

ABSTRACT
We have shown that the herpes simplex virus 1 (HSV-1) gK gene is essential for efficient replication and spread in the corneal epithelium and trigeminal ganglion neuroinvasion in mice (A. T. David, A. Baghian, T. P. Foster, V. N. Chouljenko, and K. G. Kousoulas, Curr. Eye Res. 33:455–467, 2008). To further investigate the role of gK in neuronal infection, we utilized a microfluidic chamber system separating neuronal cell bodies and axonal termini. HSV-1 (McKrae) engineered virus constitutively expressing enhanced green fluorescence protein (GFP) was efficiently transmitted in both a retrograde and an anterograde manner. These results were corroborated by expression of virion structural proteins in either chamber, as well as detection of viral genomes and infectious viruses. In contrast, efficient infection of either chamber with a gK-null virus did not result in infection of the apposed chamber. These results show that gK is an important determinant in virion axonal infection. Moreover, the inability of the gK-null virus to be transmitted in an anterograde manner suggests that virions acquire cytoplasmic envelopes prior to entering axons.
IMPORTANCE
Herpes simplex virus 1 (HSV-1) enters mucosal epithelial cells and neurons via fusion of the viral envelope with cellular membranes, mediated by viral glycoprotein B (gB) in cooperation with other viral glycoproteins. Retrograde transport of virions to neuronal cell bodies (somata) establishes lifelong latent infection in ganglionic neurons. We have previously reported that gK binds gB and is required for gB-mediated membrane fusion (Jambunatathan et al., J. Virol. 85:12910–12918, 2011; V. N. Chouljenko, A. V. Iyer, S. Chowdhury, J. Kim, and K. G. Kousoulas, J. Virol. 84:8596–8606, 2010). In the current study, we constructed a recombinant virus with the gK gene deleted in the highly virulent ocular HSV-1 strain McKrae. This recombinant virus failed to infect rat ganglionic neuronal axons alone or cocultured with Vero cells in microfluidic chambers. In addition, lack of gK expression prevented anterograde transmission of virions. These results suggest that gK is a critical determinant for neuronal infection and transmission.
Introduction
Herpes simplex virus 1 (HSV-1) is an important human pathogen that typically causes mucocutaneous lesions in facial and genital epithelial/mucosal surfaces. The hallmark of the HSV-1 life cycle is infection of sensory neurons, where the virus establishes a latent infection for the life of the host (1). HSV-1 neuronal infection can occasionally cause acute encephalitis (2), while ocular HSV-associated disease is one of the most common infectious causes of blindness in developed countries (3, 4).
HSV-1 enters neuronal cells via a pH-independent fusion of the viral envelope with neuronal plasma membranes (5, 6), while it can enter a wide range of nonneuronal cells via either pH-independent or pH-dependent endocytosis (7). Virus entry into all cells involves the coordinated functions of the glycoproteins gD, gB, gH, gL, and gC (8). Fusion of the viral envelope with cellular membranes causes deposition of the viral capsid into the cytoplasm, which is subsequently transported to the cell nucleus. This retrograde transport of capsids to the neurons is highly dependent on the cellular microtubule network and is most likely mediated via direct interactions by one or more tegument and capsid proteins with the dynein motor and the dynein cofactor dynactin (reviewed in references 9 to 11).
Infectious virus production is initiated in the nucleus of cells, where capsids assemble. The virus acquires an initial viral envelope by budding of capsids into perinuclear spaces. Fusion of the viral envelope with the outer nuclear membrane is thought to deliver partially tegumented capsids into the cytoplasm. These capsids acquire additional tegument proteins before acquiring their final envelopes by budding into intracellular vesicles, most likely originating from the trans-Golgi network (TGN). Fully matured virions are finally released through an exocytosis process that appears to involve the kinesin family of microtubular network-associated motors (reviewed in references 9 to 11).
The mechanism of anterograde transport of alphaherpesvirus in neuronal cells is highly controversial, to a large extent because of disparate results obtained with pseudorabies virus (PRV) and HSV-1. Two principal mechanisms of axonal transport have been proposed: (i) independent axonal transport of viral glycoproteins and capsids and assembly of the enveloped virions in the plasma membrane and (ii) transport of fully enveloped virions that have formed via budding of capsids into TGN-derived membranes. A number of viral glycoproteins have been implicated in efficient egress of virions from infected nonneuronal cells (reviewed in reference 10).
HSV-1 gK is a structural component of the virion particle and functions in virus entry into epithelial cells (12, 13), cytoplasmic virion envelopment, virion egress, and virus-induced cell fusion (14). Recently, we showed that HSV-1 gK and UL20 physically bind to gB and gH and modulate gB-mediated membrane fusion (15, 16). Also, we reported that gK was essential for virus spread in the cornea of mice, neuroinvasiveness, and establishment of latency in ganglionic neurons (17). In this work, we utilized a microfluidic chamber to show that gK is required for infection of neuronal axons and anterograde transport in neurons.
RESULTS
Replication characteristics of recombinant viruses on rat dorsal root ganglionic neurons.
We have previously described the construction of recombinant HSV-1 (McKrae) viruses constitutively expressing the enhanced green fluorescence protein (GFP) under the human cytomegalovirus immediate-early promoter (HCMV-IE) (17). To determine the role of glycoprotein K (gK) in neuronal replication, a set of recombinant viruses with intact or deleted gK genes was constructed and studied. This set of viruses included the following: (i) the wild-type McKrae strain, compared to a mutant virus lacking the gK gene (ΔgK/GFP−) and a virus in which the deleted gK gene was rescued to wild type (R/GFP−) (Fig. 1A to C, respectively); and (ii) a set of McKrae-derived viruses that have an HCMV-IE-GFP gene cassette replacing the gK gene sequence (ΔgK/GFP+) and a derivative virus in which the missing gK gene is replaced, placing the HCMV-IE-GFP gene cassette adjacent to the gK gene (R/GFP+) (Fig. 1D and E, respectively).
FIG 1 .

