

Abstract
Purpose
Waardenburg syndrome (WS) is characterized by sensorineural hearing loss and pigmentation defects of the eye, skin, and hair. It is caused by mutations in one of the following genes: PAX3 (paired box 3), MITF (microphthalmia-associated transcription factor), EDNRB (endothelin receptor type B), EDN3 (endothelin 3), SNAI2 (snail homolog 2, Drosophila) and SOX10 (SRY-box containing gene 10). Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder caused by mutations in the DMD gene. The purpose of this study was to identify the genetic causes of WS and DMD in an Indian family with two patients: one affected with WS and DMD, and another one affected with only WS.
Methods
Blood samples were collected from individuals for genomic DNA isolation. To determine the linkage of this family to the eight known WS loci, microsatellite markers were selected from the candidate regions and used to genotype the family. Exon-specific intronic primers for EDN3 were used to amplify and sequence DNA samples from affected individuals to detect mutations. A mutation in DMD was identified by multiplex PCR and multiplex ligation-dependent probe amplification method using exon-specific probes.
Results
Pedigree analysis suggested segregation of WS as an autosomal recessive trait in the family. Haplotype analysis suggested linkage of the family to the WS4B (EDN3) locus. DNA sequencing identified a novel missense mutation p.T98M in EDN3. A deletion mutation was identified in DMD.
Conclusions
This study reports a novel missense mutation in EDN3 and a deletion mutation in DMD in the same Indian family. The present study will be helpful in genetic diagnosis of this family and increases the mutation spectrum of EDN3.
Introduction
Waardenburg syndrome (WS) is a hereditary auditory-pigmentary syndrome characterized by pigmentary abnormalities of the hair, including a white forelock and premature graying, pigmentary changes of the iris such as heterochromia irides and brilliant blue eyes, lateral displacement of the medial canthi, and lacrimal points, congenital deafness, and intestinal abnormalities [1]. The association of hearing loss, pigmentary abnormalities, and intestinal malformation results from an abnormal proliferation, survival, migration, or differentiation of neural crest derived melanocytes of skin and inner ear, glia, and neurons of the peripheral and enteric nervous system [2]. The incidence of WS is around 1 in 42,000 in the general population. However, it occurs in 5%–6% of deaf individuals and is considered to be the most common autosomal-dominant form of syndromic deafness [3]. It is clinically and genetically heterogeneous and is classified into four types (viz., type I to IV) based on the presence of variable clinical characteristics and additional symptoms. WS type I is characterized by the presence of dystopia canthorum (lateral displacement of inner canthi). WS type II is distinguished from type I by the absence of dystopia canthorum. WS type III patients have limb hypoplasia in addition to dystopia canthorum. WS type IV has an additional feature of Hirschsprung disease [2]. Genetic analysis has identified at least eight loci for WS: WS1/WS3 on chromosome 2q36.1, WS2A on chromosome 3p14.1-p12.3, WS2B on chromosome 1p21-p13.3, WS2C on chromosome 8p23, WS2D on chromosome 8q11.21, WS2E/WS4C on chromosome 22q13.1, WS4A on chromosome 13q22.3, and WS4B on chromosome 20q13.2-q13.3 (OMIM). The genes for six loci are known: WS1/WS3-PAX3 (paired box 3), WS2A-MITF (micropthalmia-associated transcription factor), WS2D-SNAI2 (snail homolog 2, Drosophila), WS2E/WS4C-SOX10 (SRY-box containing gene 10), WS4A-EDNRB (endothelin receptor type B) and WS4B-EDN3 (endothelin 3) [4-12].
Duchenne muscular dystrophy (DMD) is the most common X-linked recessive disease. It is estimated to affect 1 in 3,500 newborn males worldwide [13] and caused by mutations in the DMD gene, encoding a cytoskeletal protein dystrophin. DMD is the largest human gene that spans >2,200 kb of DNA and is composed of 79 exons [14]. Deletions account for approximately 65% of DMD mutations, duplications occur in approximately 6%–10% of cases and the remaining 30%–35% of mutations consist of small deletions, insertions, point mutations or splicing mutations, most of which introduce a premature stop codon [14-23].
