

Abstract
The prefrontal cortex (PFC), a region responsible for high-order cognitive functions, such as decision-making, attention and working memory, is highly influenced by stress and corticosteroid stress hormones. Recently it has been shown that acute stress affects PFC functions by potentiating glutamatergic transmission via a mechanism dependent on glucocorticoid receptor (GR) and its downstream target, serum and glucocorticoid-inducible kinase (SGK). To identify the key regulators of stress responses, we examined the role of histone deacetylase 6 (HDAC6), a unique member of the HDAC family that could regulate the GR chaperone protein heat shock protein 90 (HSP90), in the synaptic action of acute stress in PFC. We found that HDAC6 inhibition or knockdown blocked the enhancement of glutamatergic transmission and glutamate receptor trafficking by acute stress in vivo or corticosterone treatment in vitro. In addition, HDAC6 inhibition blocked the up-regulation of SGK in animals exposed to acute stress. HSP90 inhibition or knockdown produced a similar blockade of the acute stress-induced enhancement of glutamatergic signalling. These findings have identified HDAC6 as a key molecule gating the effects of acute stress on synaptic functions in the PFC.
Key points
Acute stress, via glucocorticoid receptors (GRs), enhances glutamatergic signalling in the prefrontal cortex, a region responsible for high-order cognitive functions.
We found that inhibition or knockdown of histone deacetylase 6 (HDAC6) blocked the enhancement of glutamatergic signalling by acute stress.
Inhibition or knockdown of the GR chaperone protein HSP90 (a substrate of HDAC6) produced a similar blockade of the acute stress-induced enhancement of glutamatergic signalling.
These results suggest that HDAC6 is a key molecule regulating the synaptic effects of acute stress in the prefrontal cortex.
Introduction
When confronted with stress, the brain activates many neural networks and hormonal mediators to adapt to the disturbance (McEwen, 2007; Joëls & Baram, 2009). Activation of the hypothalamic–pituitary–adrenal (HPA) axis and subsequent release of adrenal cortisol is one such network and plays a major role in the stress response (de Kloet et al. 2005). The acute response to stress is necessary for survival and adaptation. However, repeated exposure to stress can be harmful and lead to the dysregulation of neuronal circuits (de Kloet et al. 2005; McEwen, 2007).
The prefrontal cortex (PFC), a brain region responsible for high-order cognitive functions such as decision-making, attention and working memory, is one of the key targets of corticosteroid stress hormones (Liston et al. 2006; Cerqueira et al. 2007; Popoli et al. 2011). We have found that acute stress, via the activation of glucocorticoid receptor (GR) and its downstream immediate early gene, serum and glucocorticoid-inducible kinase (SGK), causes a delayed and sustained potentiation of glutamatergic signalling in the PFC, therefore facilitating working memory (Yuen et al. 2009, 2011). In this study, we set out to determine what could regulate the GR signalling and stress response.
In the absence of ligand, GR is confined in the cytoplasm where it complexes with heat shock protein 90 (HSP90) and other co-chaperones (Cadepond et al. 1991). The association of GR with HSP90 facilitates its maturation and stabilizes it in a conformation required for high affinity ligand binding. Upon binding to ligand, GR is released from the HSP90 chaperone complex and translocates to the nucleus to initiate gene transcription (Dittmar et al. 1997; Pratt & Toft, 2003). Previous studies have identified histone deacetylase 6 (HDAC6) as a key regulator of HSP90 (Bali et al. 2005; Kovacs et al. 2005). Hyperacetylation of HSP90 from inactivation of HDAC6 disrupts the chaperone function of HSP90, resulting in a non-functional GR that is unable to bind ligand and so unable to translocate to the nucleus and activate transcription (Bali et al. 2005; Kovacs et al. 2005). Thus, HDAC6 deacetylation of HSP90 is critical for maintaining the HSP90 cochaperone complex and its stabilizing association with GR (Aoyagi & Archer, 2005).
Emerging evidence suggests that HDAC family proteins, which generally exert their epigenetic regulation of gene transcription via chromatic remodelling, play a pivotal role in cognitive and emotional processes (Tsankava et al. 2007). HDAC6 is a unique member in the HDAC family because it actively resides in the cytoplasm (Verdel et al. 2000) and has limited substrates (Hubbert et al. 2002; Kovacs et al. 2005; Valenzuela-Fernandez et al. 2008). HDAC6 contains two functional deacetylase regions followed by a ubiquitin-binding domain at the C-terminus, and its regulation of cytoskeletal processes and protein folding has implicated HDAC6 as a potential therapeutic target for various neurodegenerative diseases (Pandey et al. 2007; Chuang et al. 2009). HDAC6 has also been shown to have neuroprotective effects and promote neuronal survival and regeneration after injury (Rivieccio et al. 2009). In this study, we demonstrate for the first time that HDAC6 activity is important for gating the acute stress-induced synaptic potentiation in vivo, suggesting that HDAC6 may be a key controller of neuronal adaptations to acute stress.
Methods
Animals and reagents
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the State University of New York at Buffalo, and our animal care procedures were in accordance with the IACUC guidelines under the Animal Welfare Act. Young Sprague–Dawley (SD) rats (p21–27) were group-housed in cages with a light–dark cycle of 12 h. Animals were exposed to a 20 min forced swim stress (in 25°C water) as previously described (Yuen et al. 2009, 2011). After stress exposure, rats were anaesthetized with halothane vapour and immediately killed for brain slicing.