Schematic representation of mutant viruses. The top line represents the prototypic arrangement of the HSV-1 genome with the unique long (UL) and unique short (US) regions flanked by the terminal repeat (TR) and internal repeat (IR) regions. (A) McKrae wild type. (B) McKrae parental strain with deletion mutation of gK and without the GFP cassette (ΔgK/GFP−). (C) Rescue of the gK deletion in the virus in part B without the GFP cassette (R/GFP−). (D) McKrae parental strain with deletion of the gK gene by replacement with a GFP cassette (ΔgK/GFP+). (E) Rescue of the virus in part D with preservation of the GFP (R/GFP+).
All wild-type-like viruses (wild-type and gK-rescued virus) produced on average similar-size plaques on both Vero and VK302 cells (Fig. 2) (17). In contrast, gK-null viruses prepared in either Vero or VK302 cells produced on average drastically smaller viral plaques on Vero cells. The gK-null defects were complemented on VK302 cells, which express the HSV-1 (KOS) gK gene (Fig. 2) (17, 18). Examination of individual viral plaques by fluorescence microscopy revealed that all viruses containing the GFP gene cassette readily expressed GFP (Fig. 2).
FIG 2 .

Representative plaque morphologies of the HSV-1 (McKrae) strain and mutants on Vero and VK302 cells. Confluent Vero or gK-complementing VK302 cells monolayers were infected with the R/GFP+, ΔgK/GFP+ (Vero), ΔgK/GFP+ (VK302), R/GFP−, ΔgK/GFP− (Vero), or ΔgK/GFP− (VK302) virus at an MOI of approximately 0.001. Viral plaques were visualized by immunohistochemistry at 72 hpi using rabbit anti-HSV-1 developed with Nova Red (Vector Labs) with contrast microscopy. Fluorescent plaques from viruses with GFP were observed using a fluorescence microscope at 72 hpi. Plaques shown are representative of average plaque sizes observed. All images were taken at a magnification of ×100.
Infection of rat dorsal root ganglionic neurons with wild-type McKrae virus resulted in rapid cytopathic effects exhibited as early as 12 h postinfection (hpi). Typically, neurons appeared rounded up and swollen, while the overall number and length of axons present before infection appeared to be markedly reduced at early times after infection (not shown). Ganglionic neurons were infected with each of the viruses, and growth was compared in viruses containing GFP (Fig. 3A) and those without GFP (Fig. 3B). The parental strain is included in both graphs for a baseline comparison. Wild-type HSV-1 (McKrae) virus and virus produced after rescue of the deleted gK gene replicated with approximately equal efficiencies in the presence (Fig. 3A) or absence (Fig. 3B) of the GFP gene. However, mutant viruses lacking the gK gene whether stocks were prepared on either Vero or the gK complementing cell line, VK302, replicated 1 to 2 logs less efficiently than the parental McKrae viruses at late times postinfection on rat neurons (Fig. 3A and B). Similar results were obtained for gK-null virus replication in Vero in comparison to those for the wild-type virus (17). Specifically, titers for the ΔgK viruses approached maximum titers ranging from 4 × 104 to 7 × 104 PFU, while the rescued viruses approached maximum titers of 4 × 105 to 5 × 105 PFU. In addition, gK-null virus titers appeared to decrease during an apparent eclipse phase of viral replication before increasing again at later times postinfection. These results show that infectious gK-null viruses were clearly produced in neurons.
FIG 3 .

Growth curves of McKrae strain virus and mutants on DRG neurons. Approximately 175,000 ganglionic cells plated in 24-well plates were infected with the parental McKrae strain or R/GFP+, ΔgK/GFP+ (Vero), ΔgK/GFP+ (VK302), R/GFP−, ΔgK/GFP− (Vero), or ΔgK/GFP− (VK302) virus at an MOI of approximately 0.1. Viral titers were obtained at indicated time points postinfection by standard titration plaque assay on Vero or VK302 cells. (A) GFP-containing viruses ([astc]/GFP+) and the McKrae parental strain for comparison. (B) Viruses that do not have GFP ([astc]/GFP−) and the McKrae parental strain for a baseline comparison. Plaque-forming unit totals represent the means of data from triplicate experiments, and error bars represent the standard errors.
Microfluidic multichamber device for neuronal transport studies.
A commercially available microfluidic device was utilized for neuronal transport studies (see Materials and Methods). This device enables the physical separation of neuronal somata and axon termini under virion-impermeable conditions (Fig. 4A). To ensure that virions could not be passively transmitted from one chamber to the other, control experiments were performed as follows: Vero cells were plated in wells on both sides of the microfluidic chamber. Wells of one side were infected with R/GFP+ virus at a multiplicity of infection (MOI) of 10. A hydrostatic pressure differential between the infected and uninfected chambers was maintained by ensuring that the uninfected fluid levels were substantially higher than those in the infected wells. The infected cells exhibited extensive cytopathicity and expression of GFP at 48 hpi. In contrast, the uninfected cells did not exhibit any cytopathic effects or fluorescence at 5 days postinfection, suggesting that virions were not able to passively leak from the infected into the uninfected wells (Fig. 4B).
FIG 4 .

Microfluidic neuron device system. (A) Photograph of the neuron device (Xona Microfluidics) that maintains a fluid pressure (left panel). The virus-impermeable barrier containing axons (middle panel) is shown. A side-view schematic shows wells, main central channel, and a microgroove barrier showing the maintenance of a fluid pressure gradient (right panel). (B) Control experiment to test for system integrity. Phase contrast and GFP fluorescence of infected Vero cells at 3 days postinfection and uninfected Vero cells at 5 days postinfection are shown. During all 5 days, the uninfected side wells were maintained at a higher fluid level than the infected side wells.
Infections of axonal termini.
