

Abstract
Leptin exerts a powerful permissive influence on neurogenic thermogenesis. During starvation and an absence of leptin, animals cannot produce thermogenic reactions to cold stress. However, thermogenesis is rescued by restoring leptin. We have previously observed a highly cooperative interaction between leptin and thyrotropin releasing hormone [TRH] to activate hindbrain generated thermogenic responses (Hermann et al., 2006). In vivo physiological studies (Rogers et al., 2009) suggested that the thermogenic impact of TRH in the hindbrain is amplified by the action of leptin through a leptin receptor-mediated production of phosphoinositiol tris phosphate [PIP3]. In turn, PIP3 can activate a tyrosine kinase whose target is the Src-SH2 regulatory site on the phospholipase C [PLC] complex. The TRH receptor signals through the PLC complex. Our immunohistochemical studies (Barnes et al., 2010) suggest that this transduction interaction between leptin and TRH occurs within neurons of the solitary nucleus [NST], though this interaction had not been verified. The present in vitro live cell calcium imaging study shows that while medial NST neurons are rarely activated by leptin alone, leptin pretreatment significantly augments NST neurons’ responsiveness to TRH. This leptin-mediated priming of NST neurons was uncoupled by pretreatment with the phosphoinositol-tris phosphate kinase [PI3K] inhibitor [wortmannin], the phospholipase C inhibitor [U73122] and the Src-SH2 antagonist [PP2]. TTX did not eliminate the synergistic response of the agonists, thus the sensitization cannot be attributed to pre-synaptic mechanisms. It seems likely that NST neurons are involved in the leptin mediated increase in BAT temperature by sensitizing the TRH-PLC-IP3-calcium release mechanism.
Introduction
Leptin is released from adipose tissue roughly in proportion to the amount of stored metabolic fuel, though its secretion is also highly sensitive to acute food deprivation and refeeding (Ahima and Flier, 2000; Marie et al., 2001). Leptin increases thermogenesis especially via activation of brown adipose tissue (Trayhurn and Bing, 2006). This effect is ultimately mediated by sympathetic inputs to adipose tissue. However, the action of leptin to produce the requisite increase in sympathetic activity occurs principally in the brain. While studies examining leptin’s neurogenic thermal effects are typically focused on the forebrain (Bamshad et al., 1999; Elmquist, 2001; Morrison, 2004), inputs from the forebrain are not always needed to produce thermogenesis (Cao et al., 2010; Harris et al., 2006; Hermann et al., 2006; Rogers et al., 2009).
Within limits, the hindbrain has the capacity to control body temperature. Decerebrate rats can increase heart rate, and to an extent, maintain core temperature in response to cold exposure (Harris et al., 2006). The hindbrain has the neural circuitry to both detect the need for heat and ameliorate that need through the release of stored fuels when metabolites are in abundance (DiRocco and Grill, 1979; I’Anson et al., 2003; Skibicka and Grill, 2009). Our studies have shown that administering leptin, alone, into the fourth ventricle did not have an effect on thermogenesis. However, if thyrotropin-releasing hormone [TRH] follows exposure to leptin, a significant increase in thermogenesis was observed in both intact and decerebrate animals (Hermann et al., 2006).
TRH-containing pathways of the hindbrain are potentially important mediators of neurogenic heat production. Raphe projections to the spinal cord are responsible for triggering sympathetic and thermogenic inputs to brown adipose tissue [BAT] (Cano et al., 2003; Nagashima et al., 2000). Several lines of evidence suggest that these raphe projections are TRHergic and are responsible for initiating neurogenic heat production (Arancibia et al., 1996; Helke et al., 1986). These raphe neurons could, themselves, be influenced by descending hypothalamic TRHergic projections commanding an increase in heat production (Lechan and Fekete, 2006). Additionally, TRHergic projections from raphe to the NST are responsible for driving increased gastric motility that occurs in cold stress (Martinez et al., 2001; Palkovits et al., 1986; Rogers et al., 1996). This raphe-NST pathway provides the means by which a signal related to need for heat production [i.e., activation of the TRHergic projection to the NST] can converge with a signal related to metabolic fuel supply [i.e., circulating leptin] on NST neurons projecting to raphe and reticular areas controlling thermal homeostasis (Blessing, 1997b; Skibicka and Grill, 2008).
While the underlying mechanism[s] for this synergistic effect of leptin and TRH on thermogenesis (Hermann et al., 2006) is not known, recent studies from our laboratory (Rogers et al., 2009) suggest that leptin amplifies the effects of TRH by positive modulation of the TRH transduction mechanism [Figure 1]. One of the signaling pathways used by leptin is the activation of phosphoinositide 3-kinase [PI3K] to generate phosphoinositol-tris phosphate [PIP3] and activate tyrosine kinases that, in turn, potently regulate the activity of phospholipase C [PLC] (Rameh et al., 1998; Rhee, 2001). TRH signal transduction also relies on activation of PLC to generate inositol tris phosphate [IP3], which, in turn, evokes the release of calcium from the endoplasmic reticulum [ER] (Barker et al., 1987; Gershengorn, 1989; Sun et al., 2003). Thus, we propose that leptin-induced generation of PIP3 activates Src-tyrosine kinase to, ultimately, enhance the activity of PLC-IP3- ER calcium transduction events activated by TRH. The end result of this interaction of signaling pathways is hypothesized to be a mechanism for the synergistic effects of hindbrain leptin plus TRH to evoke BAT thermogenesis that we have previously described (Hermann et al., 2006; Rogers et al., 2009).
Figure 1. Schematic of experimental design to investigate potential intracellular interactions between leptin and TRH to augment neuronal activation.