Here we report on the genetic analysis of a consanguineous family from the south Indian state of Karnataka with members affected with two disorders: Waardenburg syndrome and Duchenne muscular dystrophy.
Methods
Patients
We ascertained a consanguineous family (Figure 1) with one individual (IV-3) affected with WS and DMD, and the other one (IV-2) affected with only WS from a south Indian state of Karnataka in the Department of Neurology, National Institute of Mental Health and Neuro Sciences, Bangalore. Both affected individuals were examined by Parayil Sankaran Bindu, Arun B. Taly and Sanjib Sinha. A detailed description of their clinical symptoms is given below.
Figure 1.

The haplotype analysis of the family with microsatellite markers from the EDN3 candidate region. The disease haplotype 2–1 is shown by black bars. Note the affected individuals (IV-2 and IV-3) are homozygous for the disease haplotype, whereas both parents III- 1 and III-2 are heterozygous for the disease haplotype and are therefore carriers for the mutation. An arrow marks the index case.
Individual IV-3
The propositus, a 9-year-old boy, was the third child born to consanguineous parents after an uneventful pregnancy and delivery. His developmental milestones were normal except for speech delay. A formal evaluation for the speech delay at the age of three years revealed profound bilateral sensorineural hearing loss. Since the age of six years, he developed gradually progressive difficulty in walking and getting up from sitting position. He had blue iris, faint white forelock, confluent eyebrows, bilateral calf hypertrophy, mild tendon Achilles contracture, exaggerated lumbar lordosis, and a waddling gait. He had symmetric weakness of proximal muscles of all four limbs and truncal muscles. All stretch reflexes were sluggish except for the ankle jerks. Gower’s sign was evident on getting up from the sitting position (Figure 2A). Ophthalmological evaluation showed retina with visible choroidal vessels and depigmented iridis (Figure 2B,G). Waardenburg index was 0.572, thus ruling out dystopia canthorum. Laboratory evaluation showed normal hemogram and routine biochemical parameters. Serum creatine phosphokinase was 3,218 IU/l (n<170 IU/l). Nerve conduction velocity studies that involved right median, ulnar and common peroneal, and sural nerve were normal. Electromyography of right biceps and vastus lateralis revealed a myopathic pattern. Audiometric evaluation showed profound sensorineural hearing loss and absence of bilateral brainstem auditory evoked responses. Magnetic resonance imaging of the brain showed normal myelination pattern. A left quadriceps biopsy revealed marked variation in fiber diameter with evidence of internal nuclei, occasional myophagocytosis, regenerating fiber and splitting, suggestive of muscular dystrophy. On immunohistochemistry with an anti-dystrophin antibody (Novocastra Laboratories, UK), there was absence of staining of dystrophin (Figure 2F). The above clinical symptoms suggested that this patient has both WS and DMD.
Figure 2.
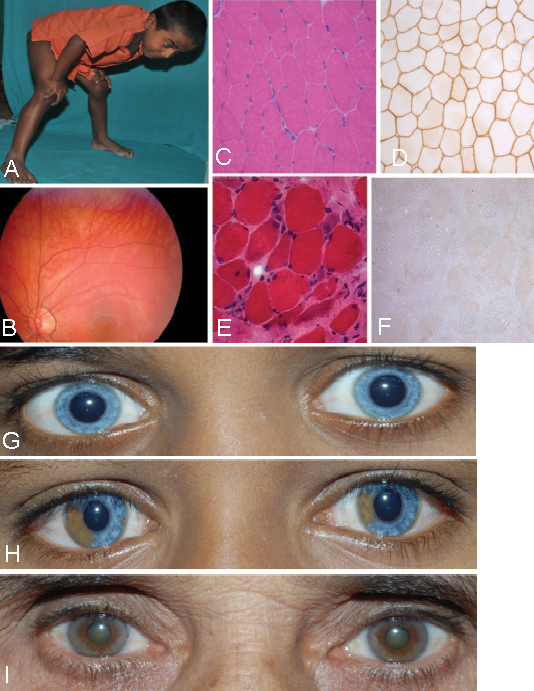
Clinical features of the affected individuals. A: Photograph of affected individual IV-3. Note the classical Gower’s sign on trying to get up from the sitting position. B: Fundus photograph of affected individual IV-3. Note the depigmented retina and underlying choroid vessels. C-F: Transverse sections of skeletal muscle: C and D from an unrelated normal individual and E and F from affected individual IV-3. Note normal polygonal myofibers with peripheral nuclei and uniform diameter in panel C (hematoxylin and eosin staining) and normal positive immunostaining of DMD protein along the sarcolemma in all the fibers in panel D. Note rounding, variation in diameter, central nuclei, regenerating fibers and fibrosis in panel E (hematoxylin and eosin staining), and total absence of DMD staining in all the fibers in panel F. G: Partial facial photograph of affected individual IV-3 showing blue iris. H: Partial facial photograph of affected individual IV-2 showing heterochromia of iris. I: Partial facial photograph of paternal grandmother II-2 showing heterochromia of iris.
