

ABSTRACT
Plant xylem fluid is considered a nutrient-poor environment, but the bacterial wilt pathogen Ralstonia solanacearum is well adapted to it, growing to 108 to 109 CFU/g tomato stem. To better understand how R. solanacearum succeeds in this habitat, we analyzed the transcriptomes of two phylogenetically distinct R. solanacearum strains that both wilt tomato, strains UW551 (phylotype II) and GMI1000 (phylotype I). We profiled bacterial gene expression at ~6 × 108 CFU/ml in culture or in plant xylem during early tomato bacterial wilt pathogenesis. Despite phylogenetic differences, these two strains expressed their 3,477 common orthologous genes in generally similar patterns, with about 12% of their transcriptomes significantly altered in planta versus in rich medium. Several primary metabolic pathways were highly expressed during pathogenesis. These pathways included sucrose uptake and catabolism, and components of these pathways were encoded by genes in the scrABY cluster. A UW551 scrA mutant was significantly reduced in virulence on resistant and susceptible tomato as well as on potato and the epidemiologically important weed host Solanum dulcamara. Functional scrA contributed to pathogen competitive fitness during colonization of tomato xylem, which contained ~300 µM sucrose. scrA expression was induced by sucrose, but to a much greater degree by growth in planta. Unexpectedly, 45% of the genes directly regulated by HrpB, the transcriptional activator of the type 3 secretion system (T3SS), were upregulated in planta at high cell densities. This result modifies a regulatory model based on bacterial behavior in culture, where this key virulence factor is repressed at high cell densities. The active transcription of these genes in wilting plants suggests that T3SS has a biological role throughout the disease cycle.
IMPORTANCE
Ralstonia solanacearum is a widespread plant pathogen that causes bacterial wilt disease. It inflicts serious crop losses on tropical farmers, with major economic and human consequences. It is also a model for the many destructive microbes that colonize the water-conducting plant xylem tissue, which is low in nutrients and oxygen. We extracted bacteria from infected tomato plants and globally identified the biological functions that R. solanacearum expresses during plant pathogenesis. This revealed the unexpected presence of sucrose in tomato xylem fluid and the pathogen’s dependence on host sucrose for virulence on tomato, potato, and the common weed bittersweet nightshade. Further, R. solanacearum was highly responsive to the plant environment, expressing several metabolic and virulence functions quite differently in the plant than in pure culture. These results reinforce the utility of studying pathogens in interaction with hosts and suggest that selecting for reduced sucrose levels could generate wilt-resistant crops.
Introduction
We know very little about the conditions and opportunities confronting xylem-dwelling pathogens such as Ralstonia solanacearum, the causal agent of bacterial wilt disease of tomato. This soilborne bacterial pathogen typically enters hosts through root wounds and colonizes the water-transporting xylem tissue, through which it spreads up into the plant stem. R. solanacearum infection reduces water transport, which eventually leads to plant wilting and death. Although xylem is considered a nutrient-limiting, low-oxygen environment (1), R. solanacearum is well adapted to it, growing to cell densities of 108 to 109 CFU/g stem while still remaining limited to xylem (2).
R. solanacearum forms a species complex, a heterogeneous group of xylem-colonizing strains with various host ranges, aggressiveness levels, and ecological traits. Genomic analyses have subclassified strains into four phylotypes that correlate with geographic origin (3). Two well-studied R. solanacearum strains, GMI1000, a broad-host-range tropical isolate, and UW551, a temperate strain with a narrower host range, both cause bacterial wilt of tomato (4-6). Strain GMI1000 belongs to phylotype I sequevar 18 and likely originated in Asia, where similar strains cause major losses to tomato growers. Strain UW551 belongs to phylotype II sequevar 1 (historically and for regulatory purposes known as race 3 biovar 2), a group that originated in the Andes and causes brown rot, a destructive disease of potato in cool tropical highlands. R. solanacearum race 3 biovar 2 is a high-concern quarantine pest in Europe and North America and is a U.S. Select Agent pathogen (7). The two strains are not closely related, with a whole-genome average nucleotide identity (ANI) around 91%, below the 95% cutoff separating bacterial species (8). Nonetheless, at tropical temperatures, strains UW551 and GMI1000 are equally aggressive on wilt-susceptible tomato plants (5).
Several quantitative virulence factors contribute to bacterial wilt disease development. These virulence factors include production of extracellular polysaccharide (EPS), a consortium of plant cell wall-degrading enzymes, twitching and swimming motility, taxis, and several dozen effectors secreted by a type 3 secretion system (T3SS) (2). R. solanacearum virulence factors are regulated by a complex interlocking cascade involving quorum sensing and undefined plant signals (9, 10). Two major virulence factors, EPS and T3SS, are controlled by a global regulator, PhcA, via the quorum-sensing molecule 3-OH-palmitic acid methyl ester (3-OH-PAME) (11, 12). At high cell densities (>5 × 108 CFU/ml) in planta and in culture, accumulated 3-OH-PAME triggers PhcA-mediated activation of EPS biosynthesis (13, 14). When the bacterium grows in culture, PhcA indirectly represses expression of hrpB, which encodes the transcriptional activator of the T3SS (12, 15, 16). It has been assumed from this in vitro result that the T3SS is active only in the early stages of host plant infection and is not needed at higher pathogen cell densities, when wilt symptoms develop (9, 12, 16). However, this model had not been validated in planta.
To better understand behavior of R. solanacearum strains and to identify the conserved traits required to wilt plants, we used microarrays to measure expression of the R. solanacearum UW551 and GMI1000 genomes at comparable cell densities in planta and in culture. This defined the R. solanacearum in planta transcriptome, the set of orthologous pathogen genes expressed during bacterial wilt of tomato. Bacteria growing in tomato stems strongly expressed many T3SS genes, even at high cell densities. Genes encoding proteins involved in metabolism of sucrose, which was not previously known to be present in tomato xylem, were expressed by both strains in planta. Functional analysis of this conserved metabolic trait revealed that R. solanacearum requires sucrose for full virulence and to succeed in tomato xylem.
RESULTS
Orthologous genes had similar expression patterns in two bacterial wilt pathogens.
We captured the transcriptional profiles of R. solanacearum strains UW551 and GMI1000 under two conditions, as summarized in Fig. 1. RNA was harvested from bacteria growing at comparable cell densities (~6 × 108 CFU per ml or per gram of stem) in rich medium and from tomato plants showing early wilt symptoms. Gene transcription was measured using microarray chips designed for each strain, with four biological replicates per strain under each condition. The resulting data had strong cross-chip correlation (R2 > 0.95) and good interarray reproducibility. Microarray expression results for selected genes were confirmed with quantitative PCR (qPCR) (see below).
FIG 1 .
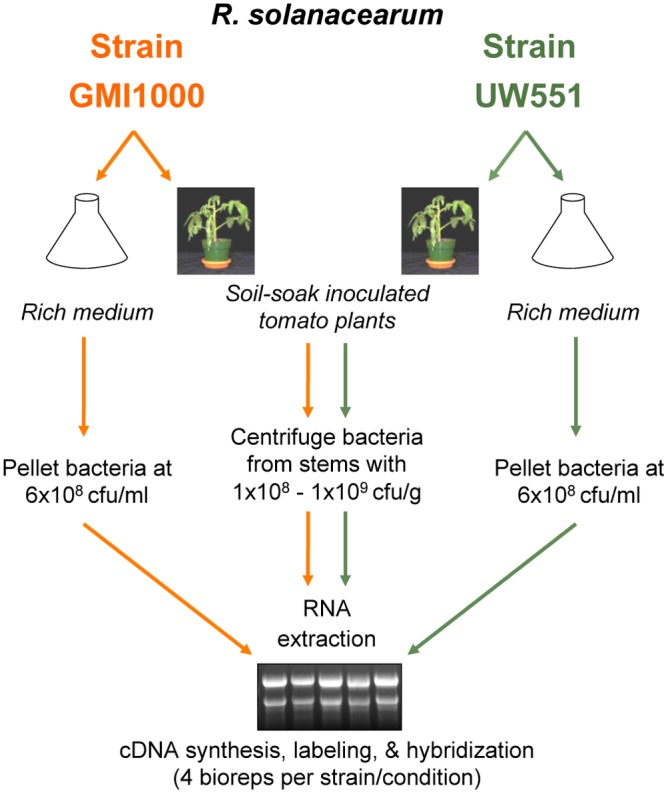
Microarray experimental design and flowchart. We compared transcriptional profiles of phylogenetically and ecologically distinct R. solanacearum strains GMI1000 (orange) and UW551 (green) during growth in tomato stems and in rich medium (CPG) at comparable cell densities. Bacteria grown in culture were treated with a transcriptional stop solution and pelleted. Bacteria were harvested from infected tomato xylem by centrifugation from cut stems into a transcriptional stop solution. Bacterial RNA was isolated from four biological replicates per strain per condition (16 replicates total).
