

Abstract
Summary: We introduce GO-Elite, a flexible and powerful pathway analysis tool for a wide array of species, identifiers (IDs), pathways, ontologies and gene sets. In addition to the Gene Ontology (GO), GO-Elite allows the user to perform over-representation analysis on any structured ontology annotations, pathway database or biological IDs (e.g. gene, protein or metabolite). GO-Elite exploits the structured nature of biological ontologies to report a minimal set of non-overlapping terms. The results can be visualized on WikiPathways or as networks. Built-in support is provided for over 60 species and 50 ID systems, covering gene, disease and phenotype ontologies, multiple pathway databases, biomarkers, and transcription factor and microRNA targets. GO-Elite is available as a web interface, GenMAPP-CS plugin and as a cross-platform application.
Availability: http://www.genmapp.org/go_elite
Contact: nsalomonis@gladstone.ucsf.edu
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The analysis of pathways, ontologies and other gene sets has become the preferred method for biologists looking to identify global trends from genomic datasets. Although a myriad of tools exist for pathway over-representation, few consider the structured nature of associated ontology data, alternative ontologies and diverse gene sets; few support a wide array of genomes or biological measurements, and they are often limited in scope (Huang da et al., 2009).
Unlike ontologies, pathways provide valuable qualitative contexts (interactions, reactions, metabolites and cellular compartments) that highlight biological relevance. Although various pathway resources now exist (Soh et al., 2010), most over-representation analysis (ORA) tools are limited to one resource that is often outdated. To address these deficiencies, GO-Elite was developed to provide an interchangeable and updatable model of pathway, ontology, species and gene ID system relationships. Using these relationships, GO-Elite performs ontology pruning to report a minimally non-redundant set of results (Fig. 1). Multiple options for running GO-Elite exist: source-code, cross-platform binaries, Opal web service (Ren et al., 2010), online interface or as extensions to the programs GenMAPP-CS (http://www.genmapp.org/) and AltAnalyze (http://www.altanalyze.org). The stand-alone versions of GO-Elite provide an intuitive user interface and command-line control. As previously shown, GO-Elite can be applied to a broad range of biological applications and data types (Hochstenbach et al., 2010; Lemay et al., 2009).
Fig. 1.
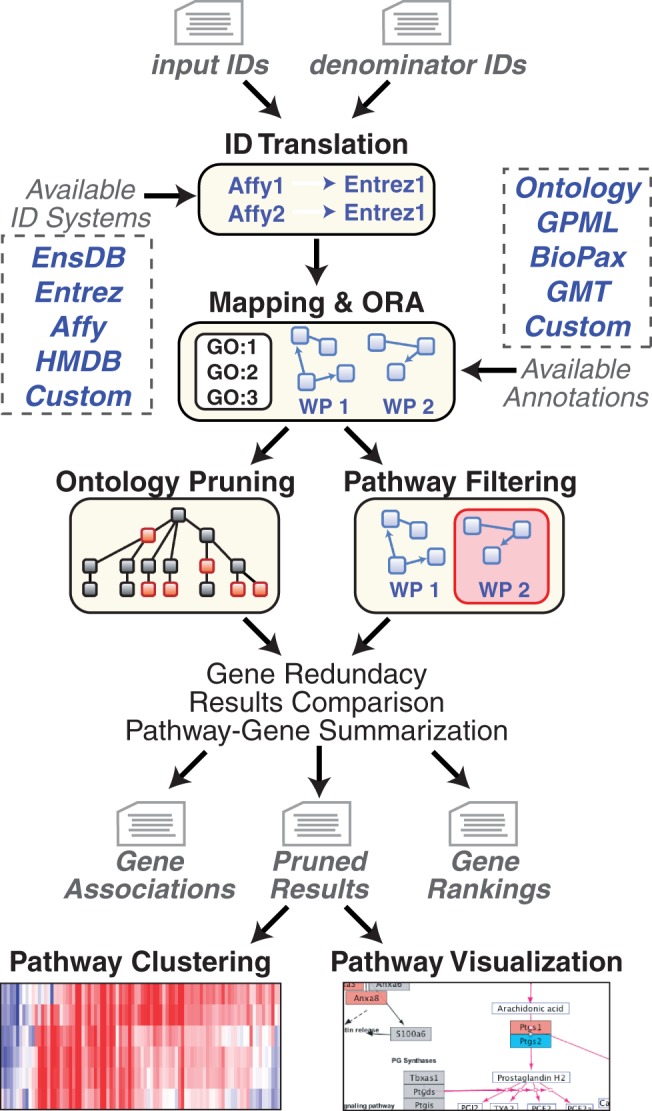
GO-Elite workflow and information sources. Before performing ORA, users create two text files containing a list of input IDs (e.g. regulated genes) and a denominator list (e.g. all genes examined), source ID type (e.g. Affymetrix) and numerical values (optional). These IDs are mapped to a primary ID system (EntrezGene, Ensembl, HMD or custom) for ORA upon pathways, ontologies or loaded gene sets. Regulated genes and metabolites can be immediately viewed on WikiPathways using the stand-alone or GenMAPP-CS interface. Pathway or ontology summarized expression values can be clustered and visualized outside of GO-Elite
2 METHODS AND IMPLEMENTATION
2.1 Database architecture
Users working with GO-Elite can create their own databases (species, ID systems, relationships) or download official GO-Elite species databases available for each release of Ensembl. The official databases are created primarily from the Ensembl database, which include all external ID systems related to Ensembl (e.g. EntrezGene, UniProt, EMBL) as well as supported microarray platforms (e.g. Affymetrix, Agilent, Codelink, Illumina). The database is augmented with relationships directly from NCBI EntrezGene and Affymetrix. Currently, relationships to multiple biological Ontology [Gene Ontology (GO), Disease and Phenotype], pathway (WikiPathways, PathwayCommons, KEGG) and gene set resources (e.g. PAZAR, Amadeus, miRanda, RNAhybrid, InterProt and Lineage Biomarkers) are supported (Supplementary Methods). In addition to gene relationships, metabolomics analyses are available for WikiPathways and KEGG. Although only a select few (ID) systems link directly to pathway and ontology annotations [Ensembl, EntrezGene and HMDB (http://www.hmdb.ca)] by default, all secondary ID systems (e.g. Affymetrix, RefSeq, MGI and Symbol) connect to these through relationship tables. Thus, users can import and analyze ID lists for dozens of supported or user added ID systems.