Individual IV-2
The 11-year-old elder brother of the index patient had a history of Hirschsprung disease that required surgery at the age of 3 months. He had iris discoloration at birth but did not report any hearing problem till the age of 8 years. On examination, he had heterochromia of iris (Figure 2H) and a normal neurologic examination. Ophthalmological evaluation revealed hypopigmented iris and choroid with visualization of the choroidal vessels. Waardenburg index was 0.4059, thus ruling out dystopia canthorum. Audiometry revealed profound hearing loss on left side with absence of auditory brainstem evoked responses. Laboratory evaluation showed normal creatine kinase levels. Nerve conduction velocity studies and magnetic resonance imaging of the brain were normal. The above clinical symptoms suggested that he has only WS, and it could be WS type IV (WS4).
A detailed clinical examination of their parents, elder sister and paternal grandmother showed heterochromic iris only in the grandmother II-2 (Figure 2I).
Mutation analysis
Following informed consent, three-five milliliter of peripheral blood sample was collected in a Vacutainer EDTATM tube (Beckton-Dickinson, Franklin Lakes, NJ) from each individual for genomic DNA isolation using a Wizard® Genomic DNA Extraction Kit (Promega, Madison, WI). This research followed the tenets of the Declaration of Helsinki and the guidelines of the Indian Council of Medical Research, New Delhi. Although the clinical features in the affected individual IV-2 suggested this family to be of WS type IV, we still went ahead and selected two microsatellite markers from each of the eight WS loci (Table 1) and used them to genotype the family, according to Kumar et al. [24].
Table 1. Microsatellite markers from the candidate regions of eight known WS Loci.
| Locus | Chromosome location | Gene | Marker* |
|---|---|---|---|
| WS1/WS3 | 2q36.1 | PAX3 | D2S2197, D2S2300 |
| WS2A | 3p14.1-p12.3 | MITF | D3S1296, D3S1566 |
| WS2B | 1p21-p13.3 | D1S495, D1S248 | |
| WS2C | 8p23 | D8S561, D8S1819 | |
| WS2D | 8q11.21 | SNAI2 | D8S1716, D8S1745 |
| WS2E/WS4C | 22q13.1 | SOX10 | D22S1045, D22S1156 |
| WS4A | 13q22.3 | EDNRB | D13S1281, D13S160 |
| WS4B | 20q13.2-q13.3 | EDN3 | D20S171, D20S173 |
*Marker order was established using the sequence map from the UCSC Genome Bioinformatics site.
For mutational analysis, the entire coding region of the EDN3 gene (GenBank NM_000114.2) was amplified using primers that amplify all coding exons and their intron-exon junctions (Table 2). Primers were designed using the gene sequence from the UCSC Genome Bioinformatics site. Sequences and PCR conditions of these primers are provided in Table 2. Mutations were identified by sequencing the PCR products from an affected individual from the family on an ABIprism A370-automated sequencer (PE Biosystems, Foster City, CA). Once a mutation was identified, all members of the family were examined for the presence of the mutation by sequencing.