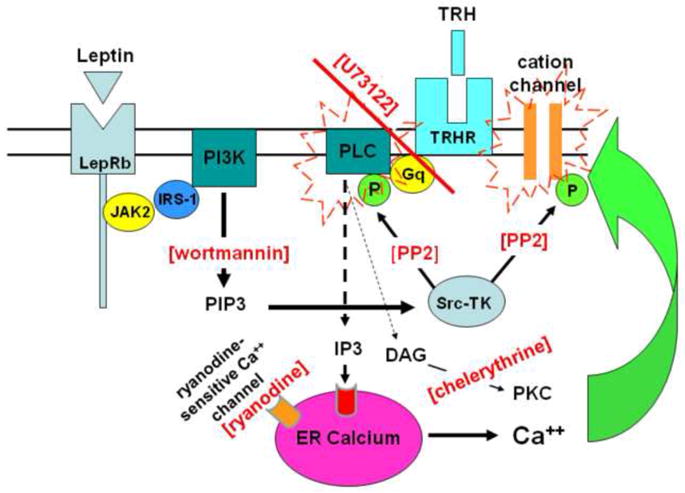
Leptin binding to its receptor causes PIP3 production through a JAK2, IRS-1 transduction mechanism (Procaccini et al., 2009). PIP3 activates Src-tyrosine kinase [Src-TK] which, in turn, can positively modulate transduction elements for other agonists such as TRH as well as cation channels which are responsible for regulating neuronal excitation. In this scheme, the sensitivity of the TRH-PLC-calcium release mechanism is strongly upregulated by the leptin-PIP3-Src-TK transduction path. A lack of activity in the leptin transduction path yields desensitization of the TRH path. Restoring leptin restores the effectiveness of TRH. Therefore, if leptin causes the augmentation of the TRH thermogenic response via these shared signal transduction elements, then this augmentation should be eliminated if Src-TK activation is blocked by wortmannin [i.e., loss of PIP3 production] or PP2 [blocks phosphorylation of PLC or cation channels]. Similarly, blocking the activation of PLC via U73122 [ultimately, eliminating the generation of IP3] should abolish the leptin-TRH synergy. In contrast, chelerythrine’s interference of the activation of phosphokinase C [PKC] or ryanodine’s blockade of calcium-induced calcium release should have little, if any, effect on the leptin-TRH synergy.
Neurons in the NST have been implicated as the targets for leptin action in the hindbrain to regulate feeding behavior and thermogenesis (Grill and Kaplan, 2001; Grill et al., 2002; Hermann et al., 2006; Rogers et al., 2009; Skibicka and Grill, 2009). Further, our recent histochemical study demonstrates that individual neurons in the NST contain both the TRH and leptin receptor (Barnes et al., 2010). Therefore, the present study used an in vitro live cell calcium imaging approach to determine if, and by what mechanism, leptin can act at NST neurons to amplify the TRH transduction mechanism. Specifically, if leptin amplifies neuronal responses to TRH via cross-talk of signaling pathways by these two agonists, then specific interference of leptin signaling should suppress this synergy as illustrated in the schematic of Figure 1.
Results
NST neurons that responded with a minimum 10% increase in relative CalciumGreen [CAG] fluorescence [(ΔF/F)%] in response to brief exposure to glutamate were considered viable candidates for leptin and/or TRH stimulation. Once an individual NST neuron was identified as “viable”, this neuron would be considered to be “responsive to agonist” [Table 1] by responding with a minimum of 1% change in CAG fluorescence [(ΔF/F)%]. Those viable NST neurons that responded with less than a 1% change were considered not responsive. Given that the premise of these studies was the possibility of crosstalk between signaling transduction mechanisms, each slice could only be used for a single experiment. Of the viable [i.e., glutamate-activated] NST neurons, we found approximately 16% of the population also responded to 10nM leptin, applied alone, with an average of 1.9% ± 0.5 change in magnitude fluorescence. Approximately 28% of the population responded to 1uM TRH, applied alone, with an average of 6.2% ± 1.0 change in magnitude fluorescence; while about 47% of the population of NST neurons responded to the stimulation of TRH after pre-exposure to leptin with an average of 14.1% ± 2.0 change in magnitude fluorescence [see Table 1 and Figure 2].
Table 1.
Summary of live cell imaging results
| # of viable NST neurons [defined as responsive to glutamate] | Agonist challenge | # of viable NST neurons responsive to agonist | % of viable NST neurons also responsive to agonist |
|---|---|---|---|
| 263 | TRH alone | 85 | 32.8 |
| 149 | TRH after leptin | 70 | 47.0 |
| 153 | Leptin alone | 24 | 15.7 |
| 117 | Leptin after TRH | 10 | 8.5 |
Individual CAG pre-labeled NST neurons were defined as “viable” based on their response [≥ 10% increase in (ΔF/F)%] to a 30sec exposure to glutamate. Viable neurons were considered to be “responsive to agonist” with ≥ 1% change in fluorescence [(ΔF/F)%] in response to 1min exposure of agonist challenge listed. Those viable NST neurons that responded with less than a 1% change were considered not responsive. Of the viable NST neurons, we found approximately 33% of the population responded to TRH, applied alone; while approximately 47% of the viable cells responded to TRH if the brain slice had been pre-treated with leptin. In those slices that were stimulated by leptin, we found approximately 16% of the population responded to leptin, applied alone; while only about 9% of the viable cells responded to leptin if the brain slice had been pre-treated with TRH.
Figure 2.
