

Abstract
Purpose
Glucocorticoids (GCs) are common anti-inflammatory agents that can cause ocular hypertension and secondary glaucoma as a consequence of impaired aqueous humor outflow through the trabecular meshwork (TM). Mechanisms of GC-signaling are complex and poorly understood. To better understand GC-signaling in the eye, we tested the hypothesis that common mechanisms of steroid responsiveness exist in TM cells from normal and glaucomatous donors.
Methods
Four primary cultures of human TM cells from normal and glaucomatous donors were treated with or without dexamethasone (Dex) for 10 days, then cellular proteins were extracted, identified and quantified by liquid chromatography tandem mass spectrometry (LC MS/MS) iTRAQ (isobaric tags for relative and absolute quantitation) technology.
Results
A total of 718 proteins were quantified. Dex-treatment significantly altered the abundance of 40 proteins in ≥3 cell samples, 37 of which have not previously been associated with GC-signaling in TM cells. Most steroid responsive proteins were changed in all four TM cells analyzed, both normal and glaucomatous. GC-induced proteomic changes support remodeling of the extracellular matrix, disorganization of the cytoskeleton/cell-cell interactions, and mitochondrial dysfunction. Such physiologic consequences appear common to those induced in TM cells by transforming growth factor-β2, another putative contributor to ocular hypertension and glaucoma pathology.
Conclusions
The results expand the repertoire of TM proteins involved in GC-signaling, demonstrate common consequences of GC-signaling in normal and glaucomatous TM cells, and reveal similarities in proteomic changes induced by steroids and TGFβ2 in normal and glaucomatous TM cells. Finally, the data contributes to a TM quantitative proteomic database.
Introduction
Glucocorticoids (GCs) are potent anti-inflammatory agents used successfully to treat a variety of diseases, but with several potentially serious side effects. In the eye, GC therapy can cause ocular hypertension and secondary open-angle glaucoma [1,2]. About 40% of the general population will develop elevated intraocular pressure (IOP) within 4–6 weeks of topical ocular administration of GCs [3]. Such “steroid responders” are more likely to develop primary open angle glaucoma (POAG) than non-responders. The trabecular meshwork (TM), located in the aqueous humor outflow pathway (Figure 1), regulates IOP through alteration of aqueous humor resistance via contractile properties, phagocytosis, cytoskeletal reorganization, cell adhesion, matrix deposition and ion channel transport [4,5]. The molecular mechanisms causing GC-induced ocular hypertension and impaired TM cell function are not well understood [5,6].
Figure 1.
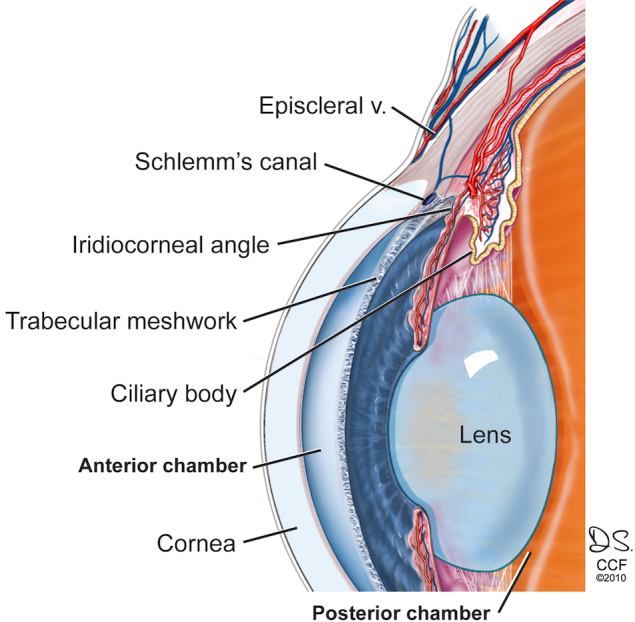
Human trabecular meshwork. Aqueous humor (AH) is actively produced by the ciliary epithelium in the posterior chamber of the eye and circulates through the pupil to the anterior chamber where it drains through the TM into Schlemm’s canal and the episcleral veins. Reproduced with copyright permission from the Cleveland Clinic. Illustration by David Schumick. All rights reserved.
GC-signaling mechanisms appear to be in part tissue-specific [7] and highly complex [8], with hundreds of gene expression changes induced in TM cell cultures by dexamethasone (Dex) [9-13]. Many of the GC-induced changes in the TM are similar to those seen in POAG [3]. GC-induced ocular hypertension occurs in both normal and glaucoma patients, although a higher percentage of glaucoma patients are steroid responsive. The GC-induced changes to the TM, the resulting elevation in IOP, and the clinical phenotype appear to be similar in these two groups. The goal of this study was to test the hypothesis that common mechanisms of steroid responsiveness exist in TM cells from normal and glaucomatous tissues. Primary TM cells cultured from carefully dissected TM tissues were used for global quantitative proteomic analysis of steroid responsiveness using liquid chromatography tandem mass spectrometry (LC MS/MS) isobaric tags for relative and absolute quantitation (iTRAQ) technology. Although cultured TM cells grow very slowly and have a limited proliferative capacity, we were able to examine GC-induced changes in protein expression in four different primary TM cell cultures, more than any previous study of GC effects on gene or protein expression in the TM. The results identfy a significant number of proteins not previously known to be steroid responsive in TM cells and show most of the GC-altered proteins were changed in all TM cell strains analyzed, both normal and glaucomatous.
Methods
TM cell cultures
All human specimens were post-mortem tissues collected with adherence to the principles expressed in the Declaration of Helsinki. Post-mortem human eyes were obtained from the Lions Eye Institute for Tissue and Research in Tampa, FL, which is accredited by the Eye Bank Association of America. TM cells were isolated from TM tissue explants derived from both open angle glaucoma and nonglaucomatous control donors. The glaucoma status was indicated from donor medical histories. The average death to preservation time was 7.7±3.3 h. Eyes were stored at 4 °C until the TM was dissected, generally within 24–36 h. Primary cultures were established and TM cell morphology and purity were characterized as previously described [14,15]. Human TM cells were grown in Dulbecco’s modified Eagle’s medium (HyClone, Logan UT) supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY), 1% penicillin-streptomycin (HyClone) and 1% L-glutamine (Thermo-Scientific Hyclone, Logan, UT) to confluency in T-25 flasks or in 6-well plates. Primary cultures of human TM cells from 2 POAG and 2 non-glaucomatous donors were treated with or without Dex (100 nM) for 10 days, yielding 4 Dex-treated and 4 untreated TM cell cultures (Table 1).