Table 2. Details of PCR primers used in mutation analysis of the EDN3 gene.
| Exon | Primer | Primer sequence (5′ to 3′) | Tm (°C) | Amplicon size (bp) |
|---|---|---|---|---|
| 1* | EN1bF | F:gaaaagcccgagccacagccggc | 64 | 379 |
| EN1bR | R:ccgcgacgcacatcttctccgcg | |||
| 2 | EN2F | F:cagacattttgcttgctccacc | 62 | 528 |
| EN2R | R:caggctctgggctaactgagc | |||
| 3 | EN3F | F:ggcggtggttctcgctccacac | 56 | 372 |
| EN3R | R:caggatgtgactgaactatccta | |||
| 4 | EN4F | F:tggggaacgcactaatgtgctca | 62 | 336 |
| EN4R | R:agaaacggtccaccaaaggcacc | |||
| 5 | EN5F | F:ttccagtctggtggtaggctcg | 56 | 458 |
| EN5R | R:gtattgttaagtggggactctttg |
Abbreviations: F, forward primer; R, reverse primer; bp, base pairs; and Tm, Annealing temperature. * Works with 5% DMSO.
The mutation analysis of the DMD gene was performed using multiplex PCR and multiplex ligation-dependent probe amplification (MLPA) technique and a kit from MRC-Holland, Amsterdam, the Netherlands according to the manufacturer’s recommendations. The kit contains specific probes for each of the 79 exons of the DMD gene on Xp21.2 and thus allows analysis of deletion and/or duplication of one or more of these exons. Briefly, 200 ng DNA was denatured and hybridized overnight at 60 °C with the probe mix. Samples were then treated with Ligase 65 for 15 min at 54 °C. The reactions were stopped by incubation at 98 °C for 5 min. Finally, PCR amplification was performed with the specific SALSA FAM PCR primers. Amplification products were run on an ABI PRISM 3730 Genetic Analyzer (PE Biosystems, Foster City, CA) and the obtained data were analyzed by using the Gene Marker 2.0 Software. Deletions of a probe’s recognition sequence on the X-chromosome will lead to a complete absence of the corresponding probe amplification product in males, whereas female heterozygotes are recognizable by a 35%–50% reduction in relative peak area.
Results and Discussion
The visual inspection of the family suggested segregation of WS as an autosomal recessive trait (Figure 1). Haplotype analysis using microsatellite markers from each of the eight known WS loci suggested linkage of the family to the WS4B locus on chromosome 20q13.2-q13.3 (Figure 1 and Table 1), indicating that WS is caused by a mutation in the EDN3 gene. The genotypes of individuals for microsatellite markers from the other seven WS loci are given in Table 3. To determine the exact nature of the mutation, the entire coding region of EDN3 from the affected individual IV-2 was amplified using 5 sets of PCR primers (Table 2) and sequenced. The results revealed a novel homozygous substitution mutation, c.293C>T in exon 2, changing codon position 98 from threonine to methionine (p.T98M; Figure 3A). The affected sibling IV-3 was also homozygous for the mutation, both parents (III-1 and III-2) and paternal grandmother (II-2) were heterozygous and the normal sibling IV-1 was homozygous for the wild-type allele (Figure 3A). The 528 bp wild-type exon 2 amplicon contains a site for the restriction endonuclease Eco72I, cleaving it into two fragments of 345 bp and 183 bp. The mutation abolishes this restriction site, thus retaining the undigested 528 bp amplicon in affected indivuduals. We therefore developed a restriction fragment length polymorphism (RFLP) analysis to determine if the mutation was present in 50 normal controls. The analysis showed absence of the mutation in 50 ethnically matched controls, further suggesting that the c.293C>T change is a mutation (data not shown). The analysis also showed segregation of the mutation in the family (Figure 3B). In addition, the pathogenic nature of the mutation also comes from the fact that threonine residue at position 98 is conserved in different EDN3 orthologs from human to zebra fish (Figure 3C). According to the Mutation Taster, the mutation is predicted to be disease causing with a p-value (probability) of 0.52. We then used two other in silico methods, PolyPhen-2 and SIFT, to see the effect of this mutation on the protein function. The effect of this mutation on EDN3 was predicted to be probably damaging by PolyPhen-2 with a score of 1 (score ranges from 0 to a positive number, where 0 is neutral, and a high positive number is damaging). SIFT analysis predicted this mutation to be damaging and intolerant with a score of zero (score ranges from 0 to 1, where 0 is damaging and 1 is neutral).