Microfluidic chambers were seeded with rat dorsal root ganglionic neurons. Neurons were allowed to propagate axons across the microgroove barrier for 10 to 15 days, or until axons and neuronal endings were visibly present across the barrier and the apposed chamber, respectively (see Materials and Methods). The neuronal ending chambers were infected with R/GFP+ and ΔgK/GFP+ viruses at an MOI of 5. Progression of infection on the neuronal soma side of the microfluidic device was monitored daily using a fluorescence microscope to detect GFP expression. GFP expression was readily detected as early as 24 hpi for the R/GFP+ virus, while infection with the ΔgK/GFP+ virus failed to produce any GFP expression as late as 5 days postinfection (Fig. 5A to E). Surprisingly, ΔgK/GFP+ virus grown on the VK302, gK-complementing, cells failed to be efficiently transmitted to the neuronal soma side of the microfluidic device, as evidenced by an occasional fluorescent cell detected in the neuronal soma chambers (Fig. 5F). In a second series of experiments, axonal compartments were coplated with a monolayer of VK302 cells prior to infection. Similarly, the R/GFP+ virus was efficiently transmitted to the soma chamber when axonal compartments containing VK302 cells were infected (Fig. 5G to J). In contrast, there was only occasional fluorescence detected in the soma chamber infected with the ΔgK/GFP+ virus despite strong replication and spread within the VK302 cell monolayer, evidenced by the level and extent of GFP-emitted fluorescence (Fig. 5K to N).
FIG 5 .
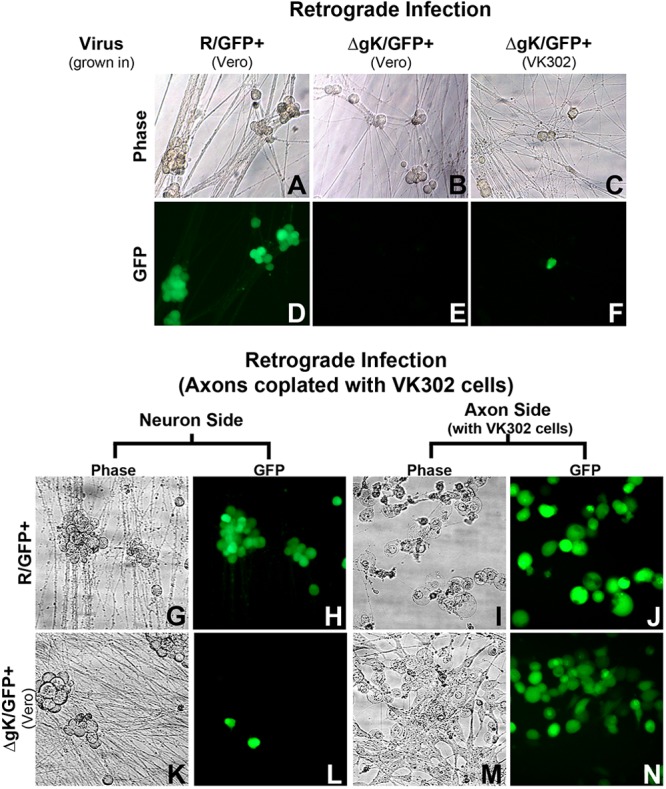
Infection of neuron cell bodies (Soma) via retrograde transport. Retrograde infection via direct infection of axons (A to F) was analyzed. Phase contrast (A to C) and GFP fluorescence (D to F) of neurons are shown. Axonal sides of chambers (not shown) were infected with ΔgK/GFP+ (VK302), ΔgK/GFP+ (Vero), and R/GFP+ (Vero) virus. Neurons potentially infected with ΔgK/GFP+ (VK302 or Vero) were photographed, using phase and fluorescence microscopy, at 5 days postinfection (dpi) to allow ample time for retrograde transport. Neurons infected with R/GFP+ were photographed, using phase and fluorescence microscopy, at 2 dpi due to the presence of ample infected neurons. Retrograde infection via infection of VK302 cells coplated on axons using phase and fluorescence microscopy (G to N) is shown. Axonal sides of chambers were infected with ΔgK/GFP+ (Vero) or R/GFP+ virus. R/GFP+ infection of neurons (G and H) and axons with VK302 cells (I and J) was photographed at 3 dpi. For ΔgK/GFP+ virus, grown in Vero cells, infection of few neurons (K and L) and infection of axons with VK302 cells (M and N) were photographed at 3 dpi. All images were taken at a magnification of ×100.
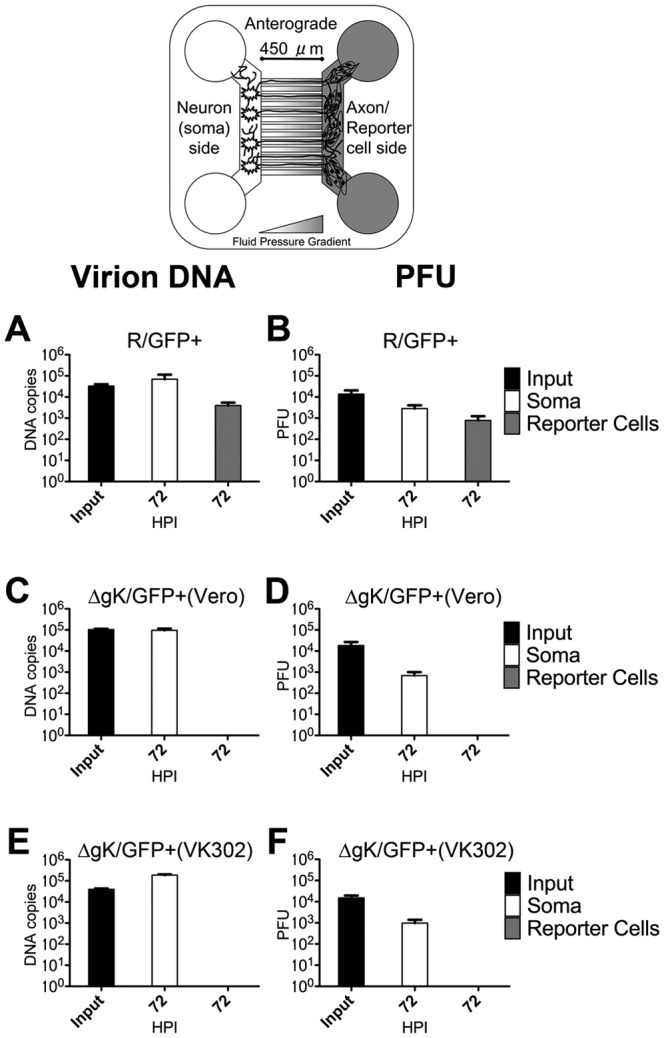
To determine whether infectious virions and/or viral DNA was transported in a retrograde manner, neuronal terminus chambers were infected with an MOI of approximately 5. The number of viral genomes was obtained by quantitative PCR, while the presence of infectious virus was assessed by determining viral titers on VK302 cells as detailed in Materials and Methods. For all three infections, there was little difference in DNA levels on the axon side over a 72-h period (Fig. 6). Examination of the number of viral genomes and PFU obtained from samples derived from the soma side of the devices revealed that the R/GFP+ virus efficiently transported and replicated in soma chambers, producing maximum viral genomes and PFU at 72 hpi. In contrast, both the ΔgK/GFP+ virus grown on Vero cells and ΔgK/GFP+ virus grown on VK302 cells failed either to enter neurite ends or to be transported through axons and subsequently replicate in soma chambers at 72 hpi (Fig. 6B, D, and F).