Pharmacological agents used include: corticosterone, trichostatin A, trapoxin A (all from Sigma-Aldrich), tubacin (gift from Ralph Mazitschek and Stuart Schreiber, Harvard University, Cambridge, MA, USA), geldanamycin (Tocris Bioscience, Ellisville, MO, USA), 17-DMAG (InvivoGen, San Diego, CA, USA).
Animal surgery
For viral expression in vivo, rats (P20–21) were first anaesthetized by s.c. injection of buprenorphine (0.05 mg kg−1), followed by i.p. injection of ketamine (70 mg kg−1) and xylazine (5 mg kg−1). Rats were then placed on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). Lentivirus was injected with a Hamilton syringe (needle gauge 31) at a speed of ∼0.2 μl min−1, and the needle was kept in place for an additional 5 min. Lentivirus was delivered bilaterally to prefrontal cortex using the following coordinates: 2.5 mm anterior to bregma; 0.75 mm lateral; 2.5 mm dorsal to ventral. The needle was extended to a depth 3.5 mm below the tissue surface and a 1–2 μl volume was injected into each side of the PFC. After surgery, all rats were monitored at least every 15 min until fully awake and ambulatory. For post-operative care, neomycin was applied topically to the incision site and carprofen (5 mg kg−1) i.p. injected once per day at the first and second day post-surgery.
ShRNA lentiviral knockdown
The shRNA oligonucleotide targeting rat HDAC6 sequence (AAAGCAAAGACAGCTAAGGCA (Rivieccio et al. 2009) was inserted to the lentiviral vector pLKO.3G (Addgene, Cambridge, MA, USA), which contains an enhanced green fluorescent protein (eGFP) marker. For the production of lentiviral particles, a mixture containing the pLKO.3G shRNA plasmid (against HDAC6), psPAX2 packaging plasmid and pMD2.G envelope plasmid (Addgene) was transfected to HEK-293FT cells using Lipofectmine 2000. The transfection reagent was removed 12–15 h later, and cells were incubated in fresh Dulbecco's modified Eagle's medium (DMEM) (containing 10% fetal bovine serum (FBS) + penicillin/streptomycin) for 24 h. The medium harvested from the cells, which contained lentiviral particles, was concentrated by centrifugation (2000 g, 20 min) with Amicon Ultra Centrifugal Filter (Ultracel-100K, Millipore). The concentrated virus was stored at –80°C. To test the knockdown effect, rat cortical cultures were infected with HDAC6 shRNA lentivirus on DIV15–17 and used for immunoblotting with anti-HDAC6 (1:500) on DIV21–23.
The shRNA oligonucleotide targeting rat HSP90α sequence (GCAAAGAAACACCTGG AGATA, Open Biosystem) was inserted in the lentiviral vector pLKO.3G (Addgene). The full-length open reading frame of HSP90 was amplified from rat brain cDNA by PCR, and a Flag tag was added to the N-terminal in frame. The PCR product was cloned to T/A vector, and then subcloned to pcDNA3.1 expression vector. The construct was verified by DNA sequencing. To test the knockdown effect, the plasmid FlagHSP90 was transfected to HEK293 cells with HSP90 shRNA plasmid. Two days after transfection, the cells were harvested and subjected to Western blotting with anti-Flag (1:1000). Actin was used as a loading control.
Electrophysiological recording in slices and cultures
All the recordings were performed on layer V pyramidal neurons in the medial PFC (prelimbic region). Synaptic currents were measured using the whole-cell voltage clamp recording technique as previously described (Yuen et al. 2009, 2011). Patch pipettes were filled with internal solution (in mm) containing: 130 Cs methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 5 EGTA, 10 Hepes, 2 QX314, 12 phosphocreatine, 5 MgATP, 0.5 Na2GTP, 0.2 leupeptin, pH 7.2–7.3, 265–270 mOsm. PFC slices were continuously perfused with oxygenated artificial cerebrospinal fluid (ACSF) containing bicuculline (10 μm) and d-APV (25 μm). Recordings were obtained with a Multiclamp 700A amplifier and Digidata 1320A data acquisition system (Axon Instruments). Membrane potential was held at –70 mV. Evoked excitatory postsynaptic currents (EPSCs) were generated with a pulse from a stimulation isolation unit controlled by a S48 pulse generator (Grass Technologies, West Warwick, RI, USA). A bipolar stimulating electrode (FHC Inc., Bowdoin, ME) was positioned ∼100 μm from the patched neuron. Recordings from control vs. stressed animals were interleaved throughout the course of all experiments. Data analyses were performed with Clampfit (Axon Instruments) and KaleidaGraph (Synergy Software, Reading, PA, USA).
Neuronal cultures were prepared from PFC tissue dissected from 18-day rat embryos as previously described (Liu et al. 2010; Yuen et al. 2011). Miniature EPSCs in cultured PFC neurons were recorded with the same internal solution used for recording evoked AMPAR-EPSCs in slices. The external solution contained (mM): 127 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 12 glucose, 10 Hepes, 0.001 TTX, pH 7.3–7.4, 300–305 mosM. Bicuculline (10 μm) and d-APV (25 μm) were added to block GABAAR and NMDAR activation. The membrane potential was held at –70 mV. Synaptic currents were analysed with Mini Analysis Program (Synaptosoft, Inc., Decatur, GA, USA).