Table 1. Trabecular meshwork cell samples.
| Experiment | Cell culture | Donor age and gender | Cell passage | Protein analyzed µg |
|---|---|---|---|---|
| 1 |
NTM416-07 |
78 / M |
P4 |
75 |
| 2 |
NTM496-05 |
82 / F |
P3 |
75 |
| 3 |
GTM304-04 |
75 / F |
P3 |
75 |
| 4 | GTM477-02 | 85 / F | P4 | 75 |
Each of the above primary TM cell cultures was treated with or without Dex then analyzed by LC MS/MS iTRAQ technology. NTM, normal trabecular meshwork; GTM, glaucomatous trabecular meshwork; M, male; F, female; P, cell passage.
iTRAQ labeling, SCX chromatography, protein identification, quantitation, and bioinformatics
Detailed methods for sample preparation, iTRAQ labeling, strong cation exchange chromatorgraphy (SCX), protein identification and quantitation and bioinformatics have been described elsewhere [16-18]. Briefly, for proteomic analyses, proteins were extracted from TM cells [18], quantified by amino acid analysis [19], reduced, alkylated, and digested with trypsin [18]. Tryptic peptides from Dex-treated TM cells were labeled with iTRAQ tag 117 and mixed with an equal amount of tryptic peptides from the corresponding untreated cell sample labeled with iTRAQ tag 115. Each peptide mixture was fractionated by strong cation exchange (SCX) chromatography and fractions collected for LC MS/MS. LC MS/MS was performed with a QTOF2 mass spectrometer equipped with a Cap LC system (Waters Corporation, Milford, MA). Protein identification used MASSLYNX 4.1 software (Waters), the Mascot search engine (Matrix Science, Boston, MA), and the SwissProtein human sequence database (version 56.0, ~20,000 total sequences). Two unique peptides per protein and Mascot peptide ion scores ≥25 were required for all protein identification and quantitation. To estimate false discovery rates, all peptide MS/MS spectra were searched (Matrix Science, version 2.2) against a randomized decoy database constructed from the above SwissProtein database with a script provided by Matrix Science [16-18]. Protein quantitation from iTRAQ labeling required ion intensities ≥10 for all iTRAQ tags and was achieved with code written in the statistical program R. To average results over multiple samples, protein ratios were normalized to the protein median per sample, then average protein ratios (unadjusted), standard errors of the mean (SEM) and p-values (two sided t-Test for the null hypothesis that the protein mean=0 in log space) were calculated. Average protein ratios were adjusted to give greater weight to data with lower SEM values then adjusted SEMs and p-values were determined from the adjusted means of the multiple measurements [16-18]. Bioinformatic analyses were performed with The Protein ANalysis THrough Evolutionary Relationships (PANTHER) Classification System, Ingenuity Pathways Analysis 8.5 (Ingenuity Systems, Redwood City, CA), and the SwissProtein database.
Western Analyses
Immunoblots were performed as previously described [18]. Briefly, TM cell cultures were washed with PBS twice, and proteins were extracted with Mammalian Protein Extraction Buffer (ThermoScientific) containing 1% protease inhibitor cocktail (Thermo Fischer Scientific, Rockford, IL). Protein concentrations in TM lysates were determined by the BioRad Dc Protein Assay (Bio-Rad Laboratories, Hercules, CA). SDS-PAGE was performed on 10% acrylamide gels with 30 µg protein applied per lane, and proteins were electrophoretically transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA). Membranes were blocked with 5% non-fat dry milk in TBST buffer then incubated overnight at 4 °C with primary antibody. The membranes were washed with TBST and probed with horseradish peroxidase-conjugated secondary antibody in 3% non-fat milk in TBST for 1 h at room temperature. Primary antibodies included mouse monoclonal anti-activated leukocyte cell adhesion molecule antigen (CD166; ALCAM; Novacastra/NCL-CD166, diluted 1:40; Abcam, Cambridge, MA) and rabbit polyclonal anti-Tplastin (Plastin-3; #ab45769, diluted 1:1,000; Abcam). Rabbit monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase was used as a loading control (#14C10, diluted 1:1,000; Cell Signaling/Millipore, Billerica, MA). Secondary antibodies included horseradish peroxidase-conjugated secondary antibody (goat anti-mouse IgG; #sc2005, diluted 1:10,000; Santa Cruz, Santa Cruz, CA) or goat anti-rabbit IgG (#7074 diluted 1:10,000; Cell Signaling). Proteins were detected using enhanced chemiluminescence and a Fluor ChemTM 8900 imager (Alpha Innotech/ProteinSimple, Santa Clara, CA). Immunoreactivity was quantified using Image J software (NIH), and statistical analyses were performed using the two-sided t-test.
Results
A total of 718 proteins were quantified by LC MS/MS iTRAQ technology from four primary cultures of human TM cells treated or not treated with Dex. TM cell sample properties are defined in Table 1. Quantitative proteomics results from each sample are presented in Appendix 1, Appendix 2, Appendix 3, and Appendix 4, including protein ratios, SDs, p-values, number of unique peptides quantified, percent sequence coverage for each protein, and peptide false discovery rates. The data from all four TM samples were of comparable quality and appropriate for averaging based on consistently low peptide false discovery (average rates, 1.6% identity, 2.7% homology) and similar distributions of protein ratios. The average relative abundance of all 718 quantified proteins is itemized in Appendix 5. The distributions of log2 mean protein ratios for all proteins, and those quantified in ≥3 TM samples (n=341), are shown in Figure 2. The normal distributions (Figure 2) support approximately equal numbers of proteins increased or decreased by Dex-treatment. Criteria for determining whether a protein was elevated or decreased by Dex-treatment included the average adjusted protein ratio and p-value with only proteins quantified in ≥3 cell samples used for comparative purposes. Proteins exhibiting average protein ratios (adjusted by SEM) above or below the mean by at least 1 SD (Figure 2) and p-values ≤0.055 were considered of higher or lower abundance.
Figure 2.
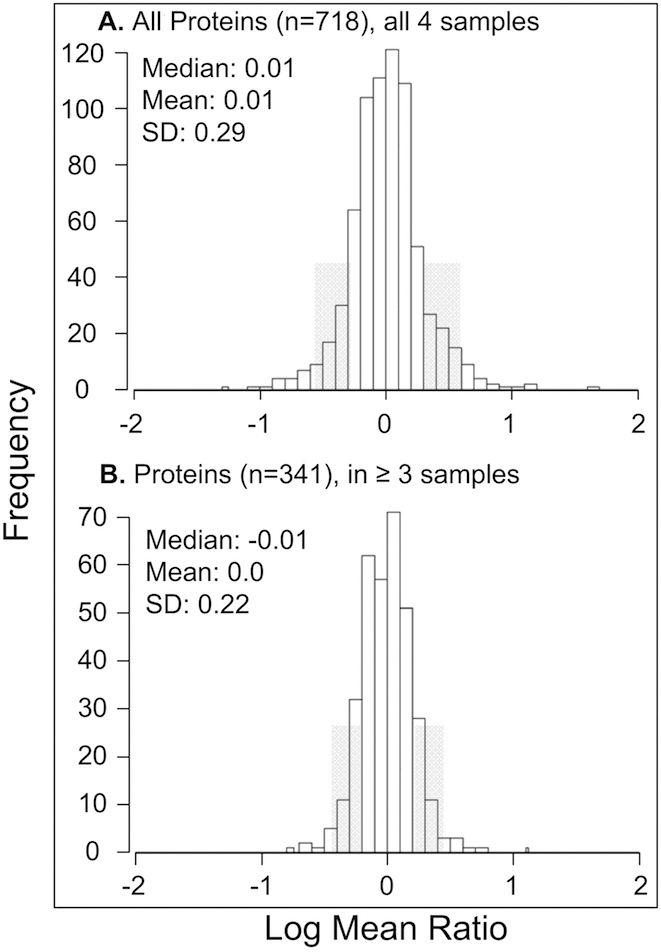
Distribution of protein ratios. The log2 mean distribution of protein ratios (Dex-treated TM/untreated TM) are shown for all 718 quantified proteins and 341 proteins quantified in ≥3 cell samples. Median, mean and SD values are indicated; protein ratios between 1 and 2 SD from the mean are shaded. The distribution of protein ratios is near-to-normal.