FIG 6 .

Determination of PFU and viral genomes recovered in retrograde virus transport experiments. Viral DNA copy number, determined by QPCR, and PFU, determined by serial titration plaque assay, are shown for samples recovered from the axon and soma sides of devices infected in a retrograde manner. Bars represent the means for 6 (DNA) or 4 (PFU) samples taken from devices at 2, 48, and 72 hpi, as well as corresponding volume samples of inocula taken at the time of infection (input). (A and B) R/GFP+ infected. (C and D) ΔgK/GFP+ (Vero) infected. (E and F) ΔgK/GFP+ (VK302) infected. Input bars are black. Bars representing the axon side (white) and soma side (gray) are shaded to correspond to the sides in the schematic. The minimum level of QPCR detection was approximately 40 genomes. A baseline threshold of 0 was set at approximately 100 genomes, since samples containing less than 100 genomes exhibited high variability among the triplicate samples.
Neuronal cell body infection and anterograde transport of virions.
For anterograde studies, after seeding of microfluidic chambers and propagation of axons, axonal side wells were coplated with a monolayer of VK302 cells. Then, the neuronal somata were infected as described for the neurite-only side in the retrograde studies. Infection of the neuronal soma side of the microfluidic chambers was also monitored daily by visualizing GFP fluorescence. All three viruses, R/GFP+, ΔgK/GFP+ (Vero), and ΔgK/GFP+ (VK302), infected neuronal somata efficiently, as evidenced by the strong GFP fluorescence (Fig. 7A). However, only the R/GFP+ virus was transported in an anterograde manner to the VK302 reporter cells on the microfluidic chambers (Fig. 7A). Similar virus axonal transport experiments were performed, but this time the cells within the soma and reporter sides of the microfluidic chambers were fixed and immune stained with antibodies specific for viral gC and neurofilament (neuron-specific stain), and viruses not expressing GFP were utilized (to allow multicolor immunofluorescence). Cells in both the soma and reporter sides of the chambers expressed relatively high levels of gC only when infected with the R/GFP− virus, while both ΔgK/GFP− (Vero) and ΔgK/GFP− (VK302) viruses expressed gC in cells in the soma side of the chambers, while no gC expression was detected on the reporter side of the chambers (Fig. 7B).
FIG 7 .

Anterograde infection and transmission. (A) Anterograde infection and transmission demonstrated by phase and fluorescence microscopy of the infected neuronal side and the uninfected reporter cell/axon side. Only the rescued virus was observed spreading in the anterograde direction to reporter cells. (B) Anterograde infection and transmission demonstrated by fluorescent antibodies. Images are stained with antineurofilament antibody with Alexa Fluor 488 secondary (green), anti-HSV-1 gC with Alexa Fluor 594 secondary (red), and DAPI (blue). The neuron sides of the microfluidic devices were infected with R/GFP−, ΔgK/GFP− (Vero), and ΔgK/GFP− (VK302) virus.
Anterograde transport of virions was also monitored by determining the number of infectious viruses produced on the reporter side of the microfluidic chamber after infection of the neuronal soma sides, as well as determining the number of viral genomes transmitted axonally to the reporter sides using quantitative PCR (QPCR). Infectious virions were retrieved only in infections with the R/GFP+ virus from the reporter cell side, while both R/GFP+ and ΔgK/GFP+ viruses were recovered from somata. In agreement with these results, viral DNA was recovered for all viruses in somata but only for R/GFP+ in the reporter cell side (Fig. 8).
FIG 8 .
Determination of PFU and viral genomes recovered in anterograde virus transport experiments. Viral genome and PFU numbers from the axon/reporter cell and soma sides of devices infected in the anterograde manner. Bars represent the means for 6 (DNA) or 4 (PFU) samples taken from devices at 72 hpi, as well as corresponding volume samples of inocula taken at the time of infection (input). (A and B) R/GFP+ infected. (C and D) ΔgK/GFP+ (Vero) infected. (E and F) ΔgK/GFP+ (VK302) infected. Input bars are black. Bars representing the soma side (white) and reporter cell side (gray) are shaded to correspond to the sides in the schematic. The minimum level of QPCR detection was approximately 40 genomes. A baseline threshold of 0 was set at approximately 100 genomes, since samples containing less than 100 genomes exhibited high variability among the triplicate samples.
DISCUSSION
HSV-1 gK functions as a protein complex with the membrane protein UL20 in membrane fusion phenomena during virus entry and virus-induced cell fusion, enabling virions to enter into cells and spread into adjacent uninfected cells, respectively (13, 15, 16, 19–21). Previously, we showed that the lack of gK prevented corneal spread in mice, transmission of the gK-null virions to the central nervous system, and establishment of latency. Here we have shown that gK is essential for infection of neuronal axons in rat dorsal root ganglionic neurons in culture, in agreement with the previous in vivo studies (17). Moreover, consistent with the role of gK in cytoplasmic envelopment and egress, gK-null virions were unable to be transported in an anterograde manner.