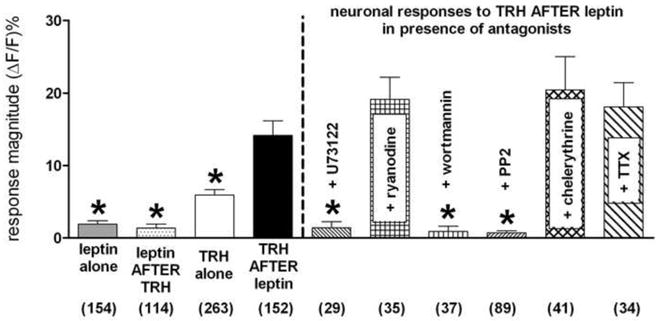
An order-sensitive synergistic relationship exists between leptin and TRH in the activation of NST neurons. Exposure to leptin [either alone or after pretreatment with TRH] yields a small response for NST neurons. However, the magnitude of response elicited by TRH exposure was doubled if the neurons had been pretreated with leptin. These in vitro effects mirrors the same results we observed in the original in vivo experiments (Rogers et al., 2009) where brown adipose tissue thermogenesis was strikingly augmented if the brainstem was exposed to leptin followed by TRH. Additionally, this leptin-mediated sensitization of NST neurons was uncoupled by pretreatment with (a) phospholipase C inhitior [U73122; ultimately resulting in loss of IP3 production]; (b) phosphoinositol-tris phosphate kinase [PI3K] inhibitor [wortmannin]; or (c) the Src-SH2 antagonist [PP2]. Neither chelerythrine [PKC blocker] nor ryanodine [calcium-induced calcium release blocker] interfered with the augmented response. Most importantly, TTX did not eliminate the synergistic response of the agonists; thus the sensitization cannot be attributed to pre-synaptic mechanisms. [Numbers at bottom of bar graphs represent the number of viable NST neurons tested in each experimental group. ANOVA: F (9,938) = 17; p<0.0001. Dunnett’s post test with “TRH after leptin” as comparison; * p<0.05]
The pretreatment of the slices with leptin combined with one of the leptin signaling blockers [i.e., PI3 kinase inhibitor (wortmannin); Src-SH2 antagonist (PP2); or potent and selective inhibitor (U73122) of IP3 action which normally causes release of ER calcium] blocked the positive modulation of the TRH response by leptin. In contrast, if the pretreatment of the slices with leptin was combined with chelerythrine [PKC antagonist], ryanodine [ryanodine endoplasmic reticular calcium-channel blocker] or TTX [block action potential-mediated communication between neurons], the enhanced calcium response to TRH was not blocked [Figure 2].
Discussion
The results from these experiments suggest that the presence of leptin in the medullary hindbrain can recruit more NST neurons to respond to TRH. Further, the responsiveness of these NST neurons to TRH is greater than in the absence of leptin. These in vitro imaging observations are in good agreement with our in vivo physiological experiments demonstrating that 4V delivery of leptin and TRH has a synergistic effect on sympathetically-controlled BAT thermogenesis (Hermann et al., 2006). This leptin-TRH synergy effect, both in vivo, and at the level of the individual NST neuron, is dependent on the order of application. Leptin works to amplify the thermogenic effect of TRH, but not vice versa. This order-effect in the in vivo experiments strongly suggests that TRH signaling is gated by components of the leptin transduction pathway (Rogers et al., 2009).
Leptin can produce rapid changes in the excitability of neurons whose activities have been correlated with decreases in feeding behavior and increases in energy expenditure (Cowley et al., 2001; Cowley, 2003; Malcher-Lopes et al., 2006; Sahu and Metlakunta, 2005). Leptin can evoke either activation or inhibition of neurons in the hypothalamic arcuate nucleus as well as neurons in the dorsal motor nucleus of the vagus dependent on the phenotype of the cell in question (Cowley et al., 2001; Cowley, 2003; Ellacott and Cone, 2004; Williams et al., 2007). Indeed, even within the NST, leptin can apparently both activate (Ellacott et al., 2006; Emond et al., 1999; Emond et al., 2001) or inhibit neurons (Williams et al., 2007). Details about the mechanisms by which leptin can cause such immediate and divergent changes in neuronal excitability are not yet completely understood. However, neuronal excitability could be rapidly modulated through the activation of phosphatidyl inositol-3 kinase [PI3K] by leptin, as reviewed below.
Studies on leptin signaling mechanisms within the brain have focused on leptin interactions with hypothalamic peptidergic neurons in the hypothalamic arcuate nucleus [see reviews, (Ellacott and Cone, 2004; Grill and Kaplan, 2002; Sahu, 2003)]. These studies show that LepRb utilizes the janus kinase 2 [JAK2] mechanism to phosphorylate the signal transducer and activator of transcription-3 [STAT3], and insulin receptor substrate-1 [IRS-1]. The phosphorylation of IRS-1 yields activation of phosphatidyl inositol-3 kinase [PI3K].
This kinase [PI3K], acting through the intermediate of the Src homolog-2 [SH2], can affect the function of cation conductances with direct effects to activate or inhibit neurons (Shanley et al., 2002; Williams et al., 2007). Phosphorylation of SH2 regulatory sites on ion channels or other transduction elements can potently regulate cellular responses to other agonists dependent on those elements (Liu and Ye, 2005; Rameh and Cantley, 1999; Rhee, 2001).
The TRH signal transduction is subject to this kind of control, i.e., phosphorylation of phospholipase C [PLC]. The TRH receptor is coupled with a Gq transduction element that activates PLC to produce inositol 1,4,5 trisphosphate [IP3] and diacyl glycerol [DAG]. IP3 causes the rapid release of calcium from the endoplasmic reticulum, while DAG activates protein kinase C [PKC] (Barker et al., 1987; Gershengorn, 1989; Sun et al., 2003). The increase in cytoplasmic calcium induced by IP3 production has been linked to the initiation of calcium oscillations that can be used to control a wide variety of cellular functions (Dekin et al., 1985; Hermann et al., 2005; Koshiya and Smith, 1999; Somogyi and Stucki, 1991; Tse and Tse, 1999; Verkhratsky, 2002). Activation of PLC and liberation of ER calcium has also been linked to the opening of non-specific cation channels and the depression of potassium currents (Bayliss et al., 1994; Ishibashi et al., 2003; Winks et al., 2005). These effects can lead to significant neuronal depolarization and excitation and have been observed in neurons in the dorsal medulla responding to TRH (Dekin et al., 1985; Livingston and Berger, 1993). TRH-PLC transduction can also inhibit DVC neurons (Browning and Travagli, 2001); the result depends on the phenotype of the cell in question.
Observations in culture systems suggest that PIP3 produced by PI3K can positively modulate the activity of PLC (Bae et al., 1998; Marshall et al., 2000; Rameh et al., 1998; Yang et al., 2001). A specific cross-talk mechanism linking PI3K and PLC activity in the brain has not been described until recently (Rogers et al., 2009). PIP3 positively modulates PLC by activating the SH2 domain of the PLC complex (Liu and Ye, 2005; Rameh and Cantley, 1999; Rhee, 2001). The present results suggest that leptin gates the action of TRH in the NST by invoking the same synergistic mechanism. Thus, as we have reported in our in vivo physiological experiments (Rogers et al., 2009), blockade of either PIP3 formation with wortmannin or SH2 activation with PP2 suppressed the order-specific potentiation of TRH effects by leptin.