Table 3. Genotypes of individuals for markers from different WS loci.
| Genotype | ||||||
|---|---|---|---|---|---|---|
| Locus | Marker | Individual III-1 | Individual III-2 | Individual IV-1 | Individual IV-2 | Individual IV-3 |
| WS1/WS3 | D2S2197 | 1 2 | 1 2 | 1 1 | 1 1 | 2 2 |
| D2S2300 | 1 1 | 1 2 | 1 1 | 1 1 | 1 2 | |
| WS2A | D3S1296 | 2 3 | 1 2 | 2 3 | 1 3 | 1 2 |
| D3S1566 | 3 4 | 1 2 | 1 3 | 2 3 | 2 4 | |
| WS2B | D1S495 | 1 3 | 2 4 | 1 2 | 2 3 | 1 4 |
| D1S248 | 1 2 | 2 2 | 2 2 | 1 2 | 2 2 | |
| WS2C | D8S561 | 1 1 | 1 2 | 1 1 | 1 1 | 1 2 |
| D8S1819 | 1 1 | 2 3 | 1 2 | 1 2 | 1 3 | |
| WS2D | D8S1716 | 2 2 | 1 3 | 1 2 | 2 3 | 1 2 |
| D8S1745 | 1 2 | 1 2 | 1 2 | 1 1 | 2 2 | |
| WS2E/WS4C | D22S1045 | 2 3 | 1 2 | 1 2 | 1 2 | 1 2 |
| D22S1156 | 2 3 | 1 3 | 2 3 | 2 3 | 2 3 | |
| WS4A | D13S1281 | 2 2 | 1 2 | 1 2 | 1 2 | 1 2 |
| D13S160 | 1 2 | 2 2 | 1 2 | 1 2 | 1 2 | |
Figure 3.

Mutation analysis of the EDN3 gene in the family. A: Sequencing chromatograms of individuals from the family. Note the homozygous change C>T in both affected individuals IV-2 and IV-3 (marked by arrows). The normal sibling IV-1 is homozygous for the wild-type allele, whereas both parents (III-2 and III-3) and grandmother (II-2) are heterozygous for the change (see double peaks marked by arrows). B: RFLP analysis to show segregation of the mutation. Note affected individuals have only 528 bp fragment due to loss of the Eco72I site, the normal sibling has 345 and 183 bp fragments, and both carrier parents have all the three fragments. C: Conservation of the threonine (T) residue in different orthologs. The threonine residues are shown in bold letters. The number refers to the position of amino acid residue.
Multiplex PCR and MLPA analyses of the affected individual IV-3 showed that he has a deletion from exons 49–51 in the DMD gene (Figure 4). MLPA analysis showed absence of the deletion in his mother (III-2), suggesting that it is a de novo mutation (data not shown). His sister (IV-1) also did not have this mutation (data not shown). This deletion causes a complete absence of the DMD protein in muscle tissues (Figure 2F) of the affected individual, and thus causing the phenotype.
Figure 4.

Deletion analysis of the DMD gene. A: Results of multiplex PCR showing the deletion of exons 49, 50 and 51 (marked by arrows) in affected individual IV-3. B: MLPA analysis of the affected individual IV-3 showing the deletion of exons 49, 50 and 51 (marked by arrows). Abbreviation: N, normal unrelated individual.
A total of 10 mutations have been reported so far in the EDN3 gene in WS patients of different ethnic origins [2,9,25-30]. The novel mutation p.T98M reported here, along with two other mutations p.H112R and p.T98K, affect one of the 21 amino acids that constitute active peptide from amino acid positions 97–117 in endothelin-3 [2,29]. Further, with the mutation reported here, the total number of mutations in the EDN3 gene is 11 (Table 4).