All resources and annotations provided by GO-Elite can be easily updated or further customized using built-in importers. These importers connect online to the various resources (e.g. WikiPathways, GO and Ensembl) or import local relationships from multiple file formats (e.g. GPML, BioPax and GMT). Alternative ontologies can also be added in GO-Elite, by specifying the URL for any OBO ontology file and importing a species-specific ontology ID relationship file through the user interface.
2.2 Optimized pathway over-representation
For ORA, ontologies, pathways and gene sets are analyzed by a method similar to the program MAPPFinder (Doniger et al., 2003s). GO-Elite ranks each analyzed term according to a Z −score, calculated with a normal approximation to the hypergeometric distribution along with a permutation or a Fisher's exact test P-value. False-discovery rate adjusted P-values are calculated using a Benjamini–Hochberg correction (Reiner et al., 2003).
The ontology ORA results from this step are further evaluated by a simple yet robust pruning method. Pruning occurs by importing these ORA statistics (Z-score, P-values and gene counts), matching user-defined or default filtering options and building all unique branch paths of these results based on the ontology tree structure. Branch paths are pruned to obtain the nodes with the largest Z-score relative to all corresponding child and parent nodes, to report the most informative, highest scoring term for a network of related terms (Supplementary Methods).
The compared scores can be optionally weighted based on the number of IDs associated with each term. This adjustment can result in more or less reported results, by favoring higher level parent nodes with more associated genes, resulting in up to an 80% reduction in the number of reported terms (Supplementary Table).
Since several alternative ORA methods exist, such as GSEA (Huang da et al., 2009), users wishing to load results from such algorithms can restrict their analysis to this pruning step.
2.3 Data representation
From these analysis steps, multiple results files are produced. The most informative of these is the pruned summary report, which includes all summary term statistics and associated gene or metabolite symbols for both ontology and non-ontology terms. Gene content redundancy between reported terms is also provided, to highlight unrelated terms with similar or identical gene content. When numerical values, such as fold changes, are included with each input ID, GO-Elite will also report mean and standard deviation ontology/pathway-level values in this summary file, analogous to GO-Quant (Yu et al., 2006), allowing for downstream pathway-level expression clustering (Supplementary Methods).
In addition, a full list of ontology and pathway statistics, associated IDs (e.g. gene symbol and associated Ensembl), comparison of reported ontology/pathway statistics between input files (where applicable) and additional gene redundancy focused files are provided. Regulated genes and metabolites can also be immediately visualized on WikiPathways in the stand-alone interface or following GenMAPP-CS analysis. Relationships between all regulated IDs and ORA terms can also be easily visualized as networks in Cytoscape using produced output files (Supplementary Methods).
This application should be of considerable interest to the genomics community, as it represents a highly customizable, simple to use and powerful framework for minimal ontology/pathway reporting. As GO-Elite is agnostic to the type of data input (e.g. gene, protein or metabolite), source ontology, pathway or gene set, we hope to rely further on community-contributed content to improve the utility of this tool in the years to come.
ACKNOWLEDGEMENTS
We thank Dr Conrad C. Huang and Dr John H. Morris for their assistance in establishing the GO-Elite web service. This work was supported by grants from the National Institutes of Health (GM080223, GM080223-06S1, HG003053) and the BioRange 1.2.4 research program of the Netherlands Bioinformatics Centre.
Conflict of Interest: none declared.
REFERENCES
- Doniger S.W., et al. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstenbach K., et al. Transcriptomic profile indicative of immunotoxic exposure: in vitro studies in peripheral blood mononuclear cells. Toxicol. Sci. 2010;118:19–30. doi: 10.1093/toxsci/kfq239. [DOI] [PubMed] [Google Scholar]
- Huang da W., et al. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay D.G., et al. The bovine lactation genome: insights into the evolution of mammalian milk. Genome Biol. 2009;10:R43. doi: 10.1186/gb-2009-10-4-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., et al. Opal web services for biomedical applications. Nucleic Acids Res. 2010;38:W724–W731. doi: 10.1093/nar/gkq503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh D., et al. Consistency, comprehensiveness, and compatibility of pathway databases. BMC Bioinformatics. 2010;11:449. doi: 10.1186/1471-2105-11-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., et al. A system-based approach to interpret dose- and time-dependent microarray data: quantitative integration of gene ontology analysis for risk assessment. Toxicol. Sci. 2006;92:560–577. doi: 10.1093/toxsci/kfj184. [DOI] [PubMed] [Google Scholar]