

Abstract
Recently developed pharmacogenetic and optogenetic approaches, with their own advantages and disadvantages, have become indispensable tools in modern neuroscience. Here, we employed a previously described knock-in mouse line (GABAARγ277Ilox) in which the γ2 subunit of the GABAA receptor (GABAAR) was mutated to become zolpidem insensitive (γ277I) and used viral vectors to swap γ277I with wild-type, zolpidem-sensitive γ2 subunits (γ277F). The verification of unaltered density and subcellular distribution of the virally introduced γ2 subunits requires their selective labelling. For this we generated six N- and six C-terminal-tagged γ2 subunits, with which cortical cultures of GABAARγ2−/− mice were transduced using lentiviruses. We found that the N-terminal AU1 tag resulted in excellent immunodetection and unimpaired synaptic localization. Unaltered kinetic properties of the AU1-tagged γ2 (AU1γ277F) channels were demonstrated with whole-cell patch-clamp recordings of spontaneous IPSCs from cultured cells. Next, we carried out stereotaxic injections of lenti- and adeno-associated viruses containing Cre-recombinase and the AU1γ277F subunit (Cre-2A-AU1γ277F) into the neocortex of GABAARγ277Ilox mice. Light microscopic immunofluorescence and electron microscopic freeze-fracture replica immunogold labelling demonstrated the efficient immunodetection of the AU1 tag and the normal enrichment of the AU1γ277F subunits in perisomatic GABAergic synapses. In line with this, miniature and action potential-evoked IPSCs whole-cell recorded from transduced cells had unaltered amplitudes, kinetics and restored zolpidem sensitivity. Our results obtained with a wide range of structural and functional verification methods reveal unaltered subcellular distributions and functional properties of γ277I and AU1γ277F GABAARs in cortical pyramidal cells. This transgenic–viral pharmacogenetic approach has the advantage that it does not require any extrinsic protein that might endow some unforeseen alterations of the genetically modified cells. In addition, this virus-based approach opens up the possibility of modifying multiple cell types in distinct brain regions and performing alternative recombination-based intersectional genetic manipulations.
Key points
We generated lenti- and adeno-associated viruses which were used to replace the zolpidem-insensitive GABAA receptors of a transgenic mouse line with wild-type, zolpidem-sensitive ones.
The virally expressed wild-type, zolpidem-sensitive GABAA receptor γ2 subunits were tagged with a small immunotag (AU1).
Light microscopic fluorescent and electron microscopic freeze-fracture immunogold labelling revealed that the virally introduced AU1-tagged γ2 subunit-containing receptors had a normal synaptic distribution on cortical pyramidal cells.
In vitro patch-clamp recordings demonstrated that the insertion of this immunotag did not alter the kinetic and pharmacological properties of the virally inserted γ2 subunits.
Our results demonstrate a novel transgenic–viral pharmacogenetic approach, which allows the selective silencing of well-defined neuronal populations in the brain.
Introduction
In recent years, a number of different approaches have been described that allow the selective pharmacological modification of the activity of genetically modified cell populations. These methods have already contributed enormously to our understanding of the role of defined cell populations in certain behaviours (Callaway, 2005). One of the first pharmacogenetic approaches was developed by Lester et al. who either virally (Slimko et al. 2002) or transgenically (Lerchner et al. 2007) introduced invertebrate ivermectin-sensitive Cl− channels into mammalian neurons and demonstrated their efficient hyperpolarization with exogenously applied low concentrations of ivermectin. A similar approach using the Drosophila G-protein-coupled allatostatin receptor (AlstR) was developed for reversible and transient inhibition of neurons (Lechner et al. 2002; Tan et al. 2006). Although allatostatin has no effect on mammalian neurons and efficiently hyperpolarizes AlstR-expressing neurons, a major weakness of this approach is the lack of penetration of the ligand through the blood–brain barrier. An alternative approach is the use of so-called designer receptors, which are mutated versions of, for example, the human muscarinic receptor (hM4D) that are made sensitive to a pharmacologically inert drug (clozapine-N-oxide: CNO; Armbruster et al. 2007; Ferguson et al. 2011) and are made insensitive to their endogenous ligands. The application of CNO exerts its effect on neuronal excitability by activating the designer receptor and consequently the endogenously expressed Kir3 K+ channels. A very similar approach was taken by Magnus et al. (2011) who modified ligand-gated ion channels to endow selectivity to inert ligands. They generated chimeras of the α7 nicotinic and glycine receptors with point mutations in the ligand-binding domains of the α7 subunit to make it insensitive to endogenous ligand and sensitive to an inert drug, the application of which produced a pronounced hyperpolarization of the chimeric receptor-expressing cells. Although these methods rely on the activation of either K+ or Cl− channels, they have one common feature: they are all based on the genetic introduction of exogenous proteins. How these proteins interfere with the translation/assembly/processing of other proteins at the level of the endoplasmic reticulum/Golgi apparatus and how they influence the neurons’ intrinsic excitability when they are present in the plasma membrane are largely unknown, but impose potential unforeseen problems.