Biochemical measurement of surface and total proteins
The surface GluR1 and GluR2 receptors were detected as previously described (Yuen et al. 2009). In brief, PFC slices were incubated with ACSF containing 1 mg ml−1 sulfo-N-hydroxysuccinimide-LC-biotin (Pierce Chemical Co., Rockford, IL, USA) for 20 min on ice. The slices were then rinsed three times in Tris-buffered saline to quench the biotin reaction, followed by homogenization in modified radioimmunoprecipitation assay buffer. The homogenates were centrifuged at 14,000 g for 15 min at 4°C. Protein (15 μg) was removed to measure total GluR1 and GluR2. For surface protein, 150 μg of protein was incubated with 100 μl of 50% NeutrAvidin Agarose (Pierce Chemical Co.) for 2 h at 4 °C, and bound proteins were resuspended in 25 μl of SDS sample buffer and boiled. Quantitative Western blots were performed on both total and biotinylated (surface) proteins using antibodies against GluR1 (1:200, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), GluR2 (1:500, Millipore, Billerica, MA, USA), and actin (1:1000, Santa Cruz). In some experiments, Western blotting was performed with anti-SGK1 (1:500; Abcam Inc., Cambridge, MA, USA) or anti-SGK3 (1:500; Abcam).
Statistics
Statistical significance between different treatment groups was analysed using student's unpaired t test or analysis of variance (ANOVA) with Tukey's post hoc test.
Results
HDAC6 regulates the acute stress-induced enhancement of synaptic glutamatergic currents
To test the involvement of HDAC6 in regulating the effects of acute stress in PFC pyramidal neurons in vivo, we injected (i.p.) SD rats (21–27 days old) with various HDAC inhibitors. One hour after the injection, rats were exposed to a 20 min forced-swim test, a well-established protocol for inducing behavioural stress (Yuen et al. 2009). Synaptic currents in the medial PFC of brain slices were measured at 1–6 h post-stress. As shown in Fig. 1A, acute stress caused a substantial enhancement of the input/output curves of AMPAR-EPSC induced by a series of stimulus intensities (80–120% increase, P < 0.001, ANOVA, n = 10), while in animals injected with trichostatin A (TSA, 0.5 mg kg−1), a pan HDAC inhibitor permeant to the blood–brain barrier (Yoshida et al. 1990; Chuang et al. 2009), the enhancing effect of acute stress was eliminated (<20% increase, P > 0.05, ANOVA, n = 11). Similarly, as shown in Fig. 1B, a significant main effect was found on AMPAR-EPSC evoked at a single intensity after exposure to acute stress (F7,74= 14.5, P < 0.001, ANOVA). Post hoc analysis indicated that injection of TSA blocked the stress-induced enhancement of AMPAR-EPSC evoked at a single intensity (vehicle: 95.5 ± 7.1 pA, n = 13; stress: 195.7 ± 19.5 pA, n = 10, P < 0.001, ANOVA; TSA: 72.1 ± 7.7 pA, n = 10; TSA+stress: 75.0 ± 6.2 pA, n = 7, P > 0.05, ANOVA), while injection of Trapoxin A (TpxA, 0.3 μg kg−1), a reagent that selectively inhibits HDACs except for HDAC6 at low doses (Furumai et al. 2001), failed to do so (Fig. 1B; TpxA: 74.8 ± 8.5 pA, n = 7; TpxA+stress: 161.8 ± 21.1 pA, n = 10, P < 0.001, ANOVA).
Figure 1. HDAC6 inhibition blocks the enhancement of AMPA receptor-mediated current by acute stress or corticosterone treatment.
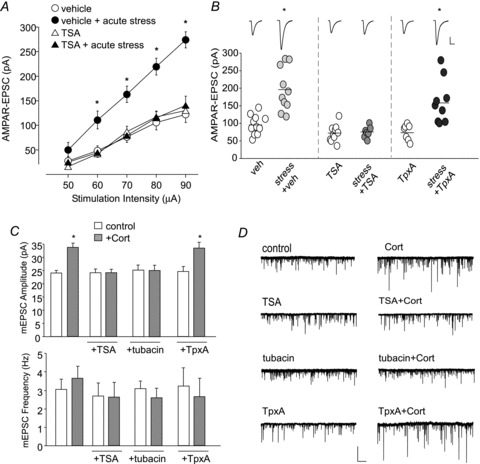
A, input–output curves of AMPAR-EPSC evoked by a series of stimulus intensities in PFC pyramidal neurons from control vs. animals exposed to a forced-swim stress with or without i.p. injection of trichostatin A (TSA, a pan-HDAC inhibitor, injected at 1 h prior to stress). *P < 0.01, ANOVA. B, dot plots showing the amplitude of AMPAR-EPSC evoked by the same stimulus in PFC pyramidal neurons from control vs. stressed animals with or without i.p. injection of TSA (0.5 mg kg−1) or trapoxin A (TpxA, 0.3 μg kg−1, a non-HDAC6 inhibitor). *P < 0.01, ANOVA. Inset: representative synaptic current traces. Scale bar: 50 pA, 20 ms. C, bar graphs (mean ± SEM) showing the mEPSC amplitude and frequency in cultured cortical neurons treated with DMSO or corticosterone (20 min; 100 nm) in the absence or presence of tubacin (20 μm, a specific HDAC6 inhibitor), TSA (1 μm) or TpxA (10 nm). *P < 0.01, ANOVA. D, representative mEPSC traces. Scale bar: 50 pA, 2 s.