Herein, we have chosen to work with the HSV-1 McKrae strain because this virus was isolated from a human ocular herpes infection (22) and shown to be highly virulent in mice and rabbits (23–25). In these studies, we utilized a set of recombinant viruses lacking gK gene expression in the presence or absence of GFP expression for comparative purposes, because constitutive expression of the GFP gene may alter viral replication characteristics (26). Wild-type-like viruses replicated to similar levels in the presence or absence of GFP expression in ganglionic neurons. In contrast, gK-null virions reached maximum viral titers that were 1 to 2 logs lower than those of their corresponding wild-type-like viruses, while GFP expression did not adversely affect viral growth. These experiments indicated that GFP expression did not alter replication characteristics of these viruses. The relative size of viral plaques is an indication of the ability of the virus to spread from infected to uninfected cells. Consistent with previous findings, the lack of gK caused the production of substantially smaller viral plaques, indicating a severe defect in virus spread. GFP expression did not have any effect on the gK-null defect in virus spread.
Microfluidic devices separating axonal termini from neuronal somata have been extensively used in herpesvirus experiments to investigate the contribution of individual genes in retrograde and anterograde axonal transport (27–30). We utilized a commercially available microfluidic device (Xona Microfluidics, LLC) originally developed from a collaboration between Lynn Enquist’s laboratory (Princeton University) and No Li Jeon’s laboratory (University of California—Irvine) (29). It is particularly important in all studies using these devices that control experiments are performed carefully to ensure that there is no leakage of fluids between the device and the coverslip on which the device is placed. Therefore, methods utilized in these studies were validated for obtaining appropriate sealed chambers prior to the onset of each experiment. Furthermore, chambers were inspected to ensure that axons were exclusively appearing within the device microgrooves.
Infections of chambers containing axonal termini showed that the wild-type-like virus (R/GFP+) was readily transported to the soma-containing chamber, while the gK-null ΔgK/GFP+ virus failed to infect neurons via their axons whether stocks were grown in Vero cells or the gK-complementing VK302 cells line. Additionally, when the ΔgK/GFP+ virus was used to infect VK302 cells cocultured with axonal endings, there was a low level of neuronal infection and transport to the somata (as demonstrated by GFP fluorescence). However, this was strikingly lower than the soma infection observed with the rescued virus, especially when considered in light of the similar and profound extent of infection of the coplated VK302 cells with either the rescued or gK-null viruses. In these experiments, GFP expression was driven by the constitutive HCMV-IE promoter, requiring entry of the viral genome into neuronal nuclei and transcription/translation of the GFP gene. Therefore, these experiments cannot differentiate whether virions entered neurons but were subsequently unable to be either transported or replicated within somata. In an attempt to address this issue, the total viral genomes and infectious virions remaining within the axonal terminus-containing chambers were determined. Viral genome numbers did not decrease in the gK-null virus, unlike the wild-type-like (rescued) infections of neurites, suggesting that the gK-null virions were unable to enter axons. This result was not corroborated by the PFU counts, most likely because of the natural decay of infectious virions with prolonged incubation at 37°C. Despite the consistent detection of few cells expressing GFP in the retrograde infections with the ΔgK/GFP+ virus within somata, there were no infectious virions recovered from these chambers even at 72 hpi. Collectively, these results indicate that the observed low levels of GFP expression may be due to transport of viral DNA fragments containing the GFP gene cassette. These gene fragments may exist in the original virus stocks used for infections or produced after degradation of endocytosed virions into neurons.
Surprisingly, ΔgK/GFP+ virus grown on the complementing cell line VK302 was also largely unable to infect neurons via their axons. This result suggests that expression of the HSV-1 (KOS) gK gene in VK302 cells may complement for infectious virus production in Vero cells; however, virions produced in VK302 cells may not be able to efficiently infect neuronal axons. We have shown that the amino terminus of gK interacts with the amino terminus of gB, most likely affecting the ability of gB to mediate fusion of the viral envelope with cellular membranes during virus entry (13, 15, 16). Furthermore, lack of gK prevented entry via fusion of the viral envelope with cellular membranes (31). Comparison of the predicted amino acid sequences of KOS and McKrae gB and gK reveal a number of amino acid differences, including amino acid changes within the amino termini of gB and gK. Thus, it is conceivable that KOS gK cannot efficiently complement the McKrae gK-null virus for entry into axons, because of altered interactions between KOS gK and McKrae gB. Moreover, KOS gK provided by the VK302 cells may not be efficiently incorporated into virions, causing reduction in neuronal infectivity.
UL20 and gK are required for cytoplasmic virion envelopment and egress in epithelial cells (14, 19, 20). Furthermore, direct comparison of the role of gK and UL20 in cytoplasmic envelopment, virion egress, and infectious virus production with similar functions provided by the carboxyl terminus of gD, gE, UL11, or gM has revealed that gK and UL20 are more important than each of these proteins alone or in combination (32). Lack of gK drastically inhibited transmission of virus in an anterograde manner. Neurons, unlike epithelial cells or fibroblasts, have an intricate distribution of cytoplasmic organelles within somata and axons. Specifically, TGN-derived membrane vesicles migrate to synapses and play important roles in synapse formation among neurons (33). Therefore, it is conceivable that cytoplasmic envelopment of virions occurs both within somata and via budding into TGN-derived vesicles proximal to synaptic membranes.
ΔgK virions grown on Vero or VK302 cells appeared to infect somata with approximately similar efficiencies, indicating that a lack of gK does not substantially alter entry of the gK-null virions via infection of neuronal cell bodies. Neurons are known to express nectin-1, which binds gD and functions in virus entry (34, 35). In agreement with these published results, we readily detected nectin-1 expression in neuronal termini, axons, and somata (not shown). Thus, altered distribution of nectin-1 between axons and somata is probably not responsible for the lack of gK-null virus infection of neuronal termini. Nectin-1 is known to primarily mediate endocytosis of virions rather than fusion of the viral envelope with cellular membranes (7). Thus, it is possible that virions enter axons primarily via membrane fusion, suggesting that gK functions in virus-cell membrane fusion utilizing cellular receptors other than nectin-1.