The majority of the determined proteome was unchanged by Dex-treatment
About 94% of all 718 proteins and ~88% of the 341 proteins quantified in three or more samples appear to be present in similar amounts in Dex-treated and untreated cells (Figure 2 and Appendix 5). Accordingly, the determined proteome largely reflects that of untreated TM cells. The quantified proteins include ~6% secreted, ~48% cytoplasmic, ~22% membrane and membrane-associated, ~15% nuclear, and ~11% mitochondrial proteins. These proportions are very similar to that obtained in quantitative proteomic analysis of the same TM cells treated with transforming growth factor beta 2 (TGFβ2) [18]. In the current study, Dex-treatment decreased the abundance of more secreted proteins (n=3) than it increased (n=0) and elevated the abundance of more mitochondrial proteins (n=6) than it decreased (n=2).
TM proteins increased by Dex
Twenty proteins were elevated ≥1 SD above the mean ratio (p<0.05) in ≥3 samples by Dex-treatment, including both normal and glaucomatous TM cells (Table 2). Six proteins were elevated ≥2 SD by Dex treatment. Elevated proteins were associated with the cytoskeleton, ECM, mitochondrial metabolism and carbohydrate metabolism. The majority of these proteins have not previously been associated with GC-signaling in TM cells.
Table 2. Proteins elevated in dexamethasone-treated TM cells.
| SwissProt accession | Protein | Subcellular source | Sample frequency total=4 | Mean protein ratio | SEM | p-value |
|---|---|---|---|---|---|---|
|
Cytoskeletal/cell-cell/cell-matrix Interactions | ||||||
|
O94875 |
Sorbin and SH3 domain-containing protein 2 |
B |
4 |
3.06* |
0.10 |
0.001 |
|
Q0ZGT2 |
Nexilin |
B |
4 |
2.03* |
0.17 |
0.026 |
|
Q13740 |
CD166 antigen |
C |
4 |
1.99* |
0.12 |
0.011 |
|
O75369 |
Filamin-B |
B |
4 |
1.64* |
0.11 |
0.020 |
|
Q15942 |
Zyxin |
BD |
4 |
1.38 |
0.04 |
0.005 |
|
Q13418 |
Integrin-linked protein kinase |
C |
4 |
1.36 |
0.09 |
0.037 |
|
Q8WX93 |
Palladin |
B |
4 |
1.31 |
0.08 |
0.044 |
|
P35613 |
Basigin |
C |
4 |
1.29 |
0.04 |
0.007 |
|
Q6NZI2 |
Polymerase I and transcript release factor |
BC |
4 |
1.26 |
0.05 |
0.020 |
|
Carbohydrate Metabolism | ||||||
|
P37837 |
Transaldolase |
B |
4 |
1.43 |
0.08 |
0.019 |
|
P52209 |
6-phosphogluconate dehydrogenase, decarboxylating |
B |
4 |
1.35 |
0.07 |
0.019 |
|
P11413 |
Glucose-6-phosphate 1-dehydrogenase |
B |
3 |
1.30 |
0.03 |
0.015 |
|
P60174 |
Triosephosphate isomerase |
B |
4 |
1.26 |
0.06 |
0.031 |
|
Mitochondrial Metabolism | ||||||
|
Q9Y6N5 |
Sulfide:quinone oxidoreductase |
E |
4 |
1.81* |
0.06 |
0.002 |
|
Q9UIJ7 |
GTP:AMP phosphotransferase |
E |
4 |
1.56* |
0.07 |
0.007 |
|
P42765 |
3-ketoacyl-CoA thiolase |
E |
4 |
1.39 |
0.06 |
0.011 |
|
Q13510 |
Acid ceramidase |
B |
3 |
1.35 |
0.05 |
0.031 |
|
P30049 |
ATP synthase subunit delta |
CE |
4 |
1.34 |
0.07 |
0.023 |
|
P61604 |
10 kDa heat shock protein |
E |
4 |
1.33 |
0.08 |
0.042 |
| P36957 | Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex | E | 3 | 1.25 | 0.03 | 0.014 |
Adjusted mean protein ratios (DEX-treated/Control), standard error of the mean (SEM), p-values and subcellular source are shown for abundant proteins (from Appendix 5). Abundant proteins exhibited mean ratios at least 1SD above the mean and p-values <0.05 (see Figure 2). All proteins were quantified in ≥3 TM cell samples (median ratio=0.99, mean=1.00, SD=0.22, n=341). Asterisks denote ratios at least 2 SD above the mean. Protein subcellular source from the Swiss Protein database: A, secreted; B, cytoplasmic; C, membrane; D, nuclear; E, mitochondrial.
TM proteins decreased by Dex
Twenty proteins were significantly decreased ≥1 SD below the mean ratio (p≤0.55) in ≥3 samples by Dex-treatment, including 4 reduced ≥2 SD (Table 3), and all were changed in both normal and glaucomatous TM cells. Decreased proteins were associated with stress response, cellular defenses, protein processing, the cytoskeleton, the extracellular Matrix (ECM), and mitochondrial metabolism. The majority of the Dex-decreased proteins have not previously been considered steroid responsive in TM cells.