The effect of acute behavioural stress can be mimicked by a brief treatment with the stress hormone corticosterone (Liu et al. 2010; Yuen et al. 2011), so we examined the role of HDAC6 in corticosterone-induced changes in glutamatergic responses in vitro. Cortical cultures were treated with various HDAC inhibitors for 1 h prior to a short corticosterone exposure (20 min, 100 nm). Miniature excitatory postsynaptic current (mEPSC), a synaptic response resulting from quantal release of single glutamate vesicles, was recorded 1–6 h afterwards. As shown in Fig. 1C and D, a significant main effect was found on mEPSC amplitude after corticosterone (CORT) treatment (F7,81= 6.8, P < 0.001, ANOVA, control: 24.1 ± 1.0 pA, n = 15; CORT: 33.4 ± 1.6 pA, n = 16, P < 0.001), while the frequency remained unchanged (control: 3.1 ± 0.6 Hz, n = 15; CORT: 3.7 ± 0.7 Hz, n = 16, P > 0.05). The enhancing effect of corticosterone on mEPSC amplitude was blocked by TSA (1 μm, TSA: 24.1 ± 1.4 pA, n = 8; TSA+CORT: 24.1 ± 1.4 pA, n = 10, P > 0.05), as well as by tubacin (20 μm), a selective HDAC6 inhibitor (Haggarty et al. 2003, tubacin: 25.3 ± 1.9 pA, n = 8; tubacin+CORT: 25.1 ± 2.1 pA, n = 11, P > 0.05). TpxA (10 nm), a non-HDAC6 inhibitor at the dose used (Furumai et al. 2001), failed to block this enhancement (TpxA: 24.6 ± 1.9 pA, n = 7; TpxA+CORT: 33.6 ± 1.9 pA, n = 7, P < 0.01). TSA, tubacin or TpxA alone did not significantly change mEPSC amplitude or frequency.
To further confirm the specific involvement of HDAC6 in regulating corticoserone-induced enhancement of glutamatergic responses, we knocked down HDAC6 expression in cultured cortical neurons using a shRNA against HDAC6 (Fig. 2A). Neurons transfected with HDAC6 shRNA were treated with DMSO or corticosterone as described above. As shown in Fig. 2B–D, HDAC6 knockdown blocked the corticosterone enhancement of mEPSC amplitude (F3,34= 10.4, P < 0.001, ANOVA, green fluorescent protein (GFP): 25.6 ± 0.9 pA, n = 8, GFP+CORT: 34.1 ± 1.9 pA, n = 10, P < 0.01; HDAC6 shRNA: 27.2 ± 0.9 pA, n = 8; HDAC6 shRNA+CORT: 25.0 ± 1.1 pA, n = 9, P > 0.05). The mEPSC frequency did not significantly change in any of the conditions (GFP: 3.1 ± 0.6 Hz, GFP+CORT: 4.5 ± 1.2 Hz; HDAC6 shRNA: 4.3 ± 0.9 Hz, HDAC6 shRNA+CORT: 3.4 ± 0.8 Hz, P > 0.05, ANOVA).
Figure 2. HDAC6 knockdown blocks the enhancement of glutmatergic signalling by corticosterone treatment or acute stress.
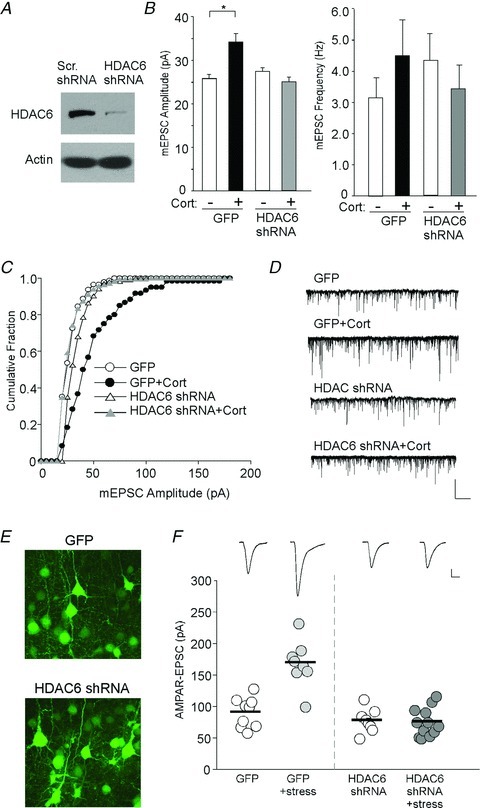
A, immunoblots of HDAC6 expression in cortical cultures infected with HDAC6 shRNA lentivirus or a scrambled control shRNA lentivirus. B, bar graphs (mean ± SEM) showing the mEPSC amplitude and frequency in GFP- or HDAC6 shRNA-transfected cortical cultures treated with DMSO or corticosterone (20 min, 100 nm). *P < 0.01, ANOVA. C and D, cumulative plots of mEPSC amplitude distribution and mEPSC traces in representative neurons under different conditions. Scale bar: 50 pA, 2 s. E, confocal images of neurons from animals with stereotaxic injection of GFP or HDAC6 shRNA lentivirus to the PFC region. F, dot plots showing the amplitude of AMPAR-EPSC in PFC pyramidal neurons from control vs. stressed animals with stereotaxic injection of GFP or HDAC6 shRNA lentivirus (injected at 1 week prior to stress). *P < 0.01, ANOVA. Inset: representative synaptic current traces. Scale bar: 50 pA, 20 ms.