Comparing expression of the genes shared between the two strains across the two conditions revealed that these orthologs were regulated similarly in planta compared to in rich medium in each strain (P < 0.001, hypergeometric distribution). About 70% (3,477) of the coding sequences in the two strains are orthologous, defined as pairwise best BLAST hits with >70% amino acid identity over >70% of the protein. About 11.6% (402 genes) of the orthologous genes were differentially expressed in the same direction (up or down) in both strains (see Table S1A and B in the supplemental material). A gene was considered differentially expressed in the two conditions if its expression level was >2-fold different with an adjusted P value of <0.05. Only 0.1% (4 genes) of the GMI1000-UW551 orthologs were differentially expressed in opposite directions in the two strains; these are all hypothetical genes and may be artifacts. COG (clusters of orthologous groups) analysis revealed that for both strains, genes of unknown function were overrepresented among those upregulated in planta (P < 0.001, hypergeometric distribution) (see Fig. S1 in the supplemental material).
Eighty-three percent of the GMI1000-UW551 orthologs were expressed during tomato pathogenesis. Direct fold change comparisons of the two strains were not possible, because the technical differences between the GMI1000 and UW551 arrays prohibited normalization across chips. We therefore determined each gene’s absolute expression (AE) value, which is the log2 of the normalized mean probe intensity of a coding sequence. This allowed us to define the R. solanacearum in planta transcriptome, a set of 2,898 orthologous genes that were expressed during pathogenesis in both strains (see Table S1 in the supplemental material). Expressed genes were conservatively defined as those with AE values of >8.0, based on a genome-wide survey of expression levels of core metabolic genes. The in planta transcriptome includes known virulence factors, conserved metabolic pathways, and many hypothetical genes (Table S1).
Type 3 secretion system and effector genes were expressed during early wilt symptom development.
As expected, many genes encoding known virulence traits such as stress tolerance (bcp, acrA, acrB, and dps), motility (pilA and fliC), cell wall-degrading enzymes (pehA, pehB, pehC, egl, and cbhA), and EPS biosynthesis (epsABCDEFP) were expressed during host infection (Fig. 2A; see also Table S1 in the supplemental material), consistent with previous functional analyses (2, 17, 18). Unexpectedly, many genes encoding components of the R. solanacearum T3SS were strongly expressed at high cell densities in planta, which was inconsistent with the current model of virulence regulation in this pathogen (9, 12, 16). T3SS genes upregulated in wilting plants included those encoding components of the secretion system structure (hrpK and hrpY), type 3-dependent effectors (popA, popB, and popC), and regulators (hrpB and hrpG) (Fig. 2A).
FIG 2 .

Gene expression heat map of experimentally determined bacterial wilt virulence and fitness factors. (A) Absolute log2 gene expression levels of known virulence genes in R. solanacearum UW551 (551) and GMI1000 (GMI) was artificially clustered with The Institute for Genomic Research (TIGR) multiple experiment viewer (MeV). Asterisks (*) designate genes previously identified in an IVET screen of R. solanacearum genes induced in tomato (28). The color indicates low (7.0 [blue]) to high (14.0 [yellow]) absolute log2 expression levels. (B and C) Absolute expression per cell (AEU) of hrpB (B) and popA (C) was quantified using qPCR at low and high cell densities both in planta and in culture. For gene expression in culture, R. solanacearum strain GMI1000 was grown in rich medium (CPG) to cell densities of 2 × 107 CFU/ml and 7 × 108 CFU/ml. The strain was also grown in minimal medium to a final cell density of 8 × 108 CFU/ml (BMM). For in planta gene expression, susceptible Bonny Best tomato plants were inoculated with strain GMI1000 via soil soak. RNA was isolated from plant stems showing early wilt symptoms and containing 1 × 109 CFU/g stem (in planta high) and from asymptomatic plants containing 3 × 107 CFU/g stem (in planta low). Purified RNA was reverse transcribed into cDNA and measured by quantitative real-time PCR. The transcript cycle threshold (CT) values of hrpB and popA were plotted on their respective cDNA standard curves, and the amount of transcript per cell was used to determine the arbitrary absolute expression unit (AEU). Each point represents the mean absolute expression value of three biological replicates; the error bars indicate the standard error (P < 0.05 by ANOVA).
To validate these microarray findings and to assess the effects of cell density on gene expression in planta, we quantified expression of the major T3SS regulator gene hrpB and the T3-secreted effector gene popA during growth of strain GMI1000 in tomato plants and in culture media. We used qPCR to measure transcripts from bacteria grown to different cell densities in rich medium (rich medium containing Casamino Acids, peptone, and glucose [CPG]), minimal medium (Boucher’s minimal medium [BMM]), and tomato stems. The qPCR data were wholly consistent with the array results. In rich culture, both hrpB and popA were expressed at low cell density (3 × 107 CFU/ml), but at high cell density (7 × 108 CFU/ml), hrpB expression dropped 71-fold and popA expression dropped 189-fold to nearly undetectable levels (Fig. 2B and C). These results are consistent with the previous observation that PhcA represses T3SS at cell densities of >5 × 108 CFU/ml (12). Also consistent with previous reports, hrpB and popA were expressed at higher levels in minimal medium than in rich medium at high cell densities (Fig. 2B and C) (12). However, during tomato wilt pathogenesis, these two genes were even more highly expressed at both cell densities, although expression was lower at high cell density. Nonetheless, the maximal expression of the two genes in planta was more than 70-fold higher than the maximal expression observed in culture (Fig. 2B and C).
Many genes in the HrpB regulon were also significantly upregulated in planta (see Table S2 in the supplemental material). In strain GMI1000, 92 of 206 predicted HrpB-regulated genes were differentially expressed in planta (Table S2) (19, 20). Of 109 genes known to be directly positively regulated by HrpB, 60 were significantly upregulated in planta. These included genes encoding parts of the secretion apparatus and 21 effectors (Table S2). Thirteen of the 40 genes positively but indirectly controlled by HrpB were also upregulated in planta; these encode hypothetical proteins as well as the major virulence regulator HrpG and HrpB itself (21) (Table S2). Strain UW551 had similar expression patterns (Table S1). Together, these results demonstrate that the T3SS and a substantial portion of the HrpB regulon are actively expressed at mid-stage bacterial wilt disease.
R. solanacearum uses a sucrose-specific PTS for sucrose uptake and catabolism.
A COG functional analysis revealed that a significant portion of the bacterial in planta transcriptome is dedicated to primary physiology, notably uptake and metabolism of carbohydrates (4.2%), amino acids (8.9%), lipids (3.6%), and ions (3.9%) (see Fig. S1 in the supplemental material). Both strains highly expressed scrRABY and scrK, which encode components of a putative sucrose-specific phosphoenolpyruvate-carbohydrate phosphotransferase system (PTS), suggesting that R. solanacearum depends on sucrose as a carbon source during xylem colonization (Fig. 3A). To test this hypothesis, we generated a UW551 mutant lacking a functional scrA gene, which encodes a predicted sucrose-6-phosphotransferase (Fig. 3). The UW551 scrA mutant multiplied as well as the wild-type (WT) strain did on glucose, but it could not grow on sucrose as the sole carbon source (Fig. 3C). Complementing UW551 scrA mutant with scrRABY in trans restored growth on sucrose. Deletion of an adjacent hypothetical T3 effector gene, RRSL_03375, did not alter UW551’s ability to metabolize sucrose (data not shown). These results confirm that this cluster plays its predicted role in sucrose metabolism (Fig. 3C).