Table 3. Proteins decreased in dexamethasone-treated TM cells.
| SwissProt accession | Protein | Subcellular source | Sample frequency total=4 | Mean protein ratio | SEM | p-value |
|---|---|---|---|---|---|---|
|
Stress Response, Cellular Defense and Protein Processing | ||||||
|
P17931 |
Galectin-3 |
ABD |
3 |
0.80 |
0.05 |
0.054 |
|
P25786 |
Proteasome subunit alpha type-1 |
BD |
3 |
0.79 |
0.06 |
0.051 |
|
Q96AY3 |
FK506-binding protein 10** |
C |
4 |
0.78 |
0.04 |
0.009 |
|
Q14697 |
Neutral alpha-glucosidase AB |
C |
3 |
0.77 |
0.06 |
0.042 |
|
P06703 |
Protein S100-A6** |
BCD |
4 |
0.75 |
0.03 |
0.003 |
|
P27797 |
Calreticulin** |
ABC |
4 |
0.73 |
0.09 |
0.039 |
|
P05388 |
60S acidic ribosomal protein P0 |
B |
3 |
0.73 |
0.02 |
0.003 |
|
O95302 |
FK506-binding protein 9** |
C |
4 |
0.71 |
0.06 |
0.010 |
|
P23284 |
Peptidyl-prolyl cis-trans isomerase B |
C |
4 |
0.69 |
0.11 |
0.045 |
|
P30040 |
Endoplasmic reticulum protein ERp29 |
C |
4 |
0.66 |
0.05 |
0.004 |
|
P50454 |
Serpin H1** |
C |
4 |
0.52* |
0.07 |
0.003 |
|
Cytoskeletal and ECM Interactions | ||||||
|
P13797 |
Plastin-3** |
B |
4 |
0.73 |
0.10 |
0.048 |
|
O15460 |
Prolyl 4-hydroxylase subunit alpha-2 |
C |
4 |
0.75 |
0.09 |
0.053 |
|
Q01995 |
Transgelin** |
B |
4 |
0.78 |
0.07 |
0.042 |
|
P13674 |
Prolyl 4-hydroxylase subunit alpha-1 |
C |
4 |
0.78 |
0.08 |
0.055 |
|
P48681 |
Nestin |
B |
3 |
0.62* |
0.04 |
0.007 |
|
P02452 |
Collagen alpha-1(I) chain |
A |
3 |
0.48* |
0.15 |
0.038 |
|
Mitochondrial Metabolism | ||||||
|
P30153 |
Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform |
D |
3 |
0.78 |
0.01 |
0.004 |
|
P22307 |
Non-specific lipid-transfer protein |
BE |
4 |
0.73 |
0.10 |
0.049 |
| O75964 | ATP synthase subunit gamma | CE | 3 | 0.61* | 0.12 | 0.054 |
Adjusted mean protein ratios (DEX-treated/Control), standard error of the mean (SEM), p-values and subcellular source are shown for less abundant proteins (from Appendix 5). Less abundant proteins exhibited mean ratios at least 1SD below the mean and p-values ≤0.055 (see Figure 2). All proteins were quantified in ≥3 TM cell samples (median ratio=0.99, mean=1.00, SD=0.21, n=341). Asterisks (*) denote ratios at least 2 SD below the mean. Double asterisks (**) denote CA++ binding proteins. Protein subcellular source from the Swiss Protein database: A, secreted; B, cytoplasmic; C, membrane; D, nuclear; E, mitochondrial.
Independent evidence supporting the iTRAQ protein quantitation
Western blot analysis (Figure 3) demonstrated Dex-increased amounts of ALCAM/CD166 antigen (p=0.02) and Dex-decreased amounts of plastin-3 (p<0.01), corroborating the iTRAQ protein quantitation. These proteins were selected only because of their presence in Table 2 and Table 3 and the availability of antibodies useful for western analysis. Dex-decreased collagen α1(1) is supported by a previous proteomic study [20] that reported Dex-down-regulation of collagen α1(1) in rat TM (Table 3). Dex-increased amounts of sorbin and SH3 domain containing protein 2 [12] and filamin B [12] are indirectly supported by gene expression studies that reported Dex-induced upregulation of these genes in TM cells.
Figure 3.

Western analyses. Western blot analysis was used to evaluate the relative amounts of proteins in Dex–treated and untreated TM cells (n=4 samples each). Immunoblot intensities were normalized to GAPDH; average densitometry ratios (Dex–treated/untreated) and p values (two-sided t-test) for CD166 antigen (ALCAM) and Plastin-3 support the quantitative data in Table 2 and Table 3.
Discussion
To better understand GC-signaling in TM cells, we quantified proteins in four primary cultures of human TM cells with and without Dex-treatment using LC MS/MS iTRAQ technology. The study goal was to test the hypothesis that common mechanisms of steroid responsiveness exist in TM cells from normal and glaucomatous tissues. Differences in the levels of glucocorticoid receptor isoforms between normal and glaucomatous cells have been suggested to contribute to steroid responsiveness in TM cells [21,22]. However, similarities in clinical phenotypes of GC-induced ocular hypertension in normal and glaucoma patients suggest common mechanisms may also contribute to steroid responsiveness. Global proteomic analysis of TM cells from both donor populations has provided evidence for common molecular consequences of GC–signaling in non-glaucomatous and glaucomatous TM cells. Notably, ~72% of Dex-altered proteins in this study were significantly changed in all four TM cell strains analyzed. The majority of the 718 quantified proteins were present in similar amounts in Dex-treated and untreated cells; therefore, the overall determined proteome reflects that of untreated TM cells. Dex-treatment significantly altered the abundance of 40 proteins; immunoblots independently corroborated two of these changes and literature reports support three other observed proteomic changes induced by Dex. Thirty-seven of the 40 altered proteins have not been previously recognized as steroid responsive in TM cells. Major biologic processes associated with the determined proteome (Figure 4) include cellular metabolism (37% of the 718 proteins), signal transduction (8%), cell structure/motility (10%), intracellular traffic (7%), and immunity and defense (5%). Ingenuity Pathway analysis of the proteome implicates protein synthesis, cell death, and post-translational modification as the highest scoring networks.
Figure 4.

Biologic functions of TM proteins. Functional analysis of the 718 proteins quantified in human TM cells was performed using the PANTHER Classification System.
Upregulated GC-responses in normal and glaucomatous TM cells
Twenty TM proteins were significantly increased in abundance following Dex-treatment (Table 2), supporting steroid-induced, upregulated expression of these polypeptides. Pathway analysis of these proteins highlights cellular movement, cell death, and cell morphology as one of the two highest scoring networks. Several cytoskeletal proteins were elevated, including filamin-B, which connects the cell membrane with the actin cytoskeleton and promotes actin filament branching. These findings are consistent with other studies showing that Dex stimulates an increase in cross-linked actin networks (CLANs) in cultured human TM cells [14]. Such morphological networks are also observed in vivo in both normal and glaucomatous TM [23]. Sorbin and SH3 domain-containing protein 2, elevated about threefold, interacts with a large number of proteins, linking signaling complexes with the cytoskeleton. Increased levels of focal adhesion proteins palladin, zyxin, integrin-linked protein kinase (ILK) and the adherens junction protein nexilin, are consistent with GC-stimulated formation of adherens junctions and tight junctions, previously observed in TM cells [24] and in mammary tumor cells [25]. Notably, ILK functions in the regulation of cell-cell and cell-matrix interactions and appears to have roles in both integrin and growth factor-signaling. Two other elevated proteins potentially impacting cell adhesion and cell motility were CD166 antigen (ALCAM) and basigin. Both are plasma membrane proteins that promote growth of cellular processes and activate leuckocytes. CD166 antigen also functions in cellular interactions in the nervous system [26] and basigin (also known as tumor cell-derived collagenase stimulatory factor) stimulates matrix metalloproteinase production and tumor progression. Finally polymerase 1 and transcript release factor was elevated ~26% by Dex treatment. This protein is essential for caveolae formation in mammals, and caveolae function in cell proliferation, cargo-specific endocytosis, and mechanosensation [27]. Overall, these 9 elevated proteins support the hypothesis that GC-treatment can alter TM cytoskeletal and cell-cell interactions that contribute to AH outflow resistance and elevated IOP [3].