To confirm the specific involvement of HDAC6 in regulating the effects of acute stress, we knocked down HDAC6 expression in vivo by stereotaxic injection of HDAC6 shRNA lentivirus into the PFC. Animals were exposed to acute stress 1 week after the viral injection to allow for sufficient knockdown. The GFP-labelled neurons, which should also express HDAC6 shRNA, exhibited normal morphological structures (Fig. 2E). As shown in Fig. 2F, acute stress significantly enhanced AMPAR-EPSC amplitude in animals injected with GFP lentivirus (F4,41= 16.5, P < 0.001, ANOVA, GFP: 91.7 ± 7.9 pA, n = 9; GFP+stress: 169.1 ± 13.2 pA, n = 8, P < 0.01), but failed to cause the enhancement in animals injected with HDAC6 shRNA lentivirus (HDAC6 shRNA: 78.6 ± 6.7 pA, n = 8; HDAC6 shRNA+stress: 76.1 ± 6.4 pA, n = 12, P > 0.05). In the non-infected neurons (GFP negative) from stressed animals injected with HDAC6 shRNA, a significant enhancement in AMPAR-EPSC amplitude was observed (188 ± 35 pA, n = 5, P < 0.001).
Our previous studies have found that NMDAR-mediated synaptic responses are also regulated by acute stress (Yuen et al. 2009, 2011), so we examined the involvement of HDAC6 in NMDAR regulation. As shown in the online Supplemental Material, Fig. S1, a significant main effect was found on NMDAR-EPSC after exposure to acute stress (F5,55= 22.4, P < 0.001, ANOVA). Post hoc analysis indicated that the stress-induced enhancement of NMDAR-EPSC was blocked by TSA, but not TpxA (vehicle: 176.2 ± 13.2 pA, n = 9; stress: 348.3 ± 25.3 pA, n = 11, P < 0.001, ANOVA; TSA: 172.8 ± 11.3 pA, n = 9; TSA+stress: 170.1 ± 11.2 pA, n = 10, P > 0.05, ANOVA; TpxA: 189.3 ± 11.6 pA, n = 9; TpxA+stress: 351.8 ± 31.3 pA, n = 8, P < 0.001, ANOVA). Taken together, these results suggest that HDAC6 is required for the acute stress/corticosterone-induced potentiation of glutamatergic transmission.
HDAC6 regulates the acute stress-induced enhancement of glutamate receptor surface expression
Our previous studies have shown that the enhancement of glutamatergic transmission by acute stress results from increased trafficking of glutamate receptors to the surface (Yuen et al. 2009, 2011). So we performed surface biotinylation and immunoblotting experiments to detect the surface and total levels of AMPAR subunits in control vs. stressed animals injected with HDAC6 inhibitors. As shown in Fig. 3A, acute stress increased the surface expression of GluR1 (3.3 ± 1.1-fold relative to control, n = 3, P < 0.01) and GluR2 (3.8 ± 0.9-fold relative to control, n = 6, P < 0.01), and this effect was completely blocked by injection with TSA (surface GluR1: 1.3 ± 0.3-fold relative to control, n = 4; surface GluR2: 1.2 ± 0.23-fold relative to control, n = 5, P > 0.05). It suggests that HDAC6 is required for the acute stress-induced enhancement of AMPAR surface expression.
Figure 3. Inhibition of HDAC6 blocks the up-regulation of surface and synaptic glutamate receptors by acute stress or corticosterone treatment.
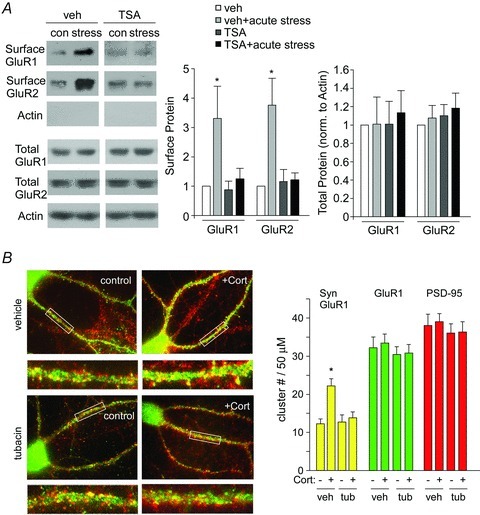
A, immunoblots and quantification analysis of surface and total GluR1 and GluR2 in lysates of PFC slices from control vs. stressed animals with or without TSA injection (0.5 mg kg−1, i.p.). *P < 0.01, ANOVA. B, immunocytochemical images and quantitative analysis of synaptic GluR1 (PSD-95 colocalized, yellow puncta), total GluR1 clusters (green puncta) and PSD-95 clusters (red puncta) along dendrites of PFC cortical cultures treatment with DMSO or corticosterone (20 min, 100 nm) in the absence or presence of tubacin (20 μm, added 1 h beforehand). *P < 0.01, ANOVA.
Because surface glutamate receptors could be synaptic and extra-synaptic, we also examined whether HDAC6 could regulate CORT-induced changes in AMPARs at synapses. To test this, cultured PFC neurons were treated with tubacin (20 μm) for 1 h prior to a short exposure to corticosterone (100 nm, 20 min). Three hours after washing off corticosterone, immunostaining was performed to detect synaptic AMPAR clusters along dendrites (GluR1 colocalized with the postsynaptic marker PSD-95). As shown in Fig. 3B, a significant main effect was found on synaptic GluR1 cluster density (no. of clusters per 50 μm dendrite) after corticosterone treatment (F3,71= 7.1, P < 0.001, ANOVA). Post hoc analysis showed that corticosterone-induced increase in the density of synaptic GluR1 clusters (yellow puncta) was blocked by tubacin (control: 12.3 ± 1.3, n = 18; CORT: 22.1 ± 1.8, n = 16, P < 0.01; tubacin: 12.7 ± 1.9, n = 19; tubacin+CORT: 13.9 ± 1.5, n = 19; P > 0.05, ANOVA). The number of total GluR1 (green puncta) and PSD-95 (red puncta) clusters was not significantly changed in any of the treatment conditions. The requirement of HDAC6 for the CORT-induced increase in AMPARs at the synaptic membrane is consistent with our electrophysiological observations.