FIG 3 .

The scrRABY sucrose metabolic cluster is induced in planta and encodes a sucrose-specific PTS. (A) R. solanacearum converts sucrose to glucose-6-phosphate (glucose-6-P) and fructose-6-P with the components of a sucrose-specific PTS encoded by the scr cluster, which was highly expressed in planta. The absolute log2 gene expression from bacteria growing in planta or in rich medium is shown in a heat map layered on the scr gene cluster. The location of the insertional mutation in scrA (scrA::Kmr or scrA) is indicated by a black triangle. (B) Relative fold change of scrA expression in R. solanacearum UW551 cells growing in culture media (CPS, broth containing Casamino Acids, peptone, and 55 mM sucrose; BMM, Boucher’s minimal medium; SUC, sucrose; GLUC, glucose) or in Bonny Best tomato stems. Gene expression was measured at a cell density of 1 × 109 CFU/ml broth or 1 × 109 CFU/g tomato stem and determined on a per-cell basis. The black bars represent the mean scrA expression fold change for each growth condition compared to the level in CPG medium, with three biological replicates for culture and four biological replicates for in planta expression. R. solanacearum UW551 scrA expression fold changes in planta were significantly different from expression levels in all culture media tested (P < 0.03 by Mann-Whitney test). (C) A UW551 scrA mutant grew as well as the wild type (WT) on minimal medium (BMM) plus glucose but failed to grow on sucrose as the sole carbon source. Growth on sucrose was restored by scrRABY in trans on plasmid pUFJ10.
Expression of scrA is induced by sucrose and especially by growth in planta.
In other bacteria, sucrose induces expression of sucrose-specific PTS gene clusters via the sucrose-binding regulator ScrR (22-24). We used qPCR to measure scrA expression by R. solanacearum cells growing in different levels of sucrose and in tomato plants. Sucrose induced scrA expression 254-fold in broth containing Casamino Acids and peptone plus 55 mM sucrose relative to the same broth plus 55 mM glucose (Fig. 3B). The addition of 300 µM sucrose to minimal medium increased scrA expression 361-fold. Increasing the sucrose concentration to 3 mM did not significantly change scrA expression (Fig. 3B). scrA expression was not affected by the presence of glucose in minimal medium or by the abundant amino acids and other complex organic nutrients present in CPG (Fig. 3B), demonstrating that this locus is specifically induced by sucrose, independent of the cell’s nutrient status. However, scrA expression in planta was significantly greater than in all culture media tested (P < 0.03 by Mann-Whitney test). Growth in planta increased scrA expression 1,724-fold, indicating that in addition to sucrose itself, a factor or condition present in tomato xylem is a strong inducer of the R. solanacearum sucrose metabolic pathway (Fig. 3B).
R. solanacearum requires a sucrose-specific PTS for virulence on multiple hosts.
To determine whether this metabolic trait contributes to bacterial wilt disease development, we used a naturalistic soil soak inoculation to measure the virulence of UW551 scrA mutant on various plant hosts: wilt-susceptible tomato and potato plants; moderately resistant tomato line Hawaii7996 (H7996); and bittersweet nightshade (Solanum dulcamara), an epidemiologically important weed host of R. solanacearum (25). The UW551 scrA mutant was delayed in disease progress on susceptible tomato plants and on bittersweet nightshade, but it did ultimately wilt all plants to wild-type levels (Fig. 4A and D) (P < 0.001 by repeated measures analysis of variance [ANOVA]). However, on resistant H7996 tomato and potato plants, the scrA mutant strain was significantly reduced in virulence and never caused wild-type wilt levels (P < 0.001) (Fig. 4B and C). An unmarked scrA mutant had similarly reduced virulence on susceptible tomato plants, showing that the kanamycin resistance gene in UW551 scrA mutant did not by itself lower strain virulence (data not shown). These findings demonstrate that the ability to use sucrose makes a quantitative contribution to bacterial wilt virulence.
FIG 4 .

Differential virulence of the R. solanacearum UW551 scrA mutant on susceptible and resistant hosts. (A to D) The wild-type R. solanacearum UW551 (WT) (blue triangles) and scrA mutant (red squares) were inoculated by soil soaking onto the unwounded roots of susceptible tomato cultivar Bonny Best (A), quantitatively resistant tomato cultivar Hawaii7996 (B), potato cultivar Russet Norkotah (C), and solanaceous nightshade (D). Plant symptoms were rated daily on a disease index scale from 0 to 4 (0 for healthy or no leaf area wilted, 1 for 1 to 25% of the leaf area wilted, 2 for 26 to 50% of the leaf area wilted, 3 for 51 to 75% of the leaf area wilted, and 4 for 76 to 100% of the leaf area wilted). Each point indicates the mean disease index for three independent inoculations (A, B, and D) or one experiment (C); the scrA mutant was significantly less virulent than the wild type on each host (P < 0.001 by repeated measures ANOVA).
Sucrose in xylem contributes to R. solanacearum success in planta.
These results suggested that R. solanacearum encounters sucrose during pathogenesis. To test this hypothesis, we measured sucrose concentrations in xylem fluid from infected tomato plants containing ~7 × 108 CFU/ml of either strain UW551 or UW551 scrA mutant, as well as in xylem fluid from water-inoculated control plants. Fluid from control plants contained 318 ± 35 µM sucrose. Xylem fluid from plants infected by the wild-type bacterium contained slightly less sucrose, 268 ± 14 µM, possibly because it was being consumed by the many bacteria growing in these plants. In contrast, xylem from plants infected with comparable populations of UW551 scrA mutant contained sucrose levels similar to those in uninfected plants, 312 ± 12 µM, consistent with the fact that these bacteria cannot utilize sucrose. No glucose was detected in xylem fluid (detection limit <2 nM), suggesting that it is not an available carbon source in this habitat. These results indicate that sucrose could be a significant nutrient for R. solanacearum in planta.
Carbohydrate-rich EPS is a major R. solanacearum virulence factor (2), so we tested the hypothesis that a scrA mutant strain had reduced virulence because it produced smaller amounts of EPS. However, strain UW551 and UW551 scrA mutant produced similar amounts of EPS as determined by an enzyme-linked immunosorbent assay (ELISA) (data not shown), suggesting that catabolism is the principal function of this cluster in bacterial wilt virulence.
When cells of R. solanacearum UW551 scrA mutant and UW551 were separately introduced directly into tomato vascular systems via a cut leaf petiole, they reached similar final population sizes (P < 0.2 by Student’s t test) and caused comparable levels of disease (P < 0.2 by repeated measures ANOVA) (Fig. 5A and B), indicating that loss of sucrose metabolism did not reduce R. solanacearum’s fundamental competence to colonize the host stem. This raised the possibility that sucrose is an important nutrient for R. solanacearum only in the host rhizosphere. To specifically measure the contribution of xylem sucrose to bacterial success, we used an in planta competition assay to determine whether the UW551 scrA mutant was subtly reduced in fitness. Tomato plants were inoculated with a 1:1 mixture of wild-type UW551 and UW551 scrA mutant cells directly into the xylem through cut petioles, and the population size of each strain was quantified in each plant after wilting symptoms appeared. Under these conditions, wild-type UW551 consistently outcompeted the UW551 scrA mutant (P < 0.006 by paired Student’s t test) (Fig. 5C), indicating that the ability to metabolize xylem sucrose provides R. solanacearum with a significant fitness advantage during bacterial wilt pathogenesis.