The results of the present calcium imaging study are entirely consistent with this view of leptin-modulation of PLC-dependent transduction processes. Leptin pretreatment of the NST leads to a significant increase in neuronal responsiveness to TRH. This increase in TRH responsiveness, measured as an increase in calcium signal, is blocked by antagonists of PI3kinase [e.g., wortmannin] and the Src tyrosine kinase [e.g., PP2] responsible for activating PLC. Blockade of transduction steps “beyond” the TRH-PLC-mediated increase in cytoplasmic calcium [e.g., using chelerythrine to block protein kinase C] had no effect on this augmentation. Similarly, blockade of intracellular Ca++ release via antagonists of transduction pathways not used by leptin [e.g., ryanodine] also had no effect on this enhanced response. Lastly, the synergistic effect was not blocked by TTX, thus discounting the possibility that the sensitization could be attributed to pre-synaptic mechanisms. The combined results suggest that leptin and TRH operate on the same NST neurons to trigger a hindbrain-mediated increase in BAT thermogenesis.
The NST is emerging as a structure in strategic position to regulate thermogenesis. Subsets of NST neurons possess the long form of the leptin receptor (Barnes et al., 2010) and leptin can gain access to those neurons in a weakened region of the blood-brain barrier (Broadwell and Sofroniew, 1993; Gross et al., 1990; Gross et al., 1991; Maolood and Meister, 2009; Rogers and McCann, 1993). The NST, therefore, receives data on the amount of fuel stored as signaled by leptin (Grill et al., 2002; Hermann et al., 2006). The same NST neurons that possess leptin receptors also have TRH receptors (Barnes et al., 2010) and the NST receives functionally important TRHergic input from the raphe pallidus. The activity of this raphe input to the NST is very likely to be proportional to the demand for heat production during cold stress (Nagashima et al., 2000; Rogers et al., 1996) (Cano et al., 2003). The NST receives vagal afferents sensitive to core temperature (Adachi and Niijima, 1982) and the presence of pyrogens (Blatteis et al., 1998). Destruction of the NST depresses thermogenesis normally induced by pyrogenic stimuli (Gordon, 2000). While the NST is clearly not involved in establishing thermocontrol setpoint, it does appear capable of integrating many signals relevant to thermal control including leptin and TRH.
While the connectional details are vague at this point, it is clear that the NST sends efferent projections to the ventrolateral medulla and the raphe (Blessing, 1997a), structures critical to controlling blood flow [hence heat distribution] and BAT thermogenesis (Bamshad et al., 1999; Cano et al., 2003). Through these connections, the NST can apparently exert significant influence over the sensitivity of the thermocontrol mechanism. Such an arrangement may explain why conditions associated with low levels of circulating leptin [e.g., food deprivation] depress thermogenesis and why supplementation of leptin restores thermogenesis (Bignall et al., 1974; Blumberg et al., 1999; Hausberg et al., 2002).
Experimental Procedure
Long-Evans rats [200–400g; 2–4 months of age] of either sex, obtained from the breeding colony located at Pennington Biomedical Research Center, were used in these studies. All animals were maintained in a room with a 12:12 hour light-dark cycle with constant temperature and humidity, and given food and water ad libitum. All experimental protocols were performed according to the guidelines set forth by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committees at the Pennington Biomedical Research Center.
Live Cell Calcium Imaging
Direct Injection of Calcium Green Dextran and SR101 into the solitary nucleus
Animals [N = 27] were deeply anesthetized with urethane [1.5g/kg, ip; ethyl carbamate, Sigma] and placed in a stereotaxic frame. Using aseptic technique, the occipital plate of the skull was removed to expose the medullary brainstem. A micropipette [30micron tip diameter; filled with 0.2% Calcium Green 1 AM [CAG; Invitrogen], 0.3% sulforhodamine 101 [SR101; Sigma Chemical] and 10% pluronic-DMSO [F-127, Invitrogen] in pH 7.2 tris-PBS buffer] was directed toward the medial solitary nucleus using a stereotaxic carrier. Four injections [40nL each] of the CAG-SR101 solution were made unilaterally into the NST at the level of calamus and 0.2, 0.4, and 0.6mm anterior to calamus; all at a depth of 300micron below surface. This injection pattern labeled the entire ipsilateral medial NST. CAG, a calcium reporter dye, labels both neurons and glia while SR101 labels only astrocytes (Nimmerjahn et al., 2004) but does not interfere with CAG fluorescence (Hermann et al., 2009). This method made it possible to easily discriminate astrocytes from neurons in a slice preparation which is important given the similarity in size of NST neuronal and astrocytic cell bodies. After 30min interval to allow for dye uptake, the anesthetized rat was decapitated and the brainstem was quickly harvested. The caudal brainstem was glued to an aluminum block and placed in cold [~4°C] carbogenated [95% O2; 5% CO2] cutting solution [recipe below]. Coronal sections [300micron thick] were cut through the medulla using a Leica VT1200 tissue slicer equipped with a sapphire knife. Slices were incubated in a carbogenated normal Krebs at 29°C for at least 30min prior to transfer to the imaging chamber.
Confocal imaging
Individual medullary slices were transferred to a temperature regulated perfusion chamber [Bioptechs, Inc, Butler, PA] and held in place with a harp-type pressure foot. Imaging was performed with a Nikon F1 upright microscope - Fast Scan confocal system equipped with heated, water-immersion objectives. The recording chamber was continuously perfused at a rate of 2ml/min with carbogenated Krebs warmed to 34°C. Identification of SR101-labeled astrocytes was done using a 568nm excitation, 607nm emission filter set. CAG fluorescence and relative intracellular calcium levels were visualized when switching to the 488nm excitation/509nm emission filter. Solenoid valves were used to switch between the normal bathing solution and the experimental solutions [recipes below].