The other high scoring network from pathway analysis highlights cell cycle, carbohydrate, and nucleic acid metabolism. Consistent with GC regulation of gluconeogenesis [28], four enzymes in the pentose-phosphate pathway were upregulated ~30%–40%, namely transaldolase, 6-phosphogluconate dehydrogenase, glucose-6-phosphate dehydrogenase, and triosephosphate isomerase. The consequences of disrupting the pentose shunt in the TM remain to be determined, but genetic defects in these 4 enzymes can cause severe disorders elsewhere in the body, including progressive neuromuscular dysfunction.
Six mitochondrial proteins and a 7th protein that could significantly alter mitochondrial functions were elevated by Dex-treatment. These mitochondrial proteins serve a variety of roles in energy production (ATP synthase δ), signal transduction (GTP:AMP phosphotransferase), oxidation reduction (sulfide:quinone oxidoreductase), chaperone activity (10 kDa heat shock protein), lipid metabolism (3-ketoacyl-CoA thiolase), and carbohydrate metabolism (component of 2-oxoglutarate dehydrogenase complex). These changes could disrupt TM homeostasis by altering oxidative phosphorylation, regulation of apoptosis by reactive oxygen species, and maintenance of intracellular calcium levels [29,30]. Acid ceramidase, the 7th elevated protein, is a cytosolic protein that functions in the sphingosine-ceramide salvage pathway in sphingolipid turnover [31]. Ceramides produced in the salvage pathway in human breast cancer cells accumulate in mitochondria, promote the relocalization of mitochondria around the nuclear envelope and have been linked to apoptosis and mediators of mitochondrial functions [32]. Impaired mitochondria in TM cells has recently been implicated in POAG [18,33,34]. The present results provide clear evidence that GC-signaling can modify mitochondrial protein expression in both normal and glaucomatous TM cells. Dysfunctional TM mitochondria could promote oxidative damage [18,35] and cellular senescence in the aqueous humor outflow pathway [36].
Down-regulated GC-responses in normal and glaucomatous TM cells
Twenty TM proteins were significantly reduced by Dex-treatment (Table 3), suggesting GC-induced down-regulation, and/or degradation. Pathway analysis of these 20 proteins highlight gene expression, free radical scavenging and molecular transport as the highest scoring networks. TM cells from POAG donors reportedly contain higher levels of reactive oxygen species and Ca2+ in the cytosol and the mitochondria [33,34]. Interestingly, ~35% (n=7) of the TM proteins reduced by Dex-treatment were calcium-binding proteins (denoted by ** in Table 3), supporting calcium dysregulation in the TM as possibly contributing to elevated IOP [33]. About 50% of the decreased proteins can be associated with cellular defense, stress response, and processing events, consistent with an anti-inflammatory response. Several of these exhibit protein-folding and chaperone functions, including FK506-binding proteins 9 and 10, calreticulin, peptidyl-prolyl cis-trans isomerase B, endoplasmic reticulum resident protein 29, and Serpin H1. Noteworthy, serpin H1 is a stress-induced, collagen-binding chaperone, and calreticulin functions in the GC receptor signaling pathway. Two damage-associated molecular pattern proteins (DAMPs) were also decreased by Dex, namely advanced glycation endproduct receptor 3 (galectin-3) and protein S100-A6. DAMPs are endogenous proteins released by damaged cells that are capable of binding and activating an inflammatory response through pattern recognition receptors such as the complement system, toll-like receptors, and receptor for advanced glycation endproducts (RAGE). Also decreased was proteasome subunit α type-1, important in the processing of MHC peptides, regulation of the cell cycle, and ubiquitin-dependent degradation processes. Others included the 60S acidic ribosomal protein P0 involved in translational elongation, and neutral α-glucosidase AB which functions in conjunction with calreticulin in the release of mono-glycosylated glycoproteins.
Six proteins impacting the cytoskeleton and/or ECM were decreased by Dex-treatment. Among this group were actin-binding proteins transgelin and plastin-3, as well as nestin, involved in intermediate filament assembly/disassembly and cellular remodeling. Altered levels of these proteins could facilitate GC-induced CLAN formation [14]. ECM protein collagen α1(1) was the most significantly reduced, down ~50%. Two enzymes that catalyze proline hydroxylation in collagen were also decreased, namely prolyl 4-hydroxylase α1 and α2. Collagen α1(1) fibrils are a major component of collagen beams in TM in vivo and at physiologic conditions require hydroxyproline to stabilize the triple-helical structure. Altered interactions between collagen fibrils and other ECM components have been reported to trigger collagen degradation and a loss of tissue-specific morphology [37,38]. Whether the reduced collagen α1(1) level we observed in TM cells is due to decreased expression or to degradation is not clear, but perturbed collagen levels in vivo would render the ECM more susceptible to collapse and debris deposition. Collectively, the observed proteomic changes could disrupt the TM cytoskeleton and associated ECM in both normal and glaucomatous TM.
Dex-treatment also decreased the abundance of proteins impacting mitochondrial metabolism (Table 3). Among these were mitochondrial proteins ATP synthase γ, important in energy production and non-specific lipid-transfer protein, functioning in lipid transport across membranes and possibly serving in steroid biosynthesis. Also decreased was serine/threonine-protein phosphatase 2A isoform PR65-α, which appears to be a regulator in cell adhesion, apoptosis and ceramide-associated processes. As noted, ceramides have been linked to apoptosis and may accumulate in the mitochondria [32]. These results are consistent with GC-inducable mitochondrial dysfunction in both normal and glaucomatous TM cells.
Comparison with previous studies of GC-signaling in TM cells
This is the most extensive quantitative proteomic analysis of GC-signaling in TM cells. A previous 2D-gel proteomic study of one immortalized TM cell line identified 163 proteins and reported Dex-down regulation of Rho GDP dissociation inhibitor (RhoGDI) [39]. In the present study, four different primary TM cell cultures were analyzed and RhoGDI was not changed by Dex treatment (Appendix 5). Only one other proteomic study of steroid signaling in the anterior segment is currently published. Proteomic changes induced by in vivo topical application of Dex to three rat eyes was investigated using peptide mass fingerprinting methods and 2D fluorescence difference gel electrophoresis [20]. Four Dex-altered TM proteins were reported, including upregulation of αA-crystallin and βA3-crystallin and down-regulation of collagens α1(1) and α2(1) [20]. We found Dex-decreased amounts of collagen α1(1), detected collagen α2(1) in 2 samples, but did not detect αA- and βA3-crystallins, although several other chaperones were decreased by Dex-treatment (Table 2). The earlier report suggested that Dex may induce impaired collagen processing [20] because only COOH-terminal propeptides were detected. This may occur but our study quantified seven internal peptides from throughout the collagen α1(1) sequence, supporting Dex-induced decreased expression of collagen α1(1).