Herpes simplex virus virions have evolved a highly complex system for optimum infection of neurons, the ultimate destination of the virus in the human host. It is conceivable that virus entry into neuronal synapses requires the presence of gK. We hypothesize that gK, known to interact with the amino terminus of gB, may modulate fusogenic activity of gB via specific gB cellular receptors. A lack of gK may result in a loss of critical regulatory functions provided by gK on gB to mediate fusion of the viral envelope with synaptic membranes, resulting in inability to enter axons. Alternatively, virions may enter but rapidly degrade within lysosomal compartments. Direct visualization of fluorescent virions may resolve these issues, which will be addressed in future experiments.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Rockville, MD) and grown and propagated in Dulbecco’s modified Eagle medium (DMEM) supplemented with 7% fetal bovine serum (FBS) and antibiotics. The gK-complementing VK302 cell line (generously provided by David Johnson, Oregon Health Sciences University, Portland, OR) was maintained in DMEM supplemented with 10% FBS and antibiotics. The clinical ocular isolate and neuroinvasive strain of HSV-1 (the parental wild type), McKrae strain, was obtained from J. M. Hill (Louisiana State University Health Sciences Center, New Orleans, LA) (Fig. 1A).
Construction of recombinant viruses.
The construction methodology for mutant viruses for this experiment is essentially similar to the techniques that were previously described for creating KOS strain mutants (17, 19, 36, 37). McKrae virus with a deleted UL53 (gK) gene (ΔgK) (Fig. 1B) was constructed using the ICP27-null (d27-1) virus and plasmid pSJ1724, as described previously for the HSV-1 (KOS) ΔgK virus (19). The ICP27-null virus has 1,627 bp of UL54 gene deleted but contains a complete UL53 gene. Plasmid pSJ1724 has a 1,068-bp deletion in UL53 but has an intact UL54. Recombination between homologous regions of d27-1 virus and plasmid pSJ1724 resulted in the ΔgK virus. This virus does not contain a GFP cassette and is here designated ΔgK/GFP−. This ΔgK virus was rescued (R/GFP−) (Fig. 1C) by transfecting Vero cells with PCR-amplified fragments of the UL53 gene from the wild-type HSV-1 (KOS) strain, followed by infection with the ΔgK virus (19). The unique long (UL) UL53 (gK) gene of HSV-1 McKrae was replaced with an enhanced GFP gene cassette under the control of the human cytomegalovirus immediate-early gene promoter (HCMV-GFP) (36). Briefly, The HCMV-GFP cassette was cloned into the plasmid pSJ1723 to create the plasmid pTF9201, as described previously (17, 36). Plasmid PTF9201 contains a GFP gene cassette under the human cytomegalovirus immediate-early promoter control (HCMV-IE) within a DNA fragment spanning the UL52–UL54 genomic region that lacks the UL53 gene. Plasmid PTF9201 was used in transfection, followed by infection with HSV-1 (McKrae), to isolate the McKrae ΔgK/GFP virus (Fig. 1D), designated ΔgK/GFP+, appearing as small fluorescent viral plaques under the fluorescence microscope (17, 36). The McKrae ΔgK/GFP virus was rescued using plasmid pTF9105, which contains a complete UL53 gene from the HSV-1 (KOS) strain, creating the virus designated R/GFP+ (Fig. 1E) (37).
Animals.
Adult female Sprague-Dawley rats, 15 to 18 days pregnant, were used (Charles River, Wilmington, MA). All procedures were approved by the Louisiana State University School of Veterinary Medicine Institutional Animal Care and Use Committee.
Dorsal root ganglion primary neuron culture.
Dorsal root ganglion (DRG) neurons were cultured under conditions that were adapted from those described previously for culturing sympathetic superior cervical ganglion-derived neurons (29). Additional modifications were adapted from previously described culture conditions for dorsal root ganglia (38, 39). Specifically, ganglia were dissected from fetal Sprague-Dawley rats at embryonic days 15 to 18. DRG were incubated at 37°C for 30 min in 250 µg/ml of trypsin (Worthington Biochemical Corp., Lakewood, NJ). Trypsin was neutralized by incubating ganglia in neuron culture medium with 10% FBS and 1 mg/ml trypsin inhibitor (Sigma-Aldrich, St. Louis, MO) for 2 min. Ganglia were rinsed with fresh neuron culture medium, and ganglionic neurons were gently triturated into a single-cell suspension using a pulled-glass Pasteur pipette. Then, cells were plated into one side of the microfluidic neuron devices (catalog no. SND450; Xona Microfluidics, LLC, Temecula, CA) adhered to glass coverslips or the surface of 35-mm plastic petri dishes. Neuron devices were assembled by placing a standard neuron microfluidic device firmly on the coverslip or petri dish to ensure a sealed barrier. Maintenance neuron culture medium consisted of neural basal medium with B-27 supplement, at the manufacturer’s recommended concentration (Invitrogen, Grand Island, NY). Medium was supplemented with 50 ng/ml neural growth factor 2.5s (Invitrogen), 2% normal rat serum (Invitrogen), 1× GlutaMAX (Invitrogen), and 0.2% Primocin (InvivoGen, San Diego, CA). To eliminate nonneuronal cells, 1 and 3 days postplating, neuronal cultures were treated with 4 µM cytosine β-d-arabinofuranoside (CAB) (Sigma-Aldrich) in neuronal medium for 24 h.
Infection of neuron devices.
Neurons were allowed to grow for 10 to 14 days postplating until robust axonal processes with abundant branching were observed on the axon side of neuron devices. Once these were present, the neuron devices were prepared for infection. For retrograde infections, medium was completely depleted from the wells of the axon side of microfluidic devices. Then, on the neuron soma side of the devices, wells were filled to maximum fluid capacity. After establishment of the fluid pressure gradient, axon-side wells were inoculated with the virus and concentration indicated in Results. Then, approximately 100 µl of neuron culture medium was placed in the infected wells to ensure mixing of the virus into the central chamber containing the axons. Once the fluid pressure gradient had been established, medium was added to the soma side only if necessary to combat dehydration and maintain an obvious fluid level difference. In the case of anterograde neuronal infection studies and a retrograde experiment (specified in Results), the axonal side was coplated with a monolayer of VK302 or Vero cells.