In Vitro Drugs and Solutions
The cutting solution contained [in mM]: 110 choline chloride, 25 NaHCO3, 2.5 KCl, 7 MgSO4-7H2O, 1.25 NaH2PO4; 10 glucose; 0.5 CaCl2-2H2O; bubbled with 95% O2/5% CO2 during the entire cutting process. Normal Krebs contained [in mM]: 124 NaCl, 25 NaHCO3, 3.0 KCl, 1 MgSO4-7H2O, 1.5 NaH2PO4; 10 glucose; 1.5 CaCl2-2H2O; bubbled with 95% O2/5% CO2, continuously; osmolality was 300 ± 10mOsm; pH = 7.3. As needed for the different experimental conditions, the following drugs were added to the Normal Krebs:
glutamate [500uM; Sigma-Aldrich; excitatory amino acid]
TRH [10uM; Bachem]
Leptin [10nM; mouse recombinant leptin; NIDDK National Hormone and Peptide Program, Torrance, CA]
U73122 [10uM; Tocris; PLC antagonist = blocks the TRH-PLC transduction step; (Bleasdale et al., 1990)]
Ryanodine [10uM; ryanodine endoplasmic reticular calcium channel blocker; (Lancaster et al., 2002)
Wortmannin [100nM; Sigma-Aldrich; PI3 kinase inhibitor; (Rameh et al., 1998)]
PP2 [10uM; (4-amino-5(4-chlorophenyl)-7-(t-butyl)pyrazolo(3,4-d) pyrimidine; CalBiochem; Src-SH2 antagonist; (Lee et al., 2004)
Chelerythrine [10uM; Sigma; PKC antagonist; (Herbert et al., 1990)]
TTX [1uM; Sigma; used to block action potential-mediated communication between neurons (Lee and Ruben, 2008)
In vitro Calcium Signal Analysis
Initially, each brainstem slice was briefly exposed (30sec) to 500uM glutamate [see Figure 3 for example of experimental design]. Relative changes in intracellular calcium levels (i.e., [Ca++]i) were monitored continuously from 10sec before glutamate exposure through 2min after the exposure to visually determine viability and basal responsiveness of NST neurons. Viable neurons are easily discriminated from glia since they do not take up the red SR101 dye and neurons demonstrate a brisk [Ca++]i calcium wave in response to 500uM glutamate while astrocytes generally do not respond (Hermann et al., 2009). Viable neurons are defined as those that generate a minimum 10% increase in relative [Ca++]i over baseline in response to glutamate challenge. Relative changes in [Ca++]i were quantified as (ΔF/F)%, where F is the fluorescence intensity within an area of interest [e.g., the outline of an NST neuron] prior to stimulation and ΔF is the change from this value resulting from stimulated cellular activity (Helmchen, 2000). Background fluorescence [i.e., non-involved areas in same field] was subtracted from both Δ and F.
Figure 3.
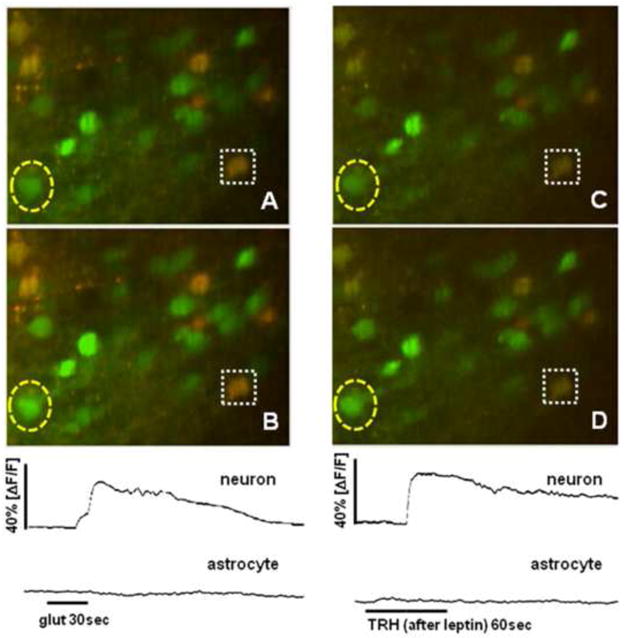
Representative example of live cell imaging experiment. NST neurons and astrocytes are pre-labeled with CalciumGreen 1AM [green only]; astrocytes [green plus red] also selectively take up SR101 as a vital dye for identification purposes. Initially, viability of NST neurons is determined by 30sec exposure to 500uM glutamate: [A] field of interest at rest; [B] same field at peak of response to bath perfusion of glutamate. Note that astrocytes are not sensitive to this concentration of glutamate (Hermann et al., 2009). Quantification of calcium response to glutamate over time by a selected neuron [outlined by yellow dashed oval] and astrocyte [outlined by white dashed box] is displayed in the panel below [B]. Relative changes in CAG fluorescence were quantified as (ΔF/F)%, where F is the fluorescence intensity within an area of interest [e.g., the outline of an NST neuron] prior to stimulation and ΔF is the change from this value resulting from stimulated cellular activity (Helmchen, 2000). Once responsive cells have been verified, brainstem slice is exposed to one of the nine experimental conditions described in Methods. In this case, the slice was bathed with Krebs plus leptin [10nM for 10min]. Then the slice was exposed to TRH [10uM for 60sec]: [C] same slice as in [A] and [B] after the leptin pre-treatment; [D] same field at peak of response to bath perfusion of TRH. Relative fluorescence changes [calcium] plots over time displayed below [D].
After the initial identification protocol for viability, the slices were exposed to one of the following ten (10) experimental protocols. To determine the basic neuronal response to either (1) leptin or (2) TRH, alone, the slice was perfused with normal Krebs solution for 10min followed by a 1min exposure to either leptin or TRH, alone. To evaluate if pre-exposure to leptin could augment neuronal response to TRH, brainstem slices were perfused with (3) normal Krebs solution plus leptin for 10min. At the end of this 10min pretreatment, the cellular responsiveness to 1min exposure to TRH was monitored. Lastly, to determine what signaling pathways may be involved in changing the neuronal responsiveness to TRH stimulation after leptin exposure, the pretreatment phase with normal Krebs plus leptin also included one of the following blockers: (4) U73122, (5) ryanodine, (6) wortmannin, (7) PP2, (8) chelerythrine, or (9) TTX. A tenth experimental group was included to evaluate if pre-exposure to TRH could augment the neuronal response to leptin. In this case, brainstem slices were perfused with normal Krebs solution plus TRH for 10min. At the end of this pretreatment, the cellular responsiveness to 1min exposure to leptin was monitored. In all experiments that involved blockers or inhibitors [e.g., U73122, ryanodine, wortmannin, PP2, chelerythrine, or TTX] slices were rewashed in normal Krebs for 10min followed by another exposure to glutamate to reassess basic viability.