Hundreds of Dex-induced transcript changes in human TM cells have been described in five gene-profiling studies [9-13]; however, only two genes were reported differentially expressed in all five studies, namely genes encoding myocilin and insulin-like growth factor binding protein 2 (both upregulated). We detected myocilin at elevated levels in two of the four TM cell samples (Appendix 1 and Appendix 3) but did not detect insulin-like growth factor binding protein 2. While myocilin is a glaucoma gene, it is not always elevated at the protein level in TM tissues in open angle glaucoma [20]. Myocilin is a secreted glycoprotein, which may account in part for variable TM levels. In our study, myocilin may have also been in the culture medium, which was not analyzed. Among transcripts reported differentially expressed in the TM by Dex [9-13], we found 27 corresponding gene products, from which reliable quantification was obtained on 11 proteins (ie, proteins found in ≥3 samples). Our proteomic data support upregulation of sorbin and SH3 domain containing protein 2 [12] and filamin B [12] but do not support upregulation of transgelin [11,12] nor the reported down-regulation of 3-ketoacyl-CoA thiolase β [9]. Seven other transcripts reported to be differentially expressed exhibited no significant change at the protein level in this study, namely lactate dehydrogenase A [11], clusterin [10], fibroblast muscle tropomyosin [9], skeletal β-tropomyosin [9], fibulin-1C [9], phosphatidylethanolamine binding protein [9], and thrombospondin [13]. The observed differences between proteomic and genomic data seem reasonable as only ~20% correlation generally exists between mRNA and corresponding protein levels in mammals [40,41]. Similar to our findings from quantitative proteomic analysis of TGFβ2-signaling in TM cells [18], very few proteases or protease inhibitors were detected in this study (Appendix 5), perhaps because they were below detection limits, and none were found significantly altered by Dex-treatment.
Additional insight to steroid responsiveness in TM cells
To further probe molecular mechanisms of steroid responsiveness, we compared Dex-induced proteomic changes with previously identified protein changes in TM cells induced by TGFβ2 [18]. TGFβ2 is elevated in the anterior segment of glaucoma patients, and while the mechanism(s) responsible for elevated expression is unknown, substantial evidence implicates TGFβ2 as a contributing factor in ocular hypertension [3,18]. Our TGFβ2 investigation [18] used the same normal and glaucomatous TM cell strains and quantitative proteomic methods as used in the present study. For comparison of Dex- and TGFβ2-induced proteomic changes, proteins quantified in ≥3 TM cell samples in both studies were sought that exhibited: (i) ratios at least 1 SD above or below the means in both data sets, and (ii) a p-value ≤0.055 in at least one of the treatments (indicating at least one of the treatments significantly changed the protein ratio). Twenty proteins meeting the above criteria were identified (Table 4), including four apparently altered by both TGFβ2 and Dex (palladin, prolyl 4-hydroxylase α1 and α2, and FK506-binding protein 10). While additional studies with larger samples sizes are warranted to validate proteins altered by both Dex and TGFβ2, these two modifiers appear to impact several common physiologic processes in both normal and glaucomatous TM cells. These processes include cytoskeletal/cell-cell interactions, cell-matrix/ECM remodeling, and mitochondrial metabolism [18]. Such common physiologic consequences suggest a molecular basis for the increased risk of ocular hypertension in steroid responders.
Table 4. Proteomic changes induced in TM cells by dexamethasone and TGFβ2.
|
|
|
Dex |
TGFβ2 |
||
|---|---|---|---|---|---|
| Swiss prot accession | Protein | Ratio | p value | Ratio | p value |
|
Cytoskeletal and Cell-Cell Interactions | |||||
|
Q8WX93 |
Palladin |
↑ 1.3 |
0.044 |
↑ 1.7 |
0.003 |
|
Q9HBL0 |
Tensin-1 |
↑ 1.7 |
0.119 |
↑ 1.7 |
0.017 |
|
P37802 |
Transgelin-2 |
↑ 1.3 |
0.127 |
↑ 1.6 |
0.012 |
|
Q0ZGT2 |
Nexilin |
↑ 2.0 |
0.026 |
↑ 1.4 |
0.152 |
|
Q01995 |
Transgelin |
↓ 0.8 |
0.042 |
↑ 1.7 |
0.151 |
|
P13797 |
Plastin-3 |
↓ 0.7 |
0.048 |
↑ 1.5 |
0.093 |
|
Cell-Matrix and ECM Remodeling | |||||
|
O15460 |
Prolyl 4-hydroxylase subunit alpha-2 |
↓ 0.7 |
0.053 |
↑ 2.1 |
0.042 |
|
P13674 |
Prolyl 4-hydroxylase subunit alpha-1 |
↓ 0.8 |
0.055 |
↑ 1.5 |
0.057 |
|
P07996 |
Thrombospondin-1 |
↓ 0.7 |
0.147 |
↑ 2.4 |
0.030 |
|
P02452 |
Collagen alpha-1(I) chain |
↓ 0.5 |
0.038 |
↑ 1.7 |
0.263 |
|
Mitochondrial Metabolism | |||||
|
P04179 |
Superoxide dismutase [Mn] |
↓ 0.8 |
0.272 |
↓ 0.5 |
0.002 |
|
Q9UIJ7 |
GTP:AMP phosphotransferase mitochondrial |
↑ 1.6 |
0.007 |
↓ 0.7 |
0.168 |
|
P42765 |
3-ketoacyl-CoA thiolase |
↑ 1.4 |
0.011 |
↓ 0.7 |
0.200 |
|
Q9Y6N5 |
Sulfide:quinone oxidoreductase |
↑ 1.8 |
0.002 |
↓ 0.7 |
0.067 |
|
P00367 |
Glutamate dehydrogenase 1 |
↑ 1.3 |
0.061 |
↓ 0.6 |
0.012 |
|
O94925 |
Glutaminase kidney isoform |
↓ 0.8 |
0.206 |
↑ 1.4 |
0.037 |
|
Stess Response and Protein Folding | |||||
|
Q96AY3 |
FK506-binding protein 10 |
↓ 0.8 |
0.009 |
↑ 1.5 |
0.005 |
|
Q15084 |
Protein disulfide-isomerase A6 |
↓ 0.8 |
0.092 |
↑ 1.4 |
0.041 |
|
Regulatory Processes | |||||
|
P50479 |
PDZ and LIM domain protein 4 |
↑ 1.3 |
0.290 |
↑ 1.7 |
0.036 |
| Q99536 | Synaptic vesicle membrane protein VAT-1 homolog | ↓ 0.8 | 0.561 | ↓ 0.7 | 0.004 |
Proteins are shown that were quanitified in both this study and a study of TGFβ2-induced proteomic changes in TM cells [18]. All proteins were quantified in ≥3 TM samples, exhibited ratios at least 1 SD above or below the means, and p-values ≤0.055 in at least one of the treatments (indicating at least one ratio significantly different than 1.0). Arrows reflect elevated or decreased expression.