Given the involvement of HDAC6 in the effect of stress on glutamatergic signalling, we further examined whether acute stress might alter HDAC6 expression or activity. As shown in Fig. S2, there was no significant difference on the level of HDAC6 in control vs. stressed animals (95 ± 10% of control, n = 6 pairs; P > 0.5, t test). The level of acetylated tubulin, a readout for HDAC6 activity, was also not significantly changed by stress (99 ± 7% of control, n = 4 pairs; P > 0.05, t test). It suggests that HDAC6 is constitutively active, and is unchanged by acute stress.
HDAC6 regulates the acute stress-induced SGK upregulation
Serum and glucocorticoid inducible kinases (SGKs) are a family of immediate early genes whose transcription is induced by GR (Lang et al. 2006). We have previously shown that the acute stress-induced potentiation of glutamatergic responses and glutamate receptor surface expression occurs through a mechanism dependent on the upregulation of SGK1/3 (Liu et al. 2010; Yuen et al. 2011). Thus, we examined whether HDAC6 could influence the effect of acute stress on SGK expression by injecting HDAC6 inhibitors to the animals. As shown in Fig. 4, a significant main effect was seen on SGK1 (F5,27= 9.8, P < 0.001, ANOVA) and SGK3 (F5,27= 11.1, P < 0.001, ANOVA) expression in vehicle-injected rats by exposure to a 20 min forced-swim stress (SGK1: 2.2 ± 0.18-fold relative to control; SGK3: 2.1 ± 0.13-fold relative to control; n = 5 pairs, P < 0.01). This stress-induced SGK up-regulation was blocked by TSA injection (SGK1: 0.98 ± 0.05-fold relative to control; SGK3: 1.0 ± 0.1-fold relative to control, n = 5 pairs, P > 0.05), but not by TpxA injection (SGK1: 2.2 ± 0.33-fold; SGK3: 1.8 ± 0.22-fold relative to control; n = 4 pairs, P < 0.01). These HDAC inhibitors alone did not have a significant effect on SGK1 (TSA: 1.1 ± 0.1-fold relative to control; TpxA: 1.2 ± 0.17-fold relative to control, P > 0.05) or SGK3 (TSA: 1.3 ± 0.13-fold relative to control; TpxA: 1.1 ± 0.09-fold relative to control, P > 0.05) expression. These results suggest that HDAC6 is required for the acute stress-induced upregulation of SGK1/3.
Figure 4. HDAC6 inhibition blocks the acute stress-induced upregulation of SGKs.
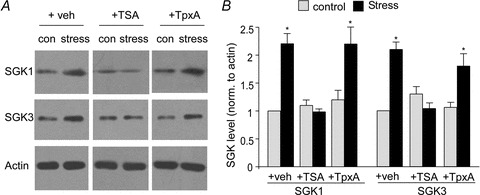
A and B, immunoblots (A) and quantitative analysis (B) of SGK1 and SGK3 expression in PFC slices from control vs. stressed animals with or without i.p. injection of TSA (0.5 mg kg−1) or TpxA (0.3 μg kg−1). *P < 0.01, ANOVA.
HSP90 is involved in HDAC6 regulation of stress effects on AMPAR receptors
Since GR signalling relies on its chaperone protein heat shock protein 90 (HSP90), whose activity is regulated via the reversible acetylation by HDAC6 (Kovacs et al. 2005; Murphy et al. 2005), we tested whether HDAC6 affects the synaptic action of acute stress by targeting its downstream substrate HSP90. As shown in Fig. 5A, i.p. injection of 17-DMAG (30 mg kg−1), a brain-permeant HSP90 inhibitor (Putcha et al. 2010), blocked the acute stress-induced potentiation of AMPAR-EPSC (F5,69= 12.1, P < 0.001, ANOVA, vehicle: 94.9 ± 6.6 pA, n = 19; vehicle+stress: 209.7 ± 24.4 pA, n = 11, P < 0.01; 17-DMAG: 88.3 ± 10.1 pA, n = 9; 17-DMAG+stress: 78.9 ± 11.1 pA, n = 11, P > 0.05). In contrast, injection of geldanamycin (2 mg kg−1), an HSP90 inhibitor that is relatively impermeant to the blood–brain barrier (Putcha et al. 2010), failed to block the enhancing effect of acute stress (geldanamycin: 88.6 ± 12.0 pA, n = 9; geldanamycin+stress: 183.1 ± 27.1 pA, n = 11, P < 0.01).
Figure 5. HSP90 inhibition or knockdown blocks the acute stress-induced regulation of AMPARs and SGK.