Table 4. Mutations reported so far in the EDN3 gene.
| Sl. # | Mutation | Exon | Nature of mutation | State of zygosity | Effect on protein | Ethnic origin of family | Reference |
|---|---|---|---|---|---|---|---|
| 1 | c.163G>T (p.E55X) | 2 | Nonsense | Homozygous | Predicted to result in complete absence of active form of peptide | French | [26] |
| 2 | c.262_263delGCinsT (p.Ala88SerfsX12) | 2 | Frameshift | Homozygous | Predicted to result in complete absence of active form of peptide | 1 French & 1 Bosnian | [2,9] |
| 3 | c.277C>G (p.R93G) | 2 | Missense | Homozygous | Predicted to impair furin-mediated proteolytic cleavage of preproendothelin | Egyptian | [30] |
| 4 | c.286C>T (p.R96C) | 2 | Missense | Homozygous | Predicted to impair furin-mediated proteolytic cleavage of preproendothelin | French | [2] |
| 5 | c.293C>A (p.T98K) | 2 | Missense | Homozygous | Predicted to result in absence of active form of protein | Indian | [2] |
| 6 | c.293C>T (p.T98M) | 2 | Missense | Homozygous | Predicted to result in absence of active form of protein | Indian | Present study |
| 7 | c.335A>G (p.H112R) | 2 | Missense | Homozygous | Could disrupt the formation of disulphide bond | Spanish | [29] |
| 8 | c.380A>G (p.Y127C) | 3 | Missense | Homozygous | Could alter disulphide bond formation in the endothelin and/or the ETlike peptide regions | Indian | [28] |
| 9 | c.476G>T (p.C159F) | 3 | Missense | Homozygous | Presumably results in less efficient cleavage or even complete failure of cleavage of prepro-endothelin | 3 Pakistani | [2,25] |
| 10 | c.507C>A (p.C169X) | 3 | Nonsense | Heterozygous | Prevents the disulphide bond formation and probably generates an inappropriately cleaved, inactive proendothelin | Yugoslavian | [27] |
| 11 | c.517T>C (p.C173R) | 3 | Missense | Compound heterozygosity with p.T98K | Prevents the disulphide bond formation | Indian | [2] |
Endothelins (EDNs) are a family of three active peptides EDN1, END2, and EDN3 that act as ligands and recognized by two G-protein coupled heptahelical receptors known as endothelin receptors, EDNRA and EDNRB. EDN3 preferentially binds the EDNRB [31]. EDN3 is produced as preproendothelin-3 encoded by the EDN3 gene. The biologically active 21 amino acid long EDN3 is produced by proteolytic cleavage of preproendothelin-3 by endopeptidases to yield proendothelin-3, which is finally cleaved by endothelin converting enzyme-1 to produce the mature active EDN3 [32,33]. Studies have shown that EDN3/EDNRB interaction is required for the proper development of neural crest derived melanocytes and enteric neurons [34-36]. Mouse models for homozygous mutations in the EDNRB or EDN3 genes show pigmentation anomaly, aganglionic megacolon and cochlear disorder [31,37]. We suggest that the disease phenotype in the present family could be due to the absence of the active form of the protein as the mutation p.T98M might impair the EDN3 activity. Although both parents (III-1 and III-2) and grandmother (II-2) are heterozygous for the mutation (Figure 3), only the grandmother showed heterochromic iris (Figure 2I). However, it is not completely unexpected as heterozygous individuals for the EDN3 mutations have been reported to show a few clinical features of WS [25,29]. The manifestation of a few of the WS clinical symptoms in heterozygous individuals could be due to environmental factors, multigenic inheritance, modifier genes or stochastic events on cell fate or cell differentiation in early embryogenesis [25,27].
In summary, we report an interesting Indian family with members affected with two different disorders, WS and DMD. The WS is caused by a novel missense mutation, whereas DMD resulted due to a deletion. The present information will be useful to provide rapid prenatal diagnosis and genetic counseling for WS to the family and their relatives using a PCR-RFLP method developed during the study.
Acknowledgments
We thank the patients and their family members for their involvement in the study. We also thank Dr. Robert Church and two anonymous reviewers for their suggestions to improve the manuscript.