Conclusions
Based on our analysIs of a small number of TM cell samples, the results expand the repertoire of proteins participating in GC-signaling and support common steroid response mechanisms in both normal and glaucomatous TM cells. While we analyzed more cell samples than in previous studies of GC effects on the TM transcriptome or proteome, the sample size is limiting and further investigations are warranted. The observed proteomic changes implicate as consequences of steroid-treatment remodeling of the extracellular matrix, disorganization of the cytoskeleton, disruption of cell-cell interactions, and mitochondrial dysfunction in the TM. These same physiologic processes appear impacted by TGFβ2-signaling in both normal and glaucomatous TM cells. Finally, the results contribute to a quantitative database of TM proteins.
Acknowledgments
Supported in part by National Institutes of Health grants EY018147, EY14239, EY15638, EY016242 (A.F.C.), a Challenge grant from Research to Prevent Blindness (RPB), American Health Assistance Foundation grant 34–0714585 to K.E.B., an RPB Senior Investigator Award to J.W.C., a Steinbach Award to J.W.C., and The Cleveland Clinic Foundation. We thank Drs Edward Rockwood, Joe Hollyfield, Andrew Schachat, and Elias Traboulsi for valuable discussions.
Appendix 1. Relative protein abundance: Trabecular meshwork sample NTM416–07.
To access the data, click or select the words “Appendix 1.” This will initiate the download of a compressed (pdf) archive that contains the file.
Appendix 2. Relative protein abundance: Trabecular meshwork sample NTM496–05.
To access the data, click or select the words “Appendix 2.” This will initiate the download of a compressed (pdf) archive that contains the file.
Appendix 3. Relative protein abundance: Trabecular meshwork sample GTM304–04.
To access the data, click or select the words “Appendix 3.” This will initiate the download of a compressed (pdf) archive that contains the file.
Appendix 4. Relative protein abundance: Trabecular meshwork sample GTM477–02.
To access the data, click or select the words “Appendix 4.” This will initiate the download of a compressed (pdf) archive that contains the file.
Appendix 5. Average relative protein abundance over all DEX-treated TM cell Samples.
To access the data, click or select the words “Appendix 5.” This will initiate the download of a compressed (pdf) archive that contains the file.
References
- 1.Kersey JP, Broadway DC. Corticosteroid-induced glaucoma: a review of the literature. Eye (Lond) 2006;20:407–16. doi: 10.1038/sj.eye.6701895. [DOI] [PubMed] [Google Scholar]
- 2.Jones R, 3rd, Rhee DJ. Corticosteroid-induced ocular hypertension and glaucoma: a brief review and update of the literature. Curr Opin Ophthalmol. 2006;17:163–7. doi: 10.1097/01.icu.0000193079.55240.18. [DOI] [PubMed] [Google Scholar]
- 3.Clark AF, Wordinger RJ. The role of steroids in outflow resistance. Exp Eye Res. 2009;88:752–9. doi: 10.1016/j.exer.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Gabelt BT, Kaufman PL. Changes in aqueous humor dynamics with age and glaucoma. Prog Retin Eye Res. 2005;24:612–37. doi: 10.1016/j.preteyeres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Tamm ER, Fuchshofer R. What increases outflow resistance in primary open-angle glaucoma? Surv Ophthalmol. 2007;52(Suppl 2):S101–4. doi: 10.1016/j.survophthal.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Tan JC, Peters DM, Kaufman PL. Recent developments in understanding the pathophysiology of elevated intraocular pressure. Curr Opin Ophthalmol. 2006;17:168–74. doi: 10.1097/01.icu.0000193079.55240.18. [DOI] [PubMed] [Google Scholar]
- 7.Viegas LR, Hoijman E, Beato M, Pecci A. Mechanisms involved in tissue-specific apopotosis regulated by glucocorticoids. J Steroid Biochem Mol Biol. 2008;109:273–8. doi: 10.1016/j.jsbmb.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Chrousos GP, Kino T. Glucocorticoid signaling in the cell. Expanding clinical implications to complex human behavioral and somatic disorders. Ann N Y Acad Sci. 2009;1179:153–66. doi: 10.1111/j.1749-6632.2009.04988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishibashi T, Takagi Y, Mori K, Naruse S, Nishino H, Yue BY, Kinoshita S. cDNA microarray analysis of gene expression changes induced by dexamethasone in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2002;43:3691–7. [PubMed] [Google Scholar]
- 10.Lo WR, Rowlette LL, Caballero M, Yang P, Hernandez MR, Borras T. Tissue differential microarray analysis of dexamethasone induction reveals potential mechanisms of steroid glaucoma. Invest Ophthalmol Vis Sci. 2003;44:473–85. doi: 10.1167/iovs.02-0444. [DOI] [PubMed] [Google Scholar]
- 11.Leung YF, Tam PO, Lee WS, Lam DS, Yam HF, Fan BJ, Tham CC, Chua JK, Pang CP. The dual role of dexamethasone on anti-inflammation and outflow resistance demonstrated in cultured human trabecular meshwork cells. Mol Vis. 2003;9:425–39. [PubMed] [Google Scholar]
- 12.Rozsa FW, Reed DM, Scott KM, Pawar H, Moroi SE, Kijek TG, Krafchak CM, Othman MI, Vollrath D, Elner VM, Richards JE. Gene expression profile of human trabecular meshwork cells in response to long-term dexamethasone exposure. Mol Vis. 2006;12:125–41. [PubMed] [Google Scholar]
- 13.Fan BJ, Wang DY, Tham CC, Lam DS, Pang CP. Gene expression profiles of human trabecular meshwork cells induced by triamcinolone and dexamethasone. Invest Ophthalmol Vis Sci. 2008;49:1886–97. doi: 10.1167/iovs.07-0414. [DOI] [PubMed] [Google Scholar]
- 14.Clark AF, Wilson K, McCartney M, Miggans S, Kunkle M, Howe W. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1994;35:281–94. [PubMed] [Google Scholar]
- 15.Wordinger RJ, Clark A, Agarwal R, Lambert W, McNatt L, Wilson S, Qu Z, Fung B. Cultured human trabecular meshwork cells express functional growth factor receptors. Invest Ophthalmol Vis Sci. 1998;39:1575–89. [PubMed] [Google Scholar]
