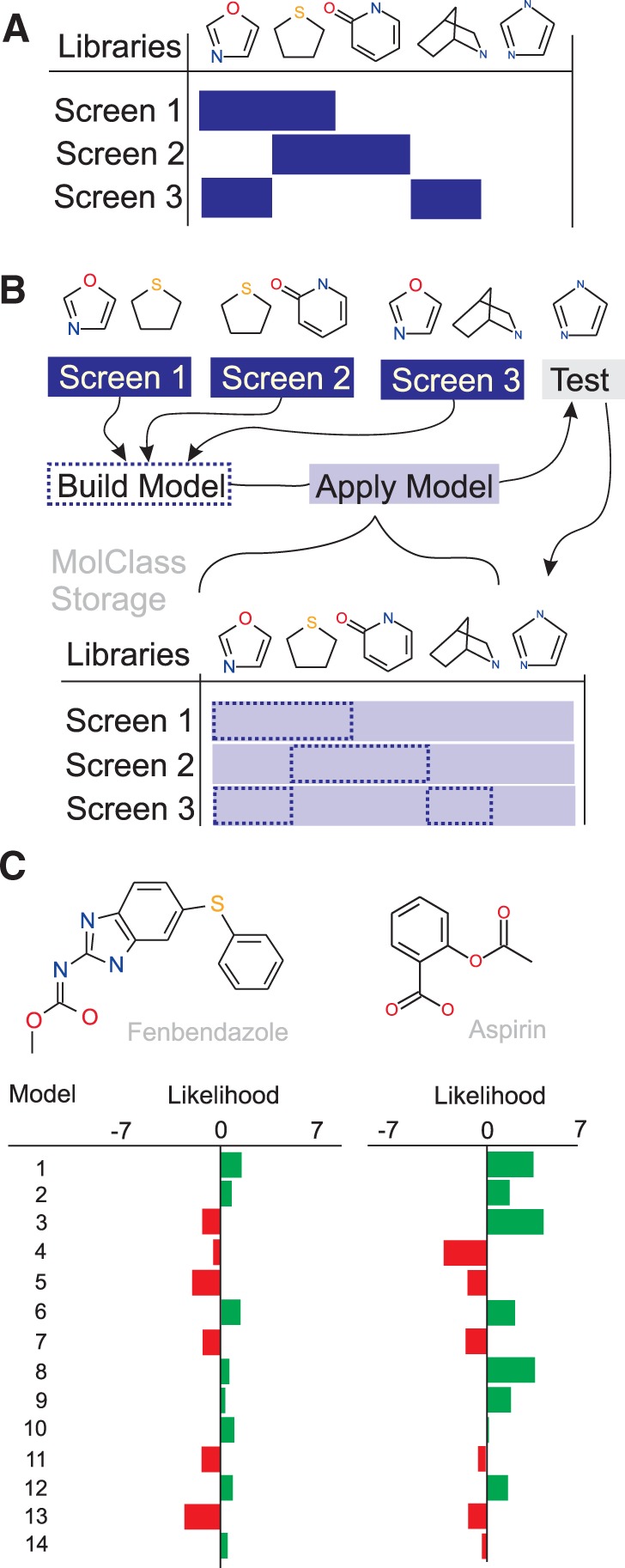
MolClass features. (A) current state of data from public resources such as PubChem and ChemBank. (B) MolClass workflow from experimental data to activity likelihoods. (C) Likelihood scores for fenbendazole and aspirin in 14 different models: (1) neurosphere proliferation, +/none (Diamandis et al. 2007); (2) Caco-2 permeation, +/− (Hou et al. 2004); (3) flucanozole synergizer, +/none (Spitzer et al. 2011); (4) Caenorhabditis elegans drug bioaccumulation, none/+ (Burns et al. 2010); (5) Ames mutagenicity benchmark, none/+ (Hansen et al. 2009); (6) mutagenicity prediction, +/none (Kazius et al. 2005); (7) blood–brain barrier penetration, +/− (Li et al. 2005); (8) PubChem AID 1828 +/none; (9) PubChem AID 595 +/− (10) ChemBank 1000423 +/− (11) ChemBank 1001644 +/− (12) ChemBank 1000359 +/− (13) autofluoresence none/+ and (14) ChEMBL TargetID CHEMBL204 none/ +. ‘+’ activating, ‘−’ inhibiting and ‘none’ no effect
3 CONCLUSION
MolClass provides a comprehensive overview of compound activity in different screens. These profiles can reveal promiscuous activities across several screens, which may reflect undesirable off-target effects. For experimental datasets, the user can discover structure activity relationships because similar structures and activities will lead to specific likelihood patterns. As the data collection is expanded by users to different biological responses and assay formats, the classification power of the portal will increase, and thereby facilitate chemical systems biology.