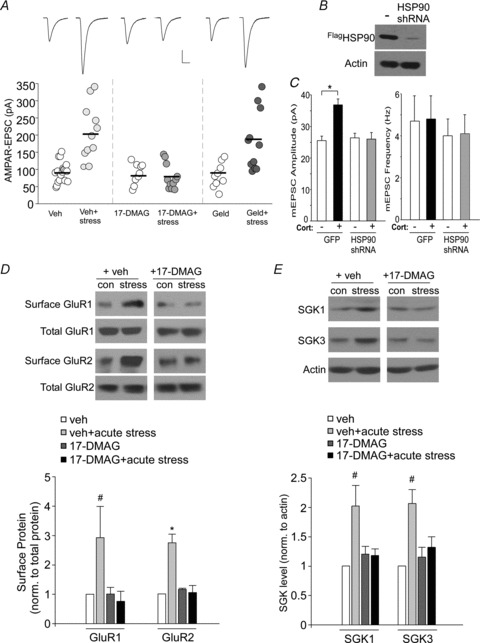
A, dot plots showing the amplitude of AMPAR-EPSC in PFC pyramidal neurons from control or stressed animals with or without i.p. injection of 17-DMAG (30 mg kg−1, a brain-permeant HSP90 inhibitor) or geldanamycin (2 mg kg−1, a non-permeant HSP90 inhibitor). *P < 0.01, ANOVA. Inset: representative synaptic current traces. Scale bar: 50 pA, 20 ms. B, representative Western blots in HEK293 cells transfected with Flag-tagged rat HSP90 in the absence or presence of a HSP90 shRNA. HSP90 was detected with anti-Flag. C, bar graphs (mean ± SEM) showing the mEPSC amplitude and frequency in GFP- or HSP90 shRNA-transfected cortical cultures treated with DMSO or corticosterone (20 min, 100 nm). *P < 0.01, ANOVA. D, immunoblots and quantification analysis of surface and total GluR1 and GluR2 in PFC slices from control vs. stressed animals with or without 17-DMAG injection (30 mg kg−1, i.p.). *P < 0.01; #P < 0.05, ANOVA. E, immunoblots and quantitative analysis of SGK1 and SGK3 expression in PFC slices from control vs. stressed animals with or without i.p. injection of 17-DMAG (30 mg kg−1). #P < 0.05, ANOVA.
To further confirm the specific involvement of HSP90, we knocked down its expression using a shRNA targeted against HSP90 (Fig. 5B), and examined the effect of GR on AMPAR responses. As shown in Fig. 5C, in contrast to GFP-transfected neuronal cultures, corticosterone (100 nm, 20 min) failed to enhance mEPSC amplitude in HSP90 shRNA-transfected cells (F3,33= 9.8, P < 0.001, ANOVA, GFP: 25.6 ± 1.4 pA, n = 8; GFP+CORT: 36.9 ± 1.8 pA, n = 9, P < 0.01; HSP90 shRNA: 26.5 ± 1.4 pA, n = 8; HSP90 shRNA+CORT: 26.1 ± 2.1 pA, n = 9; P > 0.05). The mEPSC frequency did not significantly change in any of the conditions (GFP: 4.7 ± 1.2 Hz; GFP+CORT: 4.8 ± 1.1 Hz; HSP90 shRNA: 4.0 ± 0.79 Hz; HSP90 shRNA+CORT: 4.1 ± 0.97 Hz, P > 0.05, ANOVA).
To complement the electrophysiological results, we also examined the impact of HSP90 inhibition on the expression of surface AMPARs and SGKs in animals exposed to a 20 min forced-swim stress. As shown in Fig. 5D, i.p. injection of 17-DMAG blocked the acute stress-induced increase of surface GluR1 (0.8 ± 0.4-fold relative to control, n = 4, P > 0.05) and surface GluR2 (1.1 ± 0.2-fold relative to control, n = 4, P > 0.05), which is in sharp contrast to vehicle-injected, stressed animals (surface GluR1: 2.9 ± 1.1-fold relative to control, n = 4, P < 0.05; surface GluR2: 2.7 ± 0.3-fold relative to control, n = 3, P < 0.01). Moreover, the acute stress-induced upregulation of SGK1 and SGK3 expression was diminished in 17-DMAG-injected rats (Fig. 5E, SGK1: 1.2 ± 0.1-fold relative to control; SGK3: 1.3 ± 0.2-fold relative to control, n = 8 pairs, P > 0.05), but not vehicle-injected rats (SGK1: 2.0 ± 0.4-fold relative to control; SGK3: 2.1 ± 0.2-fold relative to control; n = 5 pairs, P < 0.05). Taken together, these data indicate that suppressing HSP90 or HDAC6 produced similar blockade of the stress effects on AMPARs, suggesting that HDAC6 acts on HSP90 to gate the stress-induced enhancement of glutamatergic signalling.
Discussion
Stress has a powerful impact on PFC synaptic transmission and PFC-mediated cognitive processes (Yuen et al. 2009, 2011; Popoli et al. 2011), but little is known about how the stress response and GR signalling can be regulated at the molecular level. In this study, we show that inhibiting HDAC6 with various pharmacological agents blocks the delayed and sustained potentiation of glutmatergic transmission in PFC caused by acute stress. Targeted knockdown of HDAC6 also blocks the enhancing effect of acute stress in vivo or corticosterone treatment in vitro. Thus we have identified HDAC6 as a key molecule involved in the gating of acute stress-induced synaptic changes in PFC pyramidal neurons.
Acute stress, via the GR downstream target SGK1/3, increases glutamate receptors at the synaptic membrane, and thereby enhances glutamatergic transmission in PFC (Liu et al. 2010; Yuen et al. 2011). In this study, we show that HDAC6 inhibition blocks the acute stress-induced increase in AMPAR surface expression and SGK1/3 upregulation. It suggests that, when HDAC6 function is suppressed, GR is defective in nuclear translocation and gene activation, leading to the failure of increasing the synaptic delivery of glutamate receptors in stressed animals.