Statistical analysis
A total of 27 animals were used for these experiments; each animal yielded approximately 5 brain slices with the necessary neurocircuitry under study. Data for each of the designated experimental groups were obtained from a minimum of two animals and three different slices. The total number of individual cells [n = individual cells] evaluated in these different experimental groups is represented in Figure 2.
Peak changes in (ΔF/F)% in response to TRH [or leptin] across all conditions were compared in one way analyses of variance. Dunnett’s post test comparisons against the TRH following leptin pretreatment condition were also performed.
Research Highlights.
medial NST neurons are rarely activated by leptin alone,
leptin pretreatment significantly augments NST neurons’ responsiveness to TRH
leptin primes TRH action on NST neurons via PI3 kinase
TTX did not eliminate synergy; sensitization not due to pre-synaptic mechanisms
Acknowledgments
This work was supported by NIH Grant NS55866, DK52142, DK56373, and NS60664
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi A, Niijima A. Thermosensitive afferent fibers in the hepatic branch of the vagus nerve in the guinea pig. J Auton Nerv Syst. 1982;5:101–9. doi: 10.1016/0165-1838(82)90031-5. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Flier JS. Leptin Annu Rev Physiol. 2000;62:413–37. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- Arancibia S, Rage F, Astier H, Tapia-Arancibia L. Neuroendocrine and autonomous mechanisms underlying thermoregulation in cold environment. Neuroendocrinology. 1996;64:257–67. doi: 10.1159/000127126. [DOI] [PubMed] [Google Scholar]
- Bae YS, Cantley LG, Chen CS, Kim SR, Kwon KS, Rhee SG. Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:4465–9. doi: 10.1074/jbc.273.8.4465. [DOI] [PubMed] [Google Scholar]
- Bamshad M, Song CK, Bartness TJ. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am J Physiol. 1999;276:R1569–78. doi: 10.1152/ajpregu.1999.276.6.R1569. [DOI] [PubMed] [Google Scholar]
- Barker JL, Dufy B, Harrison NL, Owen DG, MacDonald JF. Signal transduction mechanisms in cultured CNS neurons and clonal pituitary cells. Neuropharmacology. 1987;26:941–55. doi: 10.1016/0028-3908(87)90073-6. [DOI] [PubMed] [Google Scholar]
- Barnes MJ, Rogers RC, Van Meter MJ, Hermann GE. Co-localization of TRHR1 and LepRb receptors on neurons in the hindbrain of the rat. Brain Res. 2010;1355:70–85. doi: 10.1016/j.brainres.2010.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss DA, Viana F, Berger AJ. Effects of thyrotropin-releasing hormone on rat motoneurons are mediated by G proteins. Brain Res. 1994;668:220–9. doi: 10.1016/0006-8993(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Bignall KE, Heggeness FW, Palmer JE. Effects of acute starvation on cold-induced thermogenesis in the preweanling rat. Am J Physiol. 1974;227:1088–93. doi: 10.1152/ajplegacy.1974.227.5.1088. [DOI] [PubMed] [Google Scholar]
- Blatteis CM, Sehic E, Li S. Afferent pathways of pyrogen signaling. Ann NY Acad Sci. 1998;856:95–107. doi: 10.1111/j.1749-6632.1998.tb08318.x. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, Bunting S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther. 1990;255:756–68. [PubMed] [Google Scholar]
- Blessing WW. The Lower Brainstem and Bodily Homeostasis. Oxford University Press; Oxford: 1997a. Anatomy of the lower brainstem; pp. 29–100. [Google Scholar]
- Blessing WW. The Lower Brainstem and Bodily Homeostasis. Oxford University Press; Oxford: 1997b. Eating and metabolism; pp. 323–372. [Google Scholar]
- Blumberg MS, Deaver K, Kirby RF. Leptin disinhibits nonshivering thermogenesis in infants after maternal separation. Am J Physiol. 1999;276:R606–10. doi: 10.1152/ajpregu.1999.276.2.R606. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol. 1993;120:245–263. doi: 10.1006/exnr.1993.1059. [DOI] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. The peptide TRH uncovers the presence of presynaptic 5-HT1A receptors via activation of a second messenger pathway in the rat dorsal vagal complex. J Physiol. 2001;531:425–35. doi: 10.1111/j.1469-7793.2001.0425i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Passerin AM, Schiltz JC, Card JP, Morrison SF, Sved AF. Anatomical substrates for the central control of sympathetic outflow to interscapular adipose tissue during cold exposure. J Comp Neurol. 2003;460:303–326. doi: 10.1002/cne.10643. [DOI] [PubMed] [Google Scholar]
- Cao WH, Madden CJ, Morrison SF. Inhibition of brown adipose tissue thermogenesis by neurons in the ventrolateral medulla and in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 2010;299:R277–R290. doi: 10.1152/ajpregu.00039.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–4. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Cowley MA. Hypothalamic melanocortin neurons integrate signals of energy state. Eur J Pharmacol. 2003;480:3–11. doi: 10.1016/j.ejphar.2003.08.087. [DOI] [PubMed] [Google Scholar]
- Dekin MS, Richerson GB, Getting PA. Thyrotropin-releasing hormone induces rhythmic bursting in neurons of the nucleus tractus solitarius. Science. 1985;229:67–9. doi: 10.1126/science.3925552. [DOI] [PubMed] [Google Scholar]
- DiRocco RJ, Grill HJ. The forebrain is not essential for sympathoadrenal hyperglycemic response to glucoprivation. Science. 1979;204:1112–4. doi: 10.1126/science.451558. [DOI] [PubMed] [Google Scholar]
- Ellacott KL, Cone RD. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Prog Horm Res. 2004;59:395–408. doi: 10.1210/rp.59.1.395. [DOI] [PubMed] [Google Scholar]