FIG 5 .

Sucrose metabolism provides R. solanacearum with a competitive advantage during xylem colonization. (A to C) Twenty-two-day-old wilt-resistant H7996 tomato plants were inoculated through a cut petiole with 4 µl of wild-type R. solanacearum UW551 (WT) or UW551 scrA::Kmr (UW551 scrA) (A and B) or with a 1:1 mixture of wild-type R. solanacearum UW551 Rifr and scrA mutant (~2,000 CFU total) (C). (A) Wilt disease progress of wild-type UW551 and scrA mutant strains on stem-inoculated tomato plants that were rated as described in the legend to Fig. 4. (B) Stem colonization ability of wild-type UW551 and scrA mutant cells stem inoculated separately on tomato plants. Ten plants displaying a disease index of 1 (corresponding to 5 days after inoculation) were sampled per treatment (UW551 WT or UW551 scrA mutant), and the bacterial population sizes in the stems were quantified by dilution plating. Each symbol represents one plant; short horizontal lines represent the mean bacterial colonization in tomato stems. (C) Competitive fitness of wild-type and scrA mutant strains. The tomato plants were sampled 5 days postinoculation, and in planta population sizes of wild-type and scrA mutant strains were quantified by dilution plating. Data represent two biological replicates containing 30 plants each. The WT strain significantly outcompeted the scrA mutant in planta (P < 0.006 by paired Student’s t test).
DISCUSSION
Technical barriers have delayed our understanding of how xylem-dwelling pathogenic bacteria regulate their virulence genes during host infection. Efforts to duplicate the host environment in vitro with “hrp-inducing” media, plant cell coculture or plant extracts (19, 21, 26) do not recreate the dynamic physical and biochemical conditions experienced by bacteria during infection and pathogenesis of a living host. Useful glimpses of this complex biological reality have come from in vivo expression technology (IVET) (27-29) and reporter gene experiments (30, 31), but whole-genome expression analysis offers a broader picture.
We profiled transcription of R. solanacearum cells at early bacterial wilt development, when the pathogen is largely confined to the xylem. Because RNA was extracted from bacteria centrifuged from xylem vessels, our transcriptomic data likely represent the planktonic subset of the bacterial population rather than bacteria tightly attached to vessel walls. However, we obtained similar numbers of CFU/g from ground stem segments and in pellets from centrifuged stem segments, suggesting that the centrifugal extraction was reasonably efficient (data not shown). In addition, the in planta transcription profile generated by our analysis is consistent with the pathogen’s biology, since the transcriptome includes essentially all genes encoding known bacterial wilt virulence factors. Finally, qPCR analyses of bacteria extracted by centrifugation and from ground whole-stem samples yielded similar gene expression trends. Together, these results indicate that bacteria harvested by the centrifugation method are biologically representative of the bacterial populations in the stem.
Microarray and qPCR analyses demonstrated that the xylem environment overrides the cell density-dependent repression of hrpB expression observed in culture. In culture media, the PhcA quorum-sensing system represses hrpB at high cell densities, and this finding generated the idea that the T3SS plays no role after the initial stages of plant infection (9, 12, 16). However, we found that the T3SS was strongly expressed during wilt disease, when pathogen cell densities are high. Similar results were observed using microscopy coupled with in planta hrpB::reporter assays (F. Monteiro and M. Valls, personal communication). Increasing cell density did reduce transcription of popA and hrpB in planta, but their mean absolute expression levels were nonetheless higher than in culture; the lowest popA expression level in planta was 5-fold greater than the highest expression levels in culture. Further, 55% of genes directly positively regulated by HrpB, the transcriptional activator of the T3SS genes, were upregulated in planta versus culture medium. Together, these results suggest that signals and/or conditions in the plant environment interact with and largely override the cell density-dependent repression observed in culture. This is consistent with the observation that coculture with plant cells induces hrpB expression, although induction levels cannot be directly compared because of methodological differences (26). We speculate that the T3SS may inject effectors into the living xylem parenchyma cells immediately adjacent to the dead xylem vessels. R. solanacearum could access these parenchyma cells through the pits in xylem tracheid walls. Additional studies are needed to determine whether these effectors suppress host defenses, increase nutrient availability, or otherwise improve success of R. solanacearum at mid-stage bacterial wilt.
Genes involved in sucrose catabolism also displayed significantly different expression profiles in planta than in culture, regardless of the presence of sucrose itself. Sucrose did induce scrA expression, probably via the LacI-type repressor, ScrR (32), but the regulatory cues that further augment scrA induction in planta remain to be identified. Similarly, studies are needed to define the plant signal(s) that induce hrpB and popA and likely many other genes in the R. solanacearum in planta transcriptome. More broadly, our results suggest that studies of plant pathogen gene expression in artificial media or biologically irrelevant host tissue (e.g., leaf tissue for xylem pathogens) should be validated under biologically relevant conditions in the host.
It has been proposed that the R. solanacearum core genome encodes the minimal set of traits needed for success as a bacterial wilt pathogen (33). These 2,820 genes are present in all R. solanacearum strains, as determined by genome sequencing and comparative genome hybridization studies (8, 33, 34). Sixty-five percent (2,250) of the 3,477 UW551-GMI1000 orthologs belong to the R. solanacearum core genome, and core genes were significantly overrepresented in the in planta transcriptome (P < 0.001, hypergeometric distribution).
During tomato pathogenesis, 2,898 orthologs were expressed (AE value of >8) by both strains; these include 2,020 core genes. The 597 orthologs that were highly expressed (AE value of >12.0) in planta in both strains included 455 core genes. The analyses presented here focused on shared orthologs rather than strain-specific genes, which will be considered separately. However, orthologs in both strains were more often differentially expressed in planta than strain-specific genes (P < 0.001, hypergeometric distribution; data not shown). Thus, R. solanacearum orthologs were more likely to respond to host environmental signals than the strain-specific traits, as would be predicted by the R. solanacearum core genome hypothesis (8, 33). Although suggestive, gene expression level is not a singular measure of relevance. We note that a transcriptomic analysis cannot account for genes that are expressed at very low levels, and some weakly expressed genes are almost certain to be biologically important.
Cell proliferation is essential for bacterial disease development, but little is known about how vascular plant pathogens like R. solanacearum grow in the carbon-limited xylem environment. As expected, the R. solanacearum in planta transcriptome included genes encoding transporters of anions, tricarboxylic acid (TCA) cycle intermediates, and amino acids known to be present in tomato xylem fluid (Fig. 6; see also Fig. S1 in the supplemental material) (2). Unexpectedly, genes for sucrose uptake and catabolism were also expressed at high levels in planta, although sucrose was not thought to be a significant component of tomato xylem fluid. Organic and amino acids, relatively abundant in xylem fluid (1, 28, 35), were considered primary carbon sources for R. solanacearum during xylem infection, but the reduced competitive fitness of a scrA mutant in tomato stems demonstrates that the pathogen exploits host sucrose as a carbon source during growth in xylem. Sucrose metabolism may also be important early during infection in the plant rhizosphere where sucrose is abundant (36).
FIG 6 .

Select biochemical pathways of the R. solanacearum core genome represented in the R. solanacearum in planta transcriptome. Bacterial processes previously not known to be active during tomato xylem colonization are shown in green. Abbreviations: PTS, phosphoenolpyruvate-carbohydrate phosphotransferase system; Suc-6-P, sucrose-6-phosphate; Fruc, fructose; G-6-P, glucose-6-phosphate; F-6-P, fructose-6-phosphate; F-1,6-P2, fructose-1,6-bisphosphate; PEP, phosphoenolpyruvate; CoA, coenzyme A; TCA, tricarboxylic acid; αKG, alpha-ketoglutarate; Ribulose-5-P, ribulose-5-phosphate; PPP, pentose phosphate pathway; EPS, exopolysaccharide; ROS, reactive oxygen species; T2SS, type 2 secretion system; CWDEs, cell wall-degrading enzymes; T3, type 3; T3SS, type 3 secretion system.