A fundamentally different pharmacogenetic approach was developed by Wulff et al. (2007) who generated a knock-in mouse line (GABAARγ277Ilox) in which the 77th amino acid of the GABAA receptor (GABAAR) γ2 subunit was mutated from phenylalanine (γ277F) to isoleucine (γ277I) and the mutated receptor gene was flanked by two LoxP sites. This mutation renders the composed GABAARs insensitive to zolpidem, a benzodiazepine (BZ) site ligand, and affects every γ2 subunit in every neuron of the brain, making the whole animal zolpidem insensitive. Importantly, this single amino acid mutation did not change the subcellular distribution or the kinetic properties of the GABAARs. Wulff et al. (2007) generated two additional transgenic mouse lines; in the first one the Purkinje cell-specific L7 promoter drove the expression of Cre-recombinase and in the second one it drove the expression of the γ277F subunit. By crossing these three genetically modified mouse lines, they swapped zolpidem-insensitive with zolpidem-sensitive GABAARs only in cerebellar Purkinje cells and showed that GABAergic inhibition was potentiated only in Purkinje cells following the administration of zolpidem in these animals. Another elegant aspect of this method is that GABAergic synaptic currents can be reduced by BZ site inverse agonists (e.g. β-carbolines DMCM and β-CCM), resulting in a selective increase in the excitability of the targeted cells. Another major advantage of this approach is the lack of exogenous protein expression in nerve cells, eliminating the possibility of unwanted effects. However, the major disadvantage of the methods described by Wulff et al. (2007) was the necessity of generating many transgenic mouse lines and potential alterations in the developmental expression of γ277F caused by the L7 promoter. If a single viral vector could accommodate both Cre-recombinase and γ277F, one could stereotaxically inject it into certain brain areas of GABAARγ277Ilox mice to enable negative and positive allosteric modulation of the transduced cells. The effects of transient and reversible negative and positive allosteric modulation of inhibition on the transduced cells with blood–brain barrier-penetrating pharmacological agents could then be analysed at the system and behavioural levels. In the present manuscript, we describe the generation of lenti- and adeno-associated viruses expressing Cre-recombinase and N-terminal AU1-tagged γ277F subunits and employ light- and electron microscopic immunolocalization as well as in vitro electrophysiological methods to verify the unaltered subcellular and synaptic locations and kinetic properties of the virally inserted GABAARs.
Methods
Tagging the GABAAR γ2 subunit
The original GABAA receptor γ2-L subunit cDNA (pRK5γ2-L, gift from Drs Peer Wulf and William Wisden) was modified by inserting a BamHI recognition site in front of the signal peptide's start codon. The enhanced green fluorescent protein (eGFP) sequence in the pFUGW (Lois et al. 2002) lentiviral backbone plasmid was replaced by inserting the BamHI-BstEII fragment of the modifed pRK5γ2-L between the BamHI and the EcoRI sites of the lentiviral plasmid, resulting in pF-U-[γ2]W. For the tag insertions, the γ2-L subunit cDNA was subcloned into pBSKSII(+), and an internal removable insert was placed between the 4th and 5th amino acid residue of the mature protein (referred to as N-terminal position) or at the C-terminus (Fig. 1). PCR primers used for N-terminal insertions were (TTGAATTCTCAGTCAGATGATGACTATGAAGATTAC ACTTCTAAT), (TTGAATTCCTCTAGTCATCATCTGAC TTTTGGCTTGTG). Those for the C-terminal insertions were, (CTGGGGAATTCTCTTTATCTGTAAAGAGGTAT GGGTTTTATTGATA) and (ATAGTGAATTCCTCATAC CTCTCAGATAAAGATAGGAGACCCAGTAAACAAGAT TGAAC). The removable inserts were then replaced with the tag coding synthetic double-stranded oligo-nucleotides and the tagged subunits were cloned back to the pF-U-[γ2]W construct. The amino acid sequences of the tags were AU1: DTYRYI; BBS (α-bungarotoxin binding site): WRYYESSLEPYPD; FLAG: DYKDDDDK; 3XFLAG: DYKDHDGDYKDHD IDYKDDDDK; HA: YP YDVPDYA; STREP II: WSHPQFEK.
Figure 1. Site-independent insertion of epitope tags into the GABAA receptor γ2 subunit cDNA.
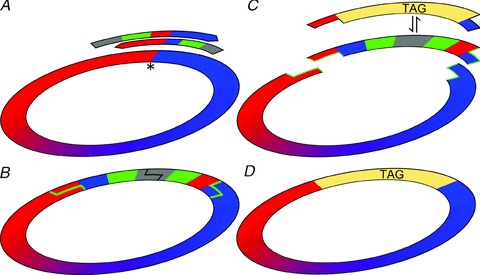
A, partially overlapping PCR primers with non-complementary 5′ tails coding for RE recognition sequences were designed to match the insertion point (*). B, both ends of the resulting PCR product were digested with EcoRI (grey) prior to self-circularization by T4-ligase. C, the sequence of the new plasmid was confirmed and then digested with BplI (green) which cuts outside of its recognition sequence (green lines), releasing the complete original plasmid sequence with protruding free ends. D, different tags, coded by double-stranded oligonucleotides with complementary protruding ends, were ligated in place of the removed insert. The recombinant tag-containing plasmids were selected by EcoRI digestion prior to transformation.