- 16.Crabb JW, Yuan X, Dvoriantchikova G, Ivanov D, Crabb JS, Shestopalov VI. Preliminary quantitative proteomic characterization of glaucomatous rat retinal ganglion cells. Exp Eye Res. 2010;91:107–10. doi: 10.1016/j.exer.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan X, Gu X, Crabb JS, Yue X, Shadrach K, Hollyfield JG, Crabb JW. Quantitative proteomics: comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol Cell Proteomics. 2010;9:1031–46. doi: 10.1074/mcp.M900523-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bollinger KE, Crabb JS, Yuan X, Putliwala T, Clark AF, Crabb JW. Qunatitative Proteomics: TGF-β2-Signaling in Trabecular Meshwork Cells. Invest Ophthalmol Vis Sci. 2011;52:8287–94. doi: 10.1167/iovs.11-8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni J, Yuan X, Gu J, Yue X, Gu X, Nagaraj RH, Crabb JW, The Clinical Genomic and Proteomic AMD Study Group Plasma protein pentosidine and carboxymethyllysine, biomarkers for age-related macular degeneration. Mol Cell Proteomics. 2009;8:1921–33. doi: 10.1074/mcp.M900127-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinzato M, Yamashiro Y, Miyara N, Iwamatsu A, Takeuchi K, Umikawa M, Bayarjargal M, Kariya K, Sawaguchi S. Proteomic analysis of the trabecular meshwork of rats in a steroid-induced ocular hypertension model: downregulation of type I collagen C-propeptides. Ophthalmic Res. 2007;39:330–7. doi: 10.1159/000109989. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Clark AF, Yorio T. Regulation of glucocorticoid responsiveness in glaucomatous trabecular meshwork cells by glucocorticoid receptor-beta. Invest Ophthalmol Vis Sci. 2005;46:4607–16. doi: 10.1167/iovs.05-0571. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Ognibene CM, Clark AF, Yorio T. Dexamethasone inhibition of trabecular meshwork cell phagocytosis and its modulation by glucocorticoid receptor beta. Exp Eye Res. 2007;84:275–84. doi: 10.1016/j.exer.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoare MJ, Grierson I, Brotchie D, Pollock N, Cracknell K, Clark AF. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Invest Ophthalmol Vis Sci. 2009;50:1255–63. doi: 10.1167/iovs.08-2706. [DOI] [PubMed] [Google Scholar]
- 24.Underwood JL, Murphy CG, Chen J, Franse-Carman L, Wood I, Epstein DL, Alvarado JA. Glucocorticoids regulate transendothelial fluid flow resistance and formation of intercellular junctions. Am J Physiol. 1999;277:C330–42. doi: 10.1152/ajpcell.1999.277.2.C330. [DOI] [PubMed] [Google Scholar]
- 25.Failor KL, Desyatnikov Y, Finger LA, Firestone GL. Glucocorticoid-induced degradation of glycogen synthase kinase-3 protein is triggered by serum- and glucocorticoid-induced protein kinase and Akt signaling and controls beta-catenin dynamics and tight junction formation in mammary epithelial tumor cells. Mol Endocrinol. 2007;21:2403–15. doi: 10.1210/me.2007-0143. [DOI] [PubMed] [Google Scholar]
- 26.Tripathi BJ, Tripathi RC. Neural crest origin of human trabecular meshwork and its implications for the pathogenesis of glaucoma. Am J Ophthalmol. 1989;107:583–90. doi: 10.1016/0002-9394(89)90253-5. [DOI] [PubMed] [Google Scholar]
- 27.Bastiani M, Liu L, Hill MM, Jedrychowski MP, Nixon SJ, Lo HP, Abankwa D, Luetterforst R, Fernandez-Rojo M, Breen MR, Gygi SP, Vinten J, Walser PJ, North KN, Hancock JF, Pilch PF, Parton RG. MURC/Cavin-4 and cavin family members form tissue-specific caveolar complexes. J Cell Biol. 2009;185:1259–73. doi: 10.1083/jcb.200903053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Exton JH. Regulation of gluconeogenesis by glucocorticoids. Monogr Endocrinol. 1979;12:535–46. doi: 10.1007/978-3-642-81265-1_28. [DOI] [PubMed] [Google Scholar]
- 29.Kong GY, Van Bergen N, Trounce I, Crowston J. Mitochondrial dysfunction and glaucoma. J Glaucoma. 2009;18:93–100. doi: 10.1097/IJG.0b013e318181284f. [DOI] [PubMed] [Google Scholar]
- 30.Osborne NN. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res. 2010;90:750–7. doi: 10.1016/j.exer.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008;20:1010–8. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitatani K, Idkowiak-Baldys J, Bielawski J, Taha TA, Jenkins RW, Senkal CE, Ogretmen B, Obeid LM, Hannun YA. Protein kinase C-induced activation of a ceramide/protein phosphatase 1 pathway leading to dephosphorylation of p38 MAPK. J Biol Chem. 2006;281:36793–802. doi: 10.1074/jbc.M608137200. [DOI] [PubMed] [Google Scholar]
- 33.He Y, Ge J, Tombran-Tink J. Mitochondrial defects and dysfunction in calcium regulation in glaucomatous trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2008;49:4912–22. doi: 10.1167/iovs.08-2192. [DOI] [PubMed] [Google Scholar]
- 34.He Y, Leung KW, Zhang YH, Duan S, Zhong XF, Jiang RZ, Peng Z, Tombran-Tink J, Ge J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: protection by antioxidants. Invest Ophthalmol Vis Sci. 2008;49:1447–58. doi: 10.1167/iovs.07-1361. [DOI] [PubMed] [Google Scholar]
- 35.Kahn MG, Giblin F, Epstein D. Glutathione in calf trabecular meshwork and its relation to aqueous humor outflow facility. Invest Ophthalmol Vis Sci. 1983;24:1283–7. [PubMed] [Google Scholar]
- 36.Liton PB, Challa P, Stinnett S, Luna C, Epstein DL, Gonzalez P. Cellular senescence in the glaucomatous outflow pathway. Exp Gerontol. 2005;40:745–8. doi: 10.1016/j.exger.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pareti FI, Niiya K, McPherson JM, Ruggeri ZM. Isolation and characterization of two domains of human von Willebrand factor that interact with fibrillar collagen types I and III. J Biol Chem. 1987;262:13835–41. [PubMed] [Google Scholar]
- 38.Marchant JK, Hahn RA, Linsenmayer TF, Birk DE. Reduction of type V collagen using a dominant-negative strategy alters the regulation of fibrillogenesis and results in the loss of corneal-specific fibril morphology. J Cell Biol. 1996;135:1415–26. doi: 10.1083/jcb.135.5.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu M, Sun J, Peng W, Chen Z, Lin X, Liu X, Li M, Wu K. Protein expression in human trabecular meshwork: downregulation of RhoGDI by dexamethasone in vitro. Mol Vis. 2010;16:213–23. [PMC free article] [PubMed] [Google Scholar]
- 40.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–30. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, Yi EC, Dai H, Thorsson V, Eng J, Goodlett D, Berger JP, Gunter B, Linseley PS, Stoughton RB, Aebersold R, Collins SJ, Hanlon WA, Hood LE. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3:960–9. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]