Diverse animal- and plant-associated bacteria use a sucrose-specific PTS to catabolize sucrose (22, 32, 37), but R. solanacearum is the only species in the betaproteobacteria with a sucrose-specific PTS (data not shown). Phylogenetic analysis revealed that R. solanacearum ScrA is most closely related to proteins in Methylobacterium sp. strain 4-45 (alphaproteobacteria) and Gram-positive Firmicutes (amino acid identity of 63% and 52 to 53%, respectively) (data not shown); the sucrose catabolic gene cluster may have been acquired by lateral gene transfer, which has played a major role in evolution of R. solanacearum (8). We hypothesize that an ancient transfer event endowed a progenitor of R. solanacearum with the fitness-increasing ability to exploit a scarce carbon resource in its plant niche, as has been speculated in another system (38). Our finding that the plant environment induces expression of this gene substantially more than the presence of sucrose suggests that additional adaptation at the regulatory level has optimized R. solanacearum’s metabolism to host conditions.
All sequenced R. solanacearum strains possess a sucrose-specific PTS, and this core metabolic trait provides the pathogen with a significant physiological advantage in planta on several hosts. Bacterial pathogens of Arabidopsis, rice, and bean manipulate host sugar transport to their benefit, suggesting that access to host sucrose is broadly important for successful pathogenesis (39, 40). We suspect that many other R. solanacearum core metabolic traits expressed in the in planta transcriptome also increase fitness during pathogenesis (Fig. 6). For example, the highly expressed cellulose-degrading enzymes endoglucanase (Egl) and cellobiohydrolase (CbhA) are known virulence factors (2); the specific mechanism by which they promote virulence may be nutritional, via release of sucrose from host cell walls. Further studies focused on pathogen primary metabolism may provide additional insights into specific growth requirements for success in the xylem and thus, successful pathogenesis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table S3 in the supplemental material. Escherichia coli strains were grown at 37°C in Luria-Bertani medium. R. solanacearum cells were grown at 28°C on morpholineethanesulfonic acid (MES)-buffered Boucher’s minimal medium (BMM) (pH 7.0), BMM with 40 mM nitrate (similar to plant xylem levels) substituted for ammonium (BMN) (pH 7.0), rich medium containing Casamino Acids, peptone, and glucose (CPG), or on tetrazolium chloride (TZC) plate (41). To determine effects of sucrose on gene expression, CPG was modified to replace the 55 mM glucose in CPG with 55 mM sucrose (CPS). When necessary, media were supplemented with gentamicin (Gm) (15 µg/ml), kanamycin (Km) (25 µg/ml), or tetracycline (Tc) (15 µg/ml).
Recombinant DNA techniques and mutagenesis.
Total genomic and plasmid DNA were isolated by standard protocols. E. coli and R. solanacearum cells were transformed as previously described (42). Primer sequences are given in Table S4 in the supplemental material. To construct an R. solanacearum UW551 scrA mutant (scrA::Kmr), the regions flanking scrA were amplified with Phusion high-fidelity DNA polymerase (Finnzymes, Vantaa, Finland), and a kanamycin resistance (Kmr) cassette amplified from pSTBlue-1 (Novagen, Madison, WI) was inserted between the two fragments via splice by overlap extension (SOE) PCR (43). The resulting SOE construct was gel purified, phosphorylated with T4 polynucleotide kinase (Promega, Madison, WI), and ligated into the EcoRV site of cloning vector pSUP202 (Cmr Tcr Apr), which cannot replicate in R. solanacearum, creating pSUP202-scrA::Kmr (44). This plasmid was transformed into wild-type (WT) R. solanacearum strain UW551 via electroporation. Candidate double-recombined transformants were screened for Kmr and Tcs to confirm proper allelic exchange and scrA disruption. Notably, double recombinations were 98% more frequent than single recombination in strain UW551 (Kmr Tcr) with pSUP202::scrA::Kmr.
RNA extraction for microarrays.
To measure gene expression of bacteria growing in culture, RNA was extracted from 30-ml cultures of R. solanacearum strain GMI1000 or UW551 grown in CPG to log phase (A600 = 0.6 to 0.7; 6 × 108 to 7 × 108 CFU/ml). To minimize RNA degradation, each sample was immediately mixed with 3.75 ml of an ice-cold transcriptional stop solution (5% [vol/vol] water-saturated phenol in ethanol). Samples were centrifuged at 20,800 × g at 4°C. The supernatant was removed, and the pellets were frozen in liquid nitrogen and stored at −80°C. Each experiment was repeated four times for strains UW551 and GMI1000, with four biological replicates per strain.
RNA was extracted using the modified hot-phenol method (44), followed by four DNase treatments. Briefly, precipitated RNA was resuspended in 400 µl DNase solution (all reagents from Ambion, Foster City, CA) containing 0.5 µl RNase inhibitor solution, 40 µl of 10× DNase I buffer, 15 µl DNase I, and 344.5 µl nuclease-free water and incubated at 37°C for 1 h. DNase was inactivated and removed by extracting first with phenol, then with phenol-chloroform, and finally twice with chloroform. RNA was precipitated with 1/10 volume of 3 M sodium acetate and 2 volumes of cold ethanol. After the final DNase treatment, RNA pellets were resuspended in 13 µl water and stored at −80°C. RNA concentration and purity was determined via microspectrophotometry (NanoDrop; Thermo Fisher Scientific, Wilmington, DE). The quality of RNA samples was assessed with the Agilent bioanalyzer 2100 nanochip system (Agilent Technologies, Santa Clara, CA). The absence of genomic DNA was verified by PCR with RS-universal primers, 759/760-specific or R3bv2-specific primers (see Table S4 in the supplemental material) (45, 46).
To measure gene expression of bacteria growing in planta, unwounded 25-day-old tomato plants (wilt-susceptible cv. Bonny Best) were inoculated by pouring a suspension of R. solanacearum strain UW551 or strain GMI1000 into the soil to a final density of 1 × 108 CFU/g soil. Bacteria were extracted from plants showing the first signs of wilting (disease index of 1 [<25% of the leaf area wilted]) 5 to 10 days postinoculation. To enrich for bacterial RNA, bacteria were centrifuged directly from cut stem segments. Individual tomato midstems (7 cm spanning the petiole) were centrifuged at 4°C and 18,500 × g for 6 min in 15-ml conical tubes containing 3 ml of 30% transcriptional stop solution in water. The resulting bacterial pellets were frozen immediately in liquid nitrogen for later extraction. Before centrifugation, a 0.1-g transverse slice from the middle of each stem segment was ground and dilution plated to determine the bacterial cell density in planta for each sample, and RNA was extracted and pooled only from the stems containing ~6 × 108 CFU/g. Each experiment was repeated four times for strains UW551 and GMI1000, for four biological replicates per strain. Each bioreplicate consisted of 10 to 15 bacterial pellets pooled from plants colonized with 1 × 108 to 1 × 109 CFU/g stem tissue. RNA was extracted from pellets and analyzed as described above, but with only three DNase treatments.
Array design.
DNA microarrays for R. solanacearum strains GMI1000 and UW551 were designed using the genome sequences available from Integrated Microbial Genomes (IMG [ http://img.jgi.doe.gov/cgi-bin/w/main.cgi]). They were custom designed at Roche Nimblegen with a target melting temperature of 78°C, and a target probe length of 40 to 70 nucleotides (nt). Each array consisted of a total of about 72,000 probes, including controls, and each 4-plex chip contained four identical arrays.
The GMI1000 gene chip consisted of 60,608 gene-specific probes representing 5,061 out of 5,206 open reading frames (ORFs), plus 9,549 probes covering the 2,213 unique intergenic regions at 50-bp intervals. The UW551 gene chip consisted of 51,664 gene-specific probes to represent 4,315 out of 4,421 ORFs and 20,036 probes covering 2,641 unique intergenic regions at 50-bp intervals. Each ORF was represented by 2 to 6 gene-specific probes, duplicated in two blocks on the array. The probes for the intergenic regions were not duplicated, and analysis from the intergenic regions is not presented here.
cDNA synthesis, labeling, and hybridization.