Generation of pF-U-[Cre]W, pF-U-[Cre-2A-AU1γ2]W and pAAV-CMV-[Cre-2A-AU1γ2]W-SV40pA viral plasmids
The nls-Cre recombinase was first removed from the pPGK-Cre-bpA plasmid (Addgene plasmid 11543) by SalI, MluI digestion and ligated into a modified pBSKSII(+) plasmid containing the MluI, HindIII, SalI, BglII sites between the EcoRI and XhoI sites of the original Multi Cloning site (MCS). Later the EcoRI, BglII fragment of the resulting plasmid was used to replace the EcoRI, BamHI fragment of the pFUGW plasmid to generate the pF-U-[Cre]W. For the generation of the pF-U-[Cre-2A-AU1γ2]W, first the PshAI,Van91I fragment of the pF-U-[Cre]W plasmid was replaced with a synthetic DNA fragment coding the last 65 amino acid residues of the Cre recombinase without the stop codon, followed by the in-frame addition of a picorna virus 2A-like peptide (KQKIVAPVKQTLNFDLLKLAGDVESNPGP) coding sequence (Donnelly et al. 2001) and the first 10 basepairs of the γ2-L cDNA. The 3′ terminus of the γ2-L cDNA was later completed by inserting the BstXI,Van91I fragment of the pF-U-[AU1γ2]W plasmid between the BstXI,Van91I sites. For AAV backbone plasmid generation the neomycin expression cassette of the pTR-eGFP plasmid (from Dr Samulski, University of North Carolina) was deleted and the eGFP coding region was replaced with the [Cre-2A-AU1γ2]W region of the pF-U-[Cre-2A-AU1γ2]W plasmid.
Virus production
Viral vectors were produced by transient transfection of cells from the human embryonic kidney cell line 293T, with a protocol modified from Naldini et al. (1996). Briefly, a total of 5 × 106 293T cells were seeded in 10 cm diameter poly-l-lysine-coated dishes in 5 ml Dulbecco's modified Eagle's medium (D-MEM)-high glucose (Sigma)–10% fetal calf serum (FCS) (Sigma) in a humidified 5% CO2 incubator at 37°C 24 h prior to transfection. The culture medium was changed with 10 ml fresh medium 1 h prior to transfection. Five micrograms of envelope plasmid (pCMV-VSV-G) and 5 μg packaging plasmid (pCMV-HIVgpΔ8.9), both kindly provided by Dr Pavel Osten at the Max Planck Institute, and 5 μg lentiviral backbone plasmid were used for each transfection. The three plasmids were mixed in water (to 870 μl final volume) and left at 4°C overnight. On the following day 130 μl of a 2 m CaCl2 solution was added to the DNA solution, mixed and 1 ml of Hepes-buffered saline (281 mm NaCl, 50 mm Hepes, 1.5 mm Na2HPO4; pH = 7.22) was slowly added during constant mixing. The freshly formed precipitate was immediately added to the cultures. After 16 h, the transfection medium was replaced with 10 ml fresh medium supplemented with 20 μl of 1 mg ml−1 forskolin (Sigma) dissolved in DMSO (Sigma). The supernatant was collected one day later, then cleared by low-speed centrifugation and filtered through a 0.45 μm PVDF membrane (Millipore). Concentration was achieved by ultracentrifugation at 50,000 g, 4°C for 2 h, followed by re-suspension in 900 μl Hank's balanced salt solution (HBSS), layering on top of 1 ml of 20% sucrose cushion and centrifuged at 53,000 g, 4°C for 2 h. The pellet was re-suspended in 100 μl calcium- and magnesium-free (CMF)-HBSS, kept in the centrifuge tube overnight at 4°C, aliquoted, snap-frozen in liquid nitrogen and stored at –80°C. The titre of some virus preparations was assayed by a HIV-1 p24 ELISA kit (PerkinElmer). One nanogram of p24 group-specific antigen (gag) protein was considered to represent 5000 infectious units (IU) and most virus preparations used in the present study had titres of >105 IU μl−1. AAV-2 virus particles pseudo-coated with AAV-9 cap proteins (4.6 × 1012 GC ml−1) were generated in the Penn Vector Core, University of Pennsylvania.
GABAARγ2−/− mice and genotyping the embryos
C57BL/6 mice heterozygous for the GABAA receptor γ2 subunit (γ2−/+; Günther et al. 1995; kindly provided by Dr Bernhard Luscher