References
- 1.Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951;3:195–253. [PMC free article] [PubMed] [Google Scholar]
- 2.Pingault V, Dorothee E, Florence D, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 3.Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34:656–65. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tassabehji M, Read AP, Newton VE, Harris R, Balling R, Gruss P, Strachan T. Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature. 1992;355:635–6. doi: 10.1038/355635a0. [DOI] [PubMed] [Google Scholar]
- 5.Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–5. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- 6.Milunsky A, Lipsky N, Sheffer R, Zlotogora J, Baldwin C. A mutation in the Waardenburg syndrome (WS-I) gene in a family with 'WS-III'. Am J Hum Genet. 1992;51(suppl.):A222. [Google Scholar]
- 7.Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am J Hum Genet. 1993;52:455–62. [PMC free article] [PubMed] [Google Scholar]
- 8.Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, Chakravarti A. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell. 1994;79:1257–66. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 9.Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, Martelli H, Bidaud C, Munnich A, Lyonnet S. Mutation of the endothelin-3 gene in Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet. 1996;12:442–4. doi: 10.1038/ng0496-442. [DOI] [PubMed] [Google Scholar]
- 10.Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu MO, Puliti A, Herbarth B, Hermans-Borgmeyer I, Legius E, Matthijs G, Amiel J, Lyonnet S, Ceccherini I, Romeo G, Smith JC, Read AP, Wegner M, Goossens M. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18:171–3. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 11.Sánchez-Martín M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet. 2002;11:3231–6. doi: 10.1093/hmg/11.25.3231. [DOI] [PubMed] [Google Scholar]
- 12.Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A, Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M, Pingault V. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet. 2007;81:1169–85. doi: 10.1086/522090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emery AEH. Population frequencies of inherited neuromuscular diseases-a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 14.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 15.Kilimann MW, Pizzuti A, Grompe M, Caskey CT. Point mutations and polymorphisms in the human dystrophin gene identified in genomic DNA sequences amplified by m-PCR. Hum Genet. 1992;89:253–8. doi: 10.1007/BF00220535. [DOI] [PubMed] [Google Scholar]
- 16.Roberts RG, Bobrow M, Bentley DR. Point mutations in dystrophin gene. Proc Natl Acad Sci USA. 1992;89:2331–5. doi: 10.1073/pnas.89.6.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prior TW, Papp AC, Snyder PJ, Burghes AHM, Bartolo C, Sedra MS, Western LM, Mendell JR. A missense mutation in the dystrophin gene in a Duchenne muscular dystrophy patient. Nat Genet. 1993;4:357–60. doi: 10.1038/ng0893-357. [DOI] [PubMed] [Google Scholar]
- 18.Nigro V, Nigro G, Esposito MG, Comi LI, Molinari AM, Puca GA, Politano L. Novel small mutations along the DMD/BMD gene associated with different phenotypes. Hum Mol Genet. 1994;3:1907–8. doi: 10.1093/hmg/3.10.1907. [DOI] [PubMed] [Google Scholar]
- 19.Kneppers ALJ, Deutz-Terlouw PP, den Dunnen JT, van Ommen GJB, Bakker E. Point mutation screening for 16 exons of the dystrophin gene by multiplex single strand conformation polymorphism analysis. Hum Mutat. 1995;5:235–42. doi: 10.1002/humu.1380050308. [DOI] [PubMed] [Google Scholar]
- 20.Tuffery S, Bareil C, Demaille J, Claustres M. Four novel dystrophin point mutations: detection by the protein truncation test and transcript analysis in lymphocytes from Duchenne muscular dystrophy patients. Eur J Hum Genet. 1996;4:143–52. doi: 10.1159/000472188. [DOI] [PubMed] [Google Scholar]