- Ellacott KL, Halatchev IG, Cone RD. Characterization of leptin-responsive neurons in the caudal brainstem. Endocrinology. 2006;147:3190–5. doi: 10.1210/en.2005-0877. [DOI] [PubMed] [Google Scholar]
- Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S78–82. doi: 10.1038/sj.ijo.0801918. [DOI] [PubMed] [Google Scholar]
- Emond M, Schwartz GJ, Ladenheim EE, Moran TH. Central leptin modulates behavioral and neural responsivity to CCK. Am J Physiol. 1999;276:R1545–9. doi: 10.1152/ajpregu.1999.276.5.R1545. [DOI] [PubMed] [Google Scholar]
- Emond M, Ladenheim EE, Schwartz GJ, Moran TH. Leptin amplifies the feeding inhibition and neural activation arising from a gastric nutrient preload. Physiol Behav. 2001;72:123–8. doi: 10.1016/s0031-9384(00)00393-0. [DOI] [PubMed] [Google Scholar]
- Gershengorn MC. Mechanism of signal transduction by TRH. Ann N Y Acad Sci. 1989;553:191–6. doi: 10.1111/j.1749-6632.1989.tb46641.x. [DOI] [PubMed] [Google Scholar]
- Gordon FJ. Effect of nucleus tractus solitarius lesions on fever produced by interleukin-1beta. Auton Neurosci. 2000;85:102–10. doi: 10.1016/s1566-0702(00)00228-9. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM. Interoceptive and integrative contributions of forebrain and brainstem to energy balance control. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S73–7. doi: 10.1038/sj.ijo.0801917. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM. The neuroanatomical axis for control of energy balance. Front Neuroendocrinol. 2002;23:2–40. doi: 10.1006/frne.2001.0224. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology. 2002;143:239–46. doi: 10.1210/endo.143.1.8589. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 1990;259:R1131–R1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Wainman DS, Shaver SW. Subregional topography of capillaries in the dorsal vagal complex of rats: II. Physiological properties J Comp Neurol. 1991;306:83–94. doi: 10.1002/cne.903060107. [DOI] [PubMed] [Google Scholar]
- Harris RB, Kelso EW, Flatt WP, Bartness TJ, Grill HJ. Energy expenditure and body composition of chronically maintained decerebrate rats in the fed and fasted condition. Endocrinology. 2006;147:1365–76. doi: 10.1210/en.2005-1156. [DOI] [PubMed] [Google Scholar]
- Hausberg M, Morgan DA, Mitchell JL, Sivitz WI, Mark AL, Haynes WG. Leptin potentiates thermogenic sympathetic responses to hypothermia: a receptor-mediated effect. Diabetes. 2002;51:2434–40. doi: 10.2337/diabetes.51.8.2434. [DOI] [PubMed] [Google Scholar]
- Helke CJ, Sayson SC, Keeler JR, Charlton CG. Thyrotropin-releasing hormone-immunoreactive neurons project from the ventral medulla to the intermediolateral cell column: partial coexistence with serotonin. Brain Res. 1986;381:1–7. doi: 10.1016/0006-8993(86)90682-7. [DOI] [PubMed] [Google Scholar]
- Helmchen F. Calibration of fluorescent calcium indicators. In: Yuste R, Lanni F, Konnerth A, editors. Imaging Neurons: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 32.1–32.9. [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–9. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Nasse JS, Rogers RC. Alpha-1 adrenergic input to solitary nucleus neurones: calcium oscillations, excitation and gastric reflex control. J Physiol. 2005;562:553–68. doi: 10.1113/jphysiol.2004.076919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Barnes MJ, Rogers RC. Leptin and thyrotropin-releasing hormone: cooperative action in the hindbrain to activate brown adipose thermogenesis. Brain Res. 2006;1117:118–24. doi: 10.1016/j.brainres.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Van Meter MJ, Rood JC, Rogers RC. Proteinase-activated receptors in the nucleus of the solitary tract: evidence for glial-neural interactions in autonomic control of the stomach. J Neurosci. 2009;29:9292–300. doi: 10.1523/JNEUROSCI.6063-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- I’Anson H, Sundling LA, Roland SM, Ritter S. Immunotoxic destruction of distinct catecholaminergic neuron populations disrupts the reproductive response to glucoprivation in female rats. Endocrinology. 2003;144:4325–31. doi: 10.1210/en.2003-0258. [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Umezu M, Jang IS, Ito Y, Akaike N. Alpha 1-adrenoceptor-activated cation currents in neurones acutely isolated from rat cardiac parasympathetic ganglia. J Physiol. 2003;548:111–20. doi: 10.1113/jphysiol.2002.033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiya N, Smith JC. Neuronal pacemaker for breathing visualized in vitro. Nature. 1999;400:360–3. doi: 10.1038/22540. [DOI] [PubMed] [Google Scholar]
- Lancaster E, Oh EJ, Gover T, Weinreich D. Calcium and calcium-activated currents in vagotomized rat primary vagal afferent neurons. J Physiol. 2002;540:543–56. doi: 10.1113/jphysiol.2001.013121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. 2006;153:209–35. doi: 10.1016/S0079-6123(06)53012-2. [DOI] [PubMed] [Google Scholar]
- Lee CH, Ruben PC. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels (Austin) 2008;2:407–12. doi: 10.4161/chan.2.6.7429. [DOI] [PubMed] [Google Scholar]
- Lee M, Kim JY, Anderson WB. Src tyrosine kinase inhibitor PP2 markedly enhances Ras-independent activation of Raf-1 protein kinase by phorbol myristate acetate and H2O2. J Biol Chem. 2004;279:48692–701. doi: 10.1074/jbc.M403132200. [DOI] [PubMed] [Google Scholar]
- Liu X, Ye K. Src homology domains in phospholipase C-gamma1 mediate its anti-apoptotic action through regulating the enzymatic activity. J Neurochem. 2005;93:892–8. doi: 10.1111/j.1471-4159.2005.03064.x. [DOI] [PubMed] [Google Scholar]