We followed NimbleGen’s standard protocols to process total prokaryotic RNAs for cDNA preparation, labeling, and hybridization. Double-stranded cDNA was synthesized from 10 or 5 µg bacterial RNA from RNA derived from bacteria grown in culture or in planta, respectively, by reverse transcription (Invitrogen SuperScript II double-stranded cDNA synthesis kit; Invitrogen, Carlsbad, CA), followed by an RNase treatment and subsequent cDNA precipitation according to NimbleGen’s protocol. After quantity and quality assessment, 10 µg of cDNA was labeled with Cy3 fluorescent dye (TriLink BioTechnologies, San Diego, CA) according to the manufacturer’s recommendations. Hybridization was carried out for 17 h at 42°C, using the NimbleGen hybridization station at the University of Wisconsin—Madison Biotechnology Center. Following hybridization, microarrays were washed and dried following recommended protocols. The arrays were scanned with a GenePix 4000B scanner at 532-nm wavelength with a photomultiplier tube (PMT) setting of 520 to achieve 1 × 105 normalized counts at the saturation (65,000-intensity) level. We imported the scanned image with NimbleScan 2.4 software to extract the raw data.
Data normalization and bioinformatic statistical analyses and MIAME (minimum information about a microarray experiment) compliance.
NimbleScan 2.4 software was used to compute expression values (gene calls) according to the robust multichip average (RMA) algorithm (45, 46). Signals from the four replicates of each hybridization experiment were normalized using quantile normalization (47) with the RMA procedure incorporated in NimbleScan 2.4. Expression data were analyzed statistically with the ArrayStar software package (DNAStar, Madison, WI). Probabilistic statistical analysis (Student’s t test) was used to identify differentially expressed (DE) genes. After selection of candidate differentially expressed genes, a statistic (referred to as “adjusted P value”) was calculated for each gene. If the adjusted P value was lower than the desired cutoff (adjusted P < 0.05), then the gene was classed as differentially expressed. The false discovery rate (FDR) (Benjamini Hochberg) adjustment for multiple testing correction was applied to the P value to reduce the number of false-positive results and to increase the chances of identifying all the differentially expressed genes. All genes with adjusted P values less than the desired cutoff were listed as differentially expressed for that data set. If necessary, the significantly differentially expressed gene list was filtered further based on fold changes or expression levels in the treatments, using the options in ArrayStar. Data were imported into a custom Microsoft Access database and analyzed further using Microsoft Excel; scatter plots and heat maps of expression profiles were generated using ArrayStar to visualize expression data within and across treatments.
Validation of microarray results.
Expression of several R. solanacearum GMI1000 genes was measured via absolute mRNA quantification to validate microarray results. To quantify gene expression of hrpB and popA, cells were grown under the following conditions: (i) in CPG at a lower cell density (2 × 107 CFU/ml), (ii) in CPG at a high cell density (7 × 108 CFU/ml), (iii) in BMM at a high cell density (8 × 108 CFU/ml), (iv) in planta at a lower cell density (3 × 107 CFU/g), and (v) in planta at a high cell density (1 × 109 CFU/g). RNA was extracted from equivalent numbers of cells grown in culture as described above but treated with at least 4 DNase steps. The plants were inoculated with strain GMI1000 by soil soak as described above. Stem tissue samples (150 mg) from asymptomatic and symptomatic (disease index of 1) tomato plants (cv. Bonny Best) colonized with ~2 × 107 CFU/g and ~1 × 109 CFU/g, respectively, were placed in Ambion RNAlater to preserve RNA integrity before storage at −80°C. Adjacent stem slice samples were dilution plated to determine bacterial cell numbers in each sample. Total RNA was extracted from ground infected stem tissue containing the target cell densities using the RNeasy plant minikit (Qiagen, Valencia, CA), followed by two DNase treatments.
For absolute quantification of scrA mRNA levels, R. solanacearum UW551 cells were grown to 1 × 109 CFU/ml in CPG or CPS. For minimal medium experiments, overnight CPG cultures were rinsed with BMN, transferred to BMN or BMN supplemented with either 300 µM or 3 mM sucrose or 3 mM glucose, and incubated at 28°C for 2.5 h. RNA was extracted as described above, with at least three DNase treatments. RNA from tomato (cv. Bonny Best) plant tissue was collected from the stems of infected tomato plants as described above.
cDNA was synthesized from this RNA by reverse transcription with SuperScript III VILO (Invitrogen) using 2.0 µg of total RNA and 50 ng of random hexamer primers, according to the manufacturer’s protocol. Absolute quantification using quantitative PCR (qPCR) reactions were performed in duplicate 25-µl reactions with Power SYBR green master mix (Applied Biosystems, Carlsbad, CA) and included 1× master mix, 400 nM forward and reverse primers for hrpB, popA, or scrA, and 50 ng template cDNA. Reactions were carried out on an ABI PRISM 7300 real-time PCR system (Applied Biosystems) with reaction parameters of 10-min polymerase activation, followed by 40 cycles, with 1 cycle consisting of 15 s at 95°C and 1 min at 57°C. Standard curves were made for absolute quantification using 10-fold dilutions (2.5 to 0.0025 µg cDNA) from RNA collected and then reverse transcribed from GMI1000 cells grown to 7 × 108 CFU/ml on BMM for hrpB and popA expression. cDNA synthesis was carried out as described above but with 2.5 µg template RNA. The standard curve for scrA absolute quantification was created using 10-fold dilutions from 2.0 µg to 0.002 ng cDNA synthesized from RNA from UW551 cells grown in BMN plus 300 µM sucrose.
qPCR via absolute quantification was used to define scrA, hrpB, and popA gene expression (48). Absolute expression was quantified in arbitrary absolute expression units (AEU) per cell by determining the amount of transcript based on each gene’s respective cDNA standard curve (described above). The fold change in expression was determined by dividing the absolute expression values on a per-cell basis for bacteria grown under the various conditions by the absolute expression on a per-cell basis of bacteria growing in CPG medium. The statistical program SAS or JMP was used to analyze the raw data.
Comparative genomics.
BLAST-based methods were used to identify homologous proteins between genomes. Orthology assignments were limited to those genes whose predicted bidirectional best hits from reciprocal BLASTP searches were filtered to eliminate incomplete and low scoring matches and retain those which shared a common backbone sequence residing in local collinear blocks (LCBs) in pairwise whole-genome alignments using the progressive alignment algorithm Progressive Mauve (http://asap.ahabs.wisc.edu/mauve/). Genes were considered orthologous if both their similarity scores and percent identity fell within cutoff values of >70% for pairwise BLASTP searches. Whole-genome alignments using Mauve allowed us to use gene context as an arbiter in cases where multiple paralogs were possible and to designate orthologs with improved specificity and precision. Downstream analyses were limited to high-confidence data sets when true orthology was critical.
Plant assays.
To measure pathogen virulence, plants were inoculated with R. solanacearum strains and mutants via a naturalistic soil soak inoculation method as previously described (49). Hosts were unwounded 17-day-old susceptible tomato (cv. Bonny Best) plants, 14- to 16-day-old quantitatively resistant tomato (cv. Hawaii7996) plants, 21-day-old susceptible potato (cv. Russet Norkotah) plants grown from minitubers, or 18- to 19-day-old Solanum dulcamara [bittersweet nightshade] plants grown from seeds collected in Madison, WI, in 2009. Each plant’s pot was soaked with a 50-ml suspension of wild-type UW551 or UW551 scrA::Kmr, or popS::Kmr strain for a final inoculum density of 1 × 108 CFU/g soil. The plants were incubated at 28°C with a 12-h light cycle and were rated daily on a disease index scale from 1 to 4 as follows: disease index of 1 for 1 to 25% of the leaf area wilted; disease index of 2 for 26 to 50% of the leaf area wilted; disease index of 3 for 51 to 75% of the leaf area wilted; and disease index of 4 for 76 to 100% of the leaf area wilted. Virulence assays contained 16 plants per treatment and were replicated three times. To measure strain competitive fitness, 22-day-old wilt-resistant H7996 tomato plants were inoculated with 4 µl of a 1:1 mixture of R. solanacearum UW551-Rifr, a spontaneous rifampin-resistant strain with wild-type fitness (WT) and scrA::Kmr (scrA mutant) (~2,000 CFU total). The plants were sampled 5 days postinoculation, and in planta population sizes of WT and scrA mutant strains were quantified by dilution plating on CPG plates with either Rif or Km. Data represent two biological replicates containing 30 plants each.