Recent studies have implicated histone hypoacetylation and transcriptional dysfunction in neurodegenerative and psychiatric disorders (Tsankava et al. 2007; Chuang et al. 2009). For example, HDAC2 is found to negatively regulate memory formation and synaptic plasticity, and the memory impairment in mice overexpressing HDAC2 is ameliorated by treatment with the HDAC inhibitor vorinostat (Guan et al. 2009). Loss of HDAC5 or HDAC2 is linked to the altered histone acetylation and global patterns of gene expression in the nucleus accumbens in animal models of drug addiction and depression, which is found to be important for the maladaptive behavioural changes after chronic exposure to cocaine or emotional stimuli (Renthal et al. 2007; Covington et al. 2009). While most of these effects of HDAC are exerted via chromatin remodelling and gene transcription in the nucleus, our findings suggest that HDAC6 regulates acute stress responses by modulating steroid hormone signalling in the cytoplasm.
HDAC6, by targeting three substrates (HSP90, tubulin and cortactin) and interacting with other proteins, regulates important and diverse cellular processes including protein ubiquitination, cell migration and microtubule dynamics (Valenzuela-Fernandez et al. 2008). For instance, the deacetylation of tublin by HDAC6 (Hubbert et al. 2002; Zhang et al. 2003) causes the recruitment of molecular motors dynein and kinesin-1 to microtubules, which influences the vesicular transport of BDNF (Dompierre et al. 2007). The deacetylation of HSP90 by HDAC6 affects the HSP90 cochaperone complex and GR nuclear translocation (Kovacs et al. 2005; Murphy et al. 2005). HDAC6 knockout mice show increased HSP90 acetylation correlating with impaired HSP90 function, while microtubule organization and stability are normal despite hyperacetylated tubulin (Zhang et al. 2008). In this study, we show that HSP90 inhibition or knockdown blocks the enhancing effect of acute stress or corticosterone treatment on AMPAR responses, similar to the blockade seen with HDAC6 inhibition. It suggests that HDAC6's ability to gate the synaptic action of acute stress is through its regulation of HSP90 acetylation and chaperone activity toward GR. Based on these results, we speculate that the stress response is blunted in HDAC6-deficient animals due to the impaired GR/HSP90 signalling.
Taken together, our findings have shed new light on the molecular mechanisms involved in regulating the synaptic effects of acute stress in vivo and implicated HDAC6 as a key controller. While this study has focused on acute stress paradigms, our findings may also have important implications for chronic stress because the actions of acute and chronic stress share interconnected and related mechanisms, such as the requirement of GR signalling. In contrast to the adaptive role of acute stress, chronic stress can lead to maladaptive alterations in neuronal functions by causing dendritic retraction, blocking synaptic plasticity and inhibiting neurogenesis (de Kloet et al. 2005; McEwen, 2007). Inhibition of HDAC6 may therefore have the potential to block the detrimental changes seen during chronic stress and provide neuroprotective effects against stress-induced mental illness. Indeed, treatment with various HDAC inhibitors has emerged as a promising new strategy for therapeutic intervention in CNS disorders (Kazantsev & Thompson, 2008).
Acknowledgments
This work was supported by NIH grants to Z.Y. We thank Xiaoqing Chen and Dr Eunice Yuen for excellent technical support.
Glossary
Abbreviations
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- EPSC
excitatory postsynaptic current
- GFP
green fluorescent protein
- GR
glucocorticoid receptor;
- GluR
glutamate receptor
- HDAC
histone deacetylase
- HSP90
heat shock protein 90
- NMDAR
N-methyl-d-aspartate receptor
- PFC
prefrontal cortex
- SGK
serum and glucocorticoid-inducible kinase
Author contributions
Conception and design of the experiments: Z.Y., J.F. and J.B.L.; collection, analysis and interpretation of data: J.B.L., J.W., W.L. and J.C.; drafting the article or revising it critically for important intellectual content: Z.Y. and J.B.L.
Supplementary material
Supplementary Figure 1
Supplementary Figure 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Aoyagi S, Archer TK. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 2005;15:565–567. doi: 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Cadepond F, Schweizer-Groyer G, Segard-Maurel I, Jibard N, Hollenberg SM, Giguère V, Evans RM, Baulieu EE. Heat shock protein 90 as a critical factor in maintaining glucocorticosteroid receptor in a nonfunctional state. J Biol Chem. 1991;266:5834–5841. [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Dittmar KD, Demady DR, Stancato LF, Krishna P, Pratt WB. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor.hsp90 heterocomplexes formed by hsp90.p60.hsp70. J Biol Chem. 1997;272:21213–21220. doi: 10.1074/jbc.272.34.21213. [DOI] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci U S A. 2001;98:87–92. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Molecular Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, Morrison JH, McEwen BS. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yuen EY, Yan Z. The stress hormone corticosterone increases synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors via serum- and glucocorticoid-inducible kinase (SGK) regulation of the GDI-Rab4 complex. J Biol Chem. 2010;285:6101–6108. doi: 10.1074/jbc.M109.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- Murphy PJ, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J Biol Chem. 2005;280:33792–33799. doi: 10.1074/jbc.M506997200. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, Hicks CD, Veal JM, Steed PM, Hyman BT, McLean PJ. Brain-permeable small-molecule inhibitors of Hsp90 prevent α-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J Pharmacol Exp Ther. 2010;332:849–857. doi: 10.1124/jpet.109.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci U S A. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Verdel A, Curtet S, Brocard MP, Rousseaux S, Lemercier C, Yoshida M, Khochbin S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol. 2000;10:747–749. doi: 10.1016/s0960-9822(00)00542-x. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A. 2009;106:14075–14079. doi: 10.1073/pnas.0906791106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16:156–170. doi: 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.