- Livingston CA, Berger AJ. Response of neurons in the dorsal motor nucleus of the vagus to thyrotropin-releasing hormone. Brain Res. 1993;621:97–105. doi: 10.1016/0006-8993(93)90302-4. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, Tasker JG. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–50. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maolood N, Meister B. Protein components of the blood-brain barrier (BBB) in the brainstem area postrema-nucleus tractus solitarius region. J Chem Neuroanat. 2009;37:182–95. doi: 10.1016/j.jchemneu.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Marie M, Findlay PA, Thomas L, Adam CL. Daily patterns of plasma leptin in sheep: effects of photoperiod and food intake. J Endocrinol. 2001;170:277–86. doi: 10.1677/joe.0.1700277. [DOI] [PubMed] [Google Scholar]
- Marshall AJ, Niiro H, Yun TJ, Clark EA. Regulation of B-cell activation and differentiation by the phosphatidylinositol 3-kinase and phospholipase Cgamma pathway. Immunol Rev. 2000;176:30–46. doi: 10.1034/j.1600-065x.2000.00611.x. [DOI] [PubMed] [Google Scholar]
- Martinez V, Wang L, Tache Y. Central TRH receptor 1 antisense blocks cold-induced gastric emptying but not brain c-Fos induction. Peptides. 2001;22:81–90. doi: 10.1016/s0196-9781(00)00359-4. [DOI] [PubMed] [Google Scholar]
- Morrison SF. Central pathways controlling brown adipose tissue thermogenesis. News Physiol Sci. 2004;19:67–74. doi: 10.1152/nips.01502.2003. [DOI] [PubMed] [Google Scholar]
- Nagashima K, Nakai S, Tanaka M, Kanosue K. Neuronal circuitries involved in thermoregulation. Auton Neurosci. 2000;85:18–25. doi: 10.1016/S1566-0702(00)00216-2. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–7. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Palkovits M, Mezey E, Eskay RL, Brownstein MJ. Innervation of the nucleus of the solitary tract and the dorsal vagal nucleus by thyrotropin-releasing hormone-containing raphe neurons. Brain Res. 1986;373:246–251. doi: 10.1016/0006-8993(86)90338-0. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Lourenco EV, Matarese G, La Cava A. Leptin signaling: A key pathway in immune responses. Curr Signal Transduct Ther. 2009;4:22–30. doi: 10.2174/157436209787048711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh LE, Rhee SG, Spokes K, Kazlauskas A, Cantley LC, Cantley LG. Phosphoinositide 3-kinase regulates phospholipase Cgamma-mediated calcium signaling. J Biol Chem. 1998;273:23750–7. doi: 10.1074/jbc.273.37.23750. [DOI] [PubMed] [Google Scholar]
- Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem. 1999;274:8347–50. doi: 10.1074/jbc.274.13.8347. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, McCann MJ. Intramedullary connections of the gastric region in the solitary nucleus: a biocytin histochemical tracing study in the rat. J Auton Nerv Syst. 1993;42:119–130. doi: 10.1016/0165-1838(93)90043-t. [DOI] [PubMed] [Google Scholar]
- Rogers RC, McTigue DM, Hermann GE. Vagal control of digestion: modulation by central neural and peripheral endocrine factors. Neurosci Biobehav Rev. 1996;20:57–66. doi: 10.1016/0149-7634(95)00040-l. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Barnes MJ, Hermann GE. Leptin “gates” thermogenic action of thyrotropin-releasing hormone in the hindbrain. Brain Res. 2009:135–41. doi: 10.1016/j.brainres.2009.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol. 2003;24:225–53. doi: 10.1016/j.yfrne.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Sahu A, Metlakunta AS. Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion. J Neuroendocrinol. 2005;17:720–6. doi: 10.1111/j.1365-2826.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, O’Malley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J Physiol. 2002;545:933–44. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibicka KP, Grill HJ. Energetic responses are triggered by caudal brainstem melanocortin receptor stimulation and mediated by local sympathetic effector circuits. Endocrinology. 2008;149:3605–16. doi: 10.1210/en.2007-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibicka KP, Grill HJ. Hindbrain leptin stimulation induces anorexia and hyperthermia mediated by hindbrain melanocortin receptors. Endocrinology. 2009;150:1705–11. doi: 10.1210/en.2008-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi R, Stucki JW. Hormone-induced calcium oscillations in liver cells can be explained by a simple one pool model. J Biol Chem. 1991;266:11068–77. [PubMed] [Google Scholar]
- Sun Y, Lu X, Gershengorn MC. Thyrotropin-releasing hormone receptors -- similarities and differences. J Mol Endocrinol. 2003;30:87–97. doi: 10.1677/jme.0.0300087. [DOI] [PubMed] [Google Scholar]
- Trayhurn P, Bing C. Appetite and energy balance signals from adipocytes. Philos Trans R Soc Lond B Biol Sci. 2006;361:1237–49. doi: 10.1098/rstb.2006.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse FW, Tse A. Regulation of exocytosis via release of Ca(2+) from intracellular stores. Bioessays. 1999;21:861–5. doi: 10.1002/(SICI)1521-1878(199910)21:10<861::AID-BIES8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium. 2002;32:393–404. doi: 10.1016/s0143416002001896. [DOI] [PubMed] [Google Scholar]
- Williams KW, Zsombok A, Smith BN. Rapid inhibition of neurons in the dorsal motor nucleus of the vagus by leptin. Endocrinology. 2007;148:1868–81. doi: 10.1210/en.2006-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ. Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci. 2005;25:3400–13. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He X, Feng L, Mizuno K, Liu XW, Russell J, Xiong WC, Lu B. PI-3 kinase and IP3 are both necessary and sufficient to mediate NT3-induced synaptic potentiation. Nat Neurosci. 2001;4:19–28. doi: 10.1038/82858. [DOI] [PubMed] [Google Scholar]