To quantify bacterial colonization in stem tissue and measure xylem fluid from mock-inoculated or infected plants, 22-day-old H7996 tomato plants were inoculated with water, R. solanacearum UW551, or UW551 scrA mutant via inoculation with ~1,000 cells per plant through a cut petiole. The plants were sampled at the onset of wilt (disease index of 1) 5 days postinoculation, and stem bacterial colonization was quantified. Plants were decapitated about 20 cm above the crown, and pooled xylem fluid was collected from plants for ~20 min. The first 100 µl of xylem fluid was discarded to remove any nonxylem metabolites released by wounding the plant. We quantified sucrose concentrations in xylem fluid by using a BioVision sucrose assay kit (Mountain View, CA) following the manufacturer’s recommendations for detecting sucrose from biological samples. Glucose concentrations were used to check for metabolite contamination from nonxylem cells; there was no detectable glucose present, suggesting little to no cellular contamination beyond xylem fluid.
Microarray data accession number.
The data from the microarray experiments described here are available at Gene Expression Omnibus (GEO [http://www.ncbi.nlm.nih.gov/geo/]) under the accession number GSE33662.
SUPPLEMENTAL MATERIAL
Clusters of orthologous groups (COG) analysis of R. solanacearum genes expressed during tomato pathogenesis. (A to D) COG categorization of the entire R. solanacearum in planta transcriptome (A), all genes differentially expressed in planta versus in rich CPG medium (fold change of >2, adjusted P < 0.05) (B), upregulated genes (fold change of >2, adjusted P < 0.05) (C), and downregulated genes (fold change of > 2, adjusted P < 0.05) (D). Download Figure S1, TIF file, 0.3 MB.
Expression patterns of Ralstonia solanacearum UW551 in planta and in rich medium.
Expression patterns of Ralstonia solanacearum GMI1000 in planta and in rich medium.
Gene expression trends in the R. solanacearum HrpB regulon in planta versus in rich medium.
Strains and plasmids used in this study.
Oligonucleotide primers used in this study.
ACKNOWLEDGMENTS
We gratefully acknowledge Wei Wei for technical assistance; Ting-Li Lin (University of Wisconsin Statistical Consulting Service), Audrey Gasch and Don Waller (University of Wisconsin) for valuable statistical advice; Gary Roberts and Yaoping Zhang (University of Wisconsin) for cloning vector pSUP202; Philippe Prior (INRA, La Réunion, France) and Benoît Remenant (University of Wisconsin) for the R. solanacearum core genome list and useful discussions; and Anne Alvarez and Gabriel Peckham (University of Hawaii) for kindly providing EPS I antibodies.
This research was supported by an APS R. J. Tarleton Award and a Storkan-Hanes-McCaslin Foundation Award to J.M.J., by USDA-CSREES Plant Biosecurity project 2006-04560, the USDA-ARS Floral and Nursery Crops Research Initiative, and by the University of Wisconsin College of Agricultural and Life Sciences.
The funding groups played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation: Jacobs JM, Babujee L, Meng F, Milling A, Allen C. 2012. The in planta transcriptome of Ralstonia solanacearum: conserved physiological and virulence strategies during bacterial wilt of tomato. mBio 3(4):e00114-12. doi:10.1128/mBio.00114-12.
REFERENCES
- 1. Pegg GF. 1985. Life in a black hole: the microenvironment of the vascular pathogen. Trans. Br. Mycol. Soc. 85:1–20 [Google Scholar]
- 2. Denny TP. 2006. Plant pathogenic Ralstonia species, p 573–644 In Plant-associated bacteria. Springer Verlag, Dordrecht, The Netherlands [Google Scholar]
- 3. Prior P, Fegan M. 2005. Recent developments in the phylogeny and classification of Ralstonia solanacearum. Acta Hort. 695:127–136 [Google Scholar]
- 4. Gabriel DW, et al. 2006. Identification of open reading frames unique to a select agent: Ralstonia solanacearum race 3 biovar 2. Mol. Plant Microbe Interact. 19:69–79 [DOI] [PubMed] [Google Scholar]
- 5. Milling A, Meng F, Denny TP, Allen C. 2009. Interactions with hosts at cool temperatures, not cold tolerance, explain the unique epidemiology of Ralstonia solanacearum race 3 biovar 2. Phytopathology 99:1127–1134 [DOI] [PubMed] [Google Scholar]
- 6. Salanoubat M, et al. 2002. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 415:497–502 [DOI] [PubMed] [Google Scholar]
- 7. Champoiseau PG, Jones JB, Allen C. 13 March 2009. Ralstonia solanacearum race 3 biovar 2 causes tropical losses and temperate anxieties. Plant Health Prog. [Google Scholar]
- 8. Remenant B, et al. 2011. Ralstonia syzygii, the blood disease bacterium and some Asian R. solanacearum strains form a single genomic species despite divergent lifestyles. PLoS One 6:e24356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mole BM, Baltrus DA, Dangl JL, Grant SR. 2007. Global virulence regulation networks in phytopathogenic bacteria. Trends Microbiol. 15:363–371 [DOI] [PubMed] [Google Scholar]
- 10. Schell MA. 2000. Control of virulence and pathogenicity genes of Ralstonia solanacearum by an elaborate sensory network. Annu. Rev. Phytopathol. 38:263–292 [DOI] [PubMed] [Google Scholar]
- 11. Flavier AB, Clough SJ, Schell MA, Denny TP. 1997. Identification of 3-hydroxypalmitic acid methyl ester as a novel autoregulator controlling virulence in Ralstonia solanacearum. Mol. Microbiol. 26:251–259 [DOI] [PubMed] [Google Scholar]
- 12. Genin S, Brito B, Denny TP, Boucher C. 2005. Control of the Ralstonia solanacearum type III secretion system (Hrp) genes by the global virulence regulator PhcA. FEBS Lett. 579:2077–2081 [DOI] [PubMed] [Google Scholar]
- 13. McGarvey JA, Denny TP, Schell MA. 1999. Spatial-temporal and quantitative analysis of growth and EPS I production by Ralstonia solanacearum in resistant and susceptible tomato cultivars. Phytopathology 89:1233–1239 [DOI] [PubMed] [Google Scholar]
- 14. McGarvey JA, Bell CJ, Denny TP, Schell MA. 1998. Analysis of extracellular polysaccharide-I in culture and in planta using immunological methods: new insights and implications, p 157–163 In Bacterial wilt disease: molecular and ecological aspects. Springer-Verlag, Berlin, Germany [Google Scholar]
- 15. Genin S, Gough CL, Zischek C, Boucher CA. 1992. Evidence that the hrpB gene encodes a positive regulator of pathogenicity genes from Pseudomonas solanacearum. Mol. Microbiol. 6:3065–3076 [DOI] [PubMed] [Google Scholar]
- 16. Yoshimochi Y, Hikichi Y, Kiba A, Ohnishi K. 2009. The global virulence regulator PhcA negatively controls the Ralstonia solanacearum hrp regulatory cascade by repressing expression of the PrhIR signaling proteins. J. Bacteriol. 191:3424–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown DG, Swanson JK, Allen C. 2007. Two host-induced Ralstonia solanacearum genes, acrA and dinF, encode multidrug efflux pumps and contribute to bacterial wilt virulence. Appl. Environ. Microbiol. 73:2777–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flores-Cruz Z, Allen C. 2009. Ralstonia solanacearum encounters an oxidative environment during tomato infection. Mol. Plant Microbe Interact. 22:773–782 [DOI] [PubMed] [Google Scholar]
- 19. Cunnac S, Occhialini A, Barberis P, Boucher C, Genin S. 2004. Inventory and functional analysis of the large Hrp regulon in Ralstonia solanacearum: identification of novel effector proteins translocated to plant host cells through the type III secretion system. Mol. Microbiol. 53:115–128 [DOI] [PubMed] [Google Scholar]