4 IMPLEMENTATION
MolClass is implemented in Java and Perl using CDK (Steinbeck et al. 2003), WEKA (Hall et al. 2009) and moldb4 (Haider, 2010). The web interface and REST service are written in PHP5, Slim and PEAR and run on a Fedora Linux 8 server, as an Apache HTTP service. The data are stored in a MySQL 5.5 database running on a separate Fedora Linux 16 server.
ACKNOWLEDGEMENTS
We thank Lutz Fischer for support on improving MolClass Java code, Michaela Spitzer for comments on this article, Ryusuke Kimura for improvements of the web interface and anonymous reviewers for helpful suggestions.
Funding: European Research Council (233457-SCG to M.T.).
Conflict of Interest: none declared.
REFERENCES
- Burns A.R., et al. A predictive model for drug bioaccumulation and bioactivity in Caenorhabditis elegans. Nat. Chem. Biol. 2010;6:549–557. doi: 10.1038/nchembio.380. [DOI] [PubMed] [Google Scholar]
- Diamandis P., et al. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat. Chem. Biol. 2007;3:268–273. doi: 10.1038/nchembio873. [DOI] [PubMed] [Google Scholar]
- Dobson C.M. Chemical space and biology. Nature. 2004;432:824–828. doi: 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- Hall M., et al. The WEKA data mining software: an update. SIGKDD Explorations. 2009;11:10–18. [Google Scholar]
- Haider N. Functionality pattern matching as an efficient complemntary structure/reaction search tool: an open-source approach. Molecules. 2010;15:5079–5092. doi: 10.3390/molecules15085079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K., et al. Benchmark data set for in silico prediction of Ames mutagenicity. J. Chem. Inf. Model. 2009;49(9):2077–2081. doi: 10.1021/ci900161g. [DOI] [PubMed] [Google Scholar]
- Hopkins L.A. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Hou T.J., et al. ADME evaluation in drug discovery. 5. Correlation of Caco-2 permeation with simple molecular properties. J. Chem. Inf. Comput. Sci. 2004;44:1585–1600. doi: 10.1021/ci049884m. [DOI] [PubMed] [Google Scholar]
- Kazius J., et al. Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem. 2005;48:312–320. doi: 10.1021/jm040835a. [DOI] [PubMed] [Google Scholar]
- Li H., et al. Effect of selection of molecular descriptors on the prediction of blood–brain barrier penetrating and nonpenetrating agents by statistical learning methods. J. Chem. Inf. Model. 2005;45:1376–1384. doi: 10.1021/ci050135u. [DOI] [PubMed] [Google Scholar]
- Nitesh V.C., et al. SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002;16:321–357. [Google Scholar]
- Spitzer M., et al. Cross-species discovery of syncretic drug combinations that potentiate the antifungal flucanozole. Mol. Syst. Biol. 2011;7:499. doi: 10.1038/msb.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck C., et al. The Chemistry Development Kit (CDK): an open-source java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003;43:493–500. doi: 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]