- 21.Sitnik R, Campiotto S, Vainzof M, Pavanello RC, Takata RI, Zatz M, Passos-Bueno MR. Novel point mutations in the dystrophin gene. Hum Mutat. 1997;10:217–22. doi: 10.1002/(SICI)1098-1004(1997)10:3<217::AID-HUMU7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 22.Eraslan S, Kayserili H, Apak MY, Kirdar B. Identification of point mutations in Turkish DMD/BMD families using multiplex-single stranded conformation analysis (SSCA). Eur J Hum Genet. 1999;7:765–70. doi: 10.1038/sj.ejhg.5200370. [DOI] [PubMed] [Google Scholar]
- 23.Wibawa T, Takeshima Y, Mitsuyoshi I, Wada H, Surono A, Nakamura H, Matsuo M. Complete skipping of exon 66 due to novel mutations of the dystrophin gene was identified in two Japanese families of Duchenne muscular dystrophy with severe mental retardation. Brain Dev. 2000;22:107–12. doi: 10.1016/s0387-7604(99)00126-6. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Becker LA, Depinet TW, Haren JM, Kurtz CL, Robin NH, Cassidy SB, Wolf DJ, Schwartz S. Molecular characterization and delineation of subtle deletions in de novo “balanced” chromosomal rearrangements. Hum Genet. 1998;103:173–8. doi: 10.1007/pl00008706. [DOI] [PubMed] [Google Scholar]
- 25.Hofstra RM, Osinga J, Tan-Sindhunata G, Wu Y, Kamsteeg EJ, Stulp RP, van Ravenswaaij-Arts C, Majoor-Krakauer D, Angrist M, Chakravarti A, Meijers C, Buys CH. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat Genet. 1996;12:445–7. doi: 10.1038/ng0496-445. [DOI] [PubMed] [Google Scholar]
- 26.Bidaud C, Salomon R, Van Camp G, Pelet A, Attie T, Eng C, Bonduelle M, Amiel J, Nihoul-Fekete C, Willems PJ, Munnich A, Lyonnet S. Endothelin-3 gene mutations in isolated and syndromic Hirschsprung disease. Eur J Hum Genet. 1997;5:247–51. [PubMed] [Google Scholar]
- 27.Pingault V, Bondurand N, Lemort N, Sancandi M, Ceccherini I, Hugot JP, Jouk PS, Goossens M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: is there a dosage effect of EDN3/EDNRB gene mutations on neurocristopathy phenotypes? J Med Genet. 2001;38:205–9. doi: 10.1136/jmg.38.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pingault V, Girard M, Bondurand N, Dorkins H, Van Maldergem L, Mowat D, Shimotake T, Verma I, Baumann C, Goossens M. SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum Genet. 2002;111:198–206. doi: 10.1007/s00439-002-0765-8. [DOI] [PubMed] [Google Scholar]
- 29.Viñuela A, Morin M, Villamar M, Morera C, Lavilla MJ, Cavalle L, Moreno-Pelayo MA, Moreno F, del Castillo I. Genetic and phenotypic heterogeneity in two novel cases of Waardenburg syndrome type IV. Am J Med Genet. 2009;149A:2296–302. doi: 10.1002/ajmg.a.33026. [DOI] [PubMed] [Google Scholar]
- 30.Shamseldin HE, Rahbeeni Z, Alkuraya FS. Perturbation of the consensus activation site of endothelin-3 leads to Waardenburg syndrome type IV. Am J Med Genet. 2010;152A:1841–3. doi: 10.1002/ajmg.a.33123. [DOI] [PubMed] [Google Scholar]
- 31.Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–85. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 32.Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, Masaki T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci USA. 1989;86:2863–7. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. ECE-1: A membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–85. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- 34.Shin MK, Levorse JM, Ingram RS, Tilghman SM. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature. 1999;402:496–501. doi: 10.1038/990040. [DOI] [PubMed] [Google Scholar]
- 35.Lee HO, Levorse JM, Shin MK. The endothelin receptor-B is required for the migration of neural crest-derived melanocyte and enteric neuron precursors. Dev Biol. 2003;259:162–75. doi: 10.1016/s0012-1606(03)00160-x. [DOI] [PubMed] [Google Scholar]
- 36.Nagy N, Goldstein AM. Endothelin-3 regulates neural crest cell proliferation and differentiation in the hindgut enteric nervous system. Dev Biol. 2006;293:203–17. doi: 10.1016/j.ydbio.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 37.Tachibana M, Kobayashi Y, Matsushima Y. Mouse models for four types of Waardenburg syndrome. Pigment Cell Res. 2003;16:448–54. doi: 10.1034/j.1600-0749.2003.00066.x. [DOI] [PubMed] [Google Scholar]