- 20. Occhialini A, Cunnac S, Reymond N, Genin S, Boucher C. 2005. Genome-wide analysis of gene expression in Ralstonia solanacearum reveals that the hrpB gene acts as a regulatory switch controlling multiple virulence pathways. Mol. Plant Microbe Interact. 18:938–949 [DOI] [PubMed] [Google Scholar]
- 21. Brito B, Marenda M, Barberis P, Boucher C, Genin S. 1999. prhJ and hrpG, two new components of the plant signal-dependent regulatory cascade controlled by PrhA in Ralstonia solanacearum. Mol. Microbiol. 31:237–251 [DOI] [PubMed] [Google Scholar]
- 22. Bogs J, Geider K. 2000. Molecular analysis of sucrose metabolism of Erwinia amylovora and influence on bacterial virulence. J. Bacteriol. 182:5351–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miller WG, Brandl MT, Quiñones B, Lindow SE. 2001. Biological sensor for sucrose availability: relative sensitivities of various reporter genes. Appl. Environ. Microbiol. 67:1308–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmid K, Schupfner M, Schmitt R. 1982. Plasmid-mediated uptake and metabolism of sucrose by Escherichia coli K-12. J. Bacteriol. 151:68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elphinstone JG, Stanford HM, Stead DE. 1998. Survival and transmission of Ralstonia solanacearum in aquatic plants Solanum dulcamara and associated surface water in England. EPPO Bull. 28:93–94 [Google Scholar]
- 26. Marenda M, et al. 1998. PrhA controls a novel regulatory pathway required for the specific induction of Ralstonia solanacearum hrp genes in the presence of plant cells. Mol. Microbiol. 27:437–453 [DOI] [PubMed] [Google Scholar]
- 27. Boch J, et al. 2002. Identification of Pseudomonas syringae pv. tomato genes induced during infection of Arabidopsis thaliana. Mol. Microbiol. 44:73–88 [DOI] [PubMed] [Google Scholar]
- 28. Brown DG, Allen C. 2004. Ralstonia solanacearum genes induced during growth in tomato: an inside view of bacterial wilt. Mol. Microbiol. 53:1641–1660 [DOI] [PubMed] [Google Scholar]
- 29. Oke V, Long SR. 1999. Bacterial genes induced within the nodule during the Rhizobium-legume symbiosis. Mol. Microbiol. 32:837–849 [DOI] [PubMed] [Google Scholar]
- 30. Dulla G, Lindow SE. 2008. Quorum size of Pseudomonas syringae is small and dictated by water availability on the leaf surface. Proc. Natl. Acad. Sci. U. S. A. 105:3082–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tans-Kersten J, Brown D, Allen C. 2004. Swimming motility, a virulence trait of Ralstonia solanacearum, is regulated by FlhDC and the plant host environment. Mol. Plant Microbe Interact. 17:686–695 [DOI] [PubMed] [Google Scholar]
- 32. Hugouvieux-Cotte-Pattat N, Charaoui-Boukerzaza S. 2009. Catabolism of raffinose, sucrose, and melibiose in Erwinia chrysanthemi 3937. J. Bacteriol. 191:6960–6967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guidot A, et al. 2007. Genomic structure and phylogeny of the plant pathogen Ralstonia solanacearum inferred from gene distribution analysis. J. Bacteriol. 189:377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Remenant B, et al. 2010. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics 11:379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bialczyk J, Lechowski Z. 1995. Chemical composition of xylem sap of tomato grown on bicarbonate containing medium. J. Plant Nutr. 18:2005–2021 [Google Scholar]
- 36. Lugtenberg BJJ, Kravchenko LV, Simons M. 1999. Tomato seed and root exudate sugars: composition, utilization by Pseudomonas biocontrol strains and role in rhizosphere colonization. Environ. Microbiol. 1:439–446 [DOI] [PubMed] [Google Scholar]
- 37. Iyer R, Camilli A. 2007. Sucrose metabolism contributes to in vivo fitness of Streptococcus pneumoniae. Mol. Microbiol. 66:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Green S, et al. 2010. Comparative genome analysis provides insights into the evolution and adaptation of Pseudomonas syringae pv. aesculi on Aesculus hippocastanum. PLoS One 5:e10224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Atkinson MM, Baker CJ. 1987. Alteration of plasmalemma sucrose transport in Phaseolus vulgaris by Pseudomonas syringae pv. syringae and its association with K+/H+ exchange. Phytopathology 77:1573–1578 [Google Scholar]
- 40. Chen L-Q, et al. 2010. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468:527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hendrick CA, Sequeira L. 1984. Lipopolysaccharide-defective mutants of the wilt pathogen Pseudomonas solanacearum. Appl. Environ. Microbiol. 48:94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ausubel F, et al. 1998. Current protocols in molecular biology. John Wiley and Sons, New York, NY. [Google Scholar]
- 43. Heckman KL, Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2:924–932 [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y, Pohlmann EL, Conrad MC, Roberts GP. 2006. The poor growth of Rhodospirillum rubrum mutants lacking PII proteins is due to an excess of glutamine synthetase activity. Mol. Microbiol. 61:497–510 [DOI] [PubMed] [Google Scholar]
- 45. Fegan M, Holoway G, Hayward AC, Timmis J. 1998. Development of a diagnostic test based on the polymerase chain reaction to identify strains of Ralstonia solanacearum exhibiting the biovar 2 genotype, p 34–43 In Bacterial wilt disease: molecular and ecological aspects. Springer Verlag, Berlin, Germany [Google Scholar]
- 46. Opina N, et al. 1997. A novel method for development of species and strain-specific DNA probes and PCR primers for identifying Burkholderia solanacearum (formerly Pseudomonas solanacearum). Asia Pac. J. Mol. Biol. Biotechnol. 5:19–30 [Google Scholar]
- 47. Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics 19:185–193 [DOI] [PubMed] [Google Scholar]
- 48. Bustin SA. 2000. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25:169–193 [DOI] [PubMed] [Google Scholar]
- 49. Tans-Kersten J, Guan Y, Allen C. 1998. Ralstonia solanacearum pectin methylesterase is required for growth on methylated pectin but not for bacterial wilt virulence. Appl. Environ. Microbiol. 64:4918–4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Swanson JK, Yao J, Tans-Kersten J, Allen C. 2005. Behavior of Ralstonia solanacearum race 3 biovar 2 during latent and active infection of geranium. Phytopathology 95:136–146 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clusters of orthologous groups (COG) analysis of R. solanacearum genes expressed during tomato pathogenesis. (A to D) COG categorization of the entire R. solanacearum in planta transcriptome (A), all genes differentially expressed in planta versus in rich CPG medium (fold change of >2, adjusted P < 0.05) (B), upregulated genes (fold change of >2, adjusted P < 0.05) (C), and downregulated genes (fold change of > 2, adjusted P < 0.05) (D). Download Figure S1, TIF file, 0.3 MB.
Expression patterns of Ralstonia solanacearum UW551 in planta and in rich medium.
Expression patterns of Ralstonia solanacearum GMI1000 in planta and in rich medium.
Gene expression trends in the R. solanacearum HrpB regulon in planta versus in rich medium.
Strains and plasmids used in this study.
Oligonucleotide primers used in this study.