Viral replication kinetics on neurons.
To determine the kinetics of viral replication on ganglionic neurons, 24-well plastic plates were plated with 175,000 ganglionic cells using the above-described harvesting protocol. Ganglionic neurons were infected at approximately a multiplicity of infection (MOI) of 0.1 with each of the viruses. Plates were prepared in triplicate, and infections were halted by freezing at 0, 12, 24, 48, and 72 hpi. Plates were frozen and thawed, and viral titers were determined for each time point by standard plaque assay on VK302 cells (for the ΔgK viruses) or Vero cells (for the parental and rescue strains). Plaques were stained with crystal violet and visualized with a dissecting microscope. Growth curves show the result of the average for the triplicate plates.
Plaque morphology.
For fluorescent plaque morphology, confluent monolayers of Vero and VK302 cells were infected at an MOI of 0.001 with the indicated viruses. GFP fluorescence of representative plaques was visualized and photographed at 72 h postinfection (hpi) with a fluorescence microscope (Olympus, Tokyo, Japan). For immunohistochemical plaque morphology, cells were fixed with 10% formalin at 72 hpi (17, 40). Immunohistochemistry was performed with primary rabbit anti-HSV antibodies (1:1,000) (Dako, Carpinteria, CA) and goat anti-rabbit horseradish peroxidase-labeled antibody (1:1,000) (Dako, Carpinteria, CA), and the reactions were developed using the NovaRed substrate (VectorLabs; Burlingame, CA). Images were taken with an inverted light microscope (Olympus) using relief contrast.
Visualization of retrograde and anterograde infections.
For direct observation by phase-contrast of the viral infection, neuronal soma sides and axon/reporter cell sides of devices were observed and imaged using an inverted microscope (Olympus) using relief contrast. GFP fluorescence was observed using the same inverted microscope set for fluorescence. For immunofluorescent antibody analysis of anterograde infections, microfluidic devices were fixed with cold 10% formalin. Then, the devices were incubated with blocking buffer (PBS containing 4% bovine serum albumin [BSA] and 2% goat serum) for 1 h at room temperature. Next, the primary antibodies were added, and devices were incubated overnight at 4°C. Primary antibodies used were mouse monoclonal anti-HSV-1 glycoprotein C IgG2a (Virusys Corp., Taneytown, MD), diluted 1:3,000, and mouse monoclonal antineurofilament IgG1 (Invitrogen), diluted 1:1,000. Next, secondary antibodies, goat anti-mouse IgG1 conjugated to Alexa Fluor 488, and goat anti-mouse IgG2a conjugated to Alexa Fluor 594 (Invitrogen), both diluted to 1:1,000, were applied. All antibodies were diluted in PBS containing 2% BSA and 1% goat serum. After 30 min, devices were washed 5 times with PBS. The microfluidic neuron devices were placed on ice and removed from the glass coverslips (per the manufacturer’s instructions). ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) was added to the coverslips, and cells were imaged with an inverted fluorescence microscope.
Sample collection for PFU and quantitative PCR (QPCR) studies.
For studies of viral DNA transmission and virus/viral DNA production, neuron devices were sampled in the following manner. The low side (axon side in retrograde studies and soma side in anterograde studies) was always aspirated first. At the appropriate time points, the medium present in the low-side wells was aspirated and collected. Then, the sampled side was flushed with a small amount of medium. Following that, medium from the higher side was collected, and that side was flushed with a small amount of medium. Subsequently, the high-side wells were filled with medium, and the devices were frozen overnight. Devices were then thawed, the low side was flushed with a small amount of medium, and this medium was collected. Finally, the high side was flushed and collected. This procedure allowed for maintenance of the fluid pressure gradient by always keeping the gradient and allowed for collection of cell-associated virus as well as any free virus in the medium. At this point, all samples were frozen. Later these samples were thawed and used for analysis of PFU or viral DNA.
Detection of PFU from anterograde and retrograde infections.
Samples of medium collected from the neuron devices were thawed and plated in serial dilutions. The number of infectious virions in the medium was determined by plaque assay on appropriate (Vero or VK302) monolayers visualized using the immunohistochemical technique described for plaque morphology.
QPCR.
For quantitative PCR, once thawed, samples were processed using the Qiagen DNeasy blood and tissue kit (Qiagen, Valencia, CA) per the manufacturer’s instructions to isolate DNA. QPCR was performed as described previously for Kaposi’s sarcoma herpesvirus (KSHV) (41, 42). Specifically, the primers and probe (6-carboxytetramethylrhodamine [TAMRA]) for the real-time PCR were designed to detect HSV-1 US6 (gD). Equal volumes of viral DNA were used for TaqMan PCR 196 analysis. Purified plasmid containing the gD gene was initially used to generate the standard curve. Samples were also tested and genome numbers were determined using validated standards provided by the Path-HSV-1-genesig real-time PCR detection kit for human herpesvirus 1 (herpes simplex type 1) (PrimerDesign, Ltd., South Hampton, United Kingdom).
ACKNOWLEDGMENTS
This work was supported by grant AI43000 from the National Institute of Allergy and Infectious Diseases to K.G.K. A.T.D. has been funded by the NIH-NCRR training grant 1T32RR021309 and BIOMMED. We acknowledge financial support by the LSU School of Veterinary Medicine to BIOMMED.
Footnotes
Citation David AT, et al. 2012. A herpes simplex virus 1 (McKrae) mutant lacking the glycoprotein K gene is unable to infect via neuronal axons and egress from neuronal cell bodies. mBio 3(4):e00144-12. doi:10.1128/mBio.00144-12.
REFERENCES
- 1. Whitley RJ, Kimberlin DW, Roizman B. 1998. Herpes simplex viruses. Clin. Infect. Dis. 26:541–555 [DOI] [PubMed] [Google Scholar]
- 2. Whitley R. 2001. Herpes simplex viruses, p 2461–2510 In Knipe D, Howley P, Fields virology. Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 3. Liesegang TJ, Melton LJ, III, Daly PJ, Ilstrup DM. 1989. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch. Ophthalmol. 107:1155–1159 [DOI] [PubMed] [Google Scholar]
- 4. Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13 [DOI] [PubMed] [Google Scholar]
- 5. Nicola AV, Hou J, Major EO, Straus SE. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609–7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qie L, Marcellino D, Herold BC. 1999. Herpes simplex virus entry is associated with tyrosine phosphorylation of cellular proteins. Virology 256:220–227 [DOI] [PubMed] [Google Scholar]
- 7. Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 18:35–51 [DOI] [PubMed] [Google Scholar]
- 10. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 11. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 12. Foster TP, Rybachuk GV, Kousoulas KG. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 75:12431–12438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jambunathan N, et al. 2011. Site-specific proteolytic cleavage of the amino terminus of herpes simplex virus glycoprotein K on virion particles inhibits virus entry. J. Virol. 85:12910–12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hutchinson L, Roop-Beauchamp C, Johnson DC. 1995. Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J. Virol. 69:4556–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chouljenko VN, Iyer AV, Chowdhury S, Chouljenko DV, Kousoulas KG. 2009. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J. Virol. 83:12301–12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 84:8596–8606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. David AT, Baghian A, Foster TP, Chouljenko VN, Kousoulas KG. 2008. The herpes simplex virus type 1 (HSV-1) glycoprotein K(gK) is essential for viral corneal spread and neuroinvasiveness. Curr. Eye Res. 33:455–467 [DOI] [PubMed] [Google Scholar]
- 18. Hutchinson L, Johnson DC. 1995. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 69:5401–5413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jayachandra S, Baghian A, Kousoulas KG. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 71:5012–5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Melancon JM, Luna RE, Foster TP, Kousoulas KG. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 82:6310–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaufman HE, Ellison ED, Waltman SR. 1969. Double-stranded RNA, an interferon inducer, in herpes simplex keratitis. Am. J. Ophthalmol. 68:486–491 [DOI] [PubMed] [Google Scholar]
- 23. Halford WP, Balliet JW, Gebhardt BM. 2004. Re-evaluating natural resistance to herpes simplex virus type 1. J. Virol. 78:10086–10095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perng GC, et al. 1996. The region of the herpes simplex virus type 1 LAT gene that is colinear with the ICP34.5 gene is not involved in spontaneous reactivation. J. Virol. 70:282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perng GC, et al. 1995. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol. 69:3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeWire SM, Money ES, Krall SP, Damania B. 2003. Rhesus monkey rhadinovirus (RRV): construction of a RRV-GFP recombinant virus and development of assays to assess viral replication. Virology 312:122–134 [DOI] [PubMed] [Google Scholar]
- 27. Ch’ng TH, Flood EA, Enquist LW. 2005. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. Methods Mol. Biol. 292:299–316 [DOI] [PubMed] [Google Scholar]
- 28. Feierbach B, Bisher M, Goodhouse J, Enquist LW. 2007. In vitro analysis of transneuronal spread of an alphaherpesvirus infection in peripheral nervous system neurons. J. Virol. 81:6846–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu WW, Goodhouse J, Jeon NL, Enquist LW. 2008. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS One 3:e2382 http://dx.doi.org/10.1371/journal.pone.0002382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Curanovic D, Enquist L. 2009. Directional transneuronal spread of α-herpesvirus infection. Future Virol. 4:591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chowdhury S, Naderi M, Chouljenko VN, Walker JD, Kousoulas KG. 13 June 2012. Amino acid differences in glycoproteins B (gB), C (gC), H (gH) and L (gL) are associated with enhanced herpes simplex virus type-1 (McKrae) entry via the paired immunoglobulin-like type-2 receptor. Virol. J. 9:112 [Epub ahead of print.] doi:10.1186/1743-422X-9-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chouljenko DV, et al. 2012. Functional hierarchy of herpes simplex virus type-1 viral glycoproteins in cytoplasmic virion envelopment and egress. J. Virol. 86:4262–4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sytnyk V, Leshchyns’ka I, Dityatev A, Schachner M. 2004. Trans-Golgi network delivery of synaptic proteins in synaptogenesis. J. Cell Sci. 117:381–388 [DOI] [PubMed] [Google Scholar]
- 34. Richart SM, et al. 2003. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J. Virol. 77:3307–3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simpson SA, et al. 2005. Nectin-1/HveC mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J. Neurovirol. 11:208–218 [DOI] [PubMed] [Google Scholar]
- 36. Foster TP, Rybachuk GV, Kousoulas KG. 1998. Expression of the enhanced green fluorescent protein by herpes simplex virus type 1 (HSV-1) as an in vitro or in vivo marker for virus entry and replication. J. Virol. Methods 75:151–160 [DOI] [PubMed] [Google Scholar]
- 37. Foster TP, Kousoulas KG. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang K, et al. 2010. Single molecule imaging of NGF axonal transport in microfluidic devices. Lab Chip 10:2566–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osakada Y, Cui B. 2011. Real-time visualization of axonal transport in neurons, p 231–243 In Roberson ED, Alzheimer’s disease and frontotemporal dementia, methods in molecular biology, vol. 670 Springer Verlag Science+Business Media LLC, New York, NY. [DOI] [PubMed] [Google Scholar]
- 40. Melancon JM, Foster TP, Kousoulas KG. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 78:7329–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Subramanian R, D’Auvergne O, Kong H, Kousoulas KG. 2008. The cytoplasmic terminus of Kaposi’s sarcoma-associated herpesvirus glycoprotein B is not essential for virion egress and infectivity. J. Virol. 82:7144–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Subramanian R, Sehgal I, D’Auvergne O, Kousoulas KG. 2010. Kaposi’s sarcoma-associated herpesvirus glycoproteins B and K8.1 regulate virion egress and synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1 cells. J. Virol. 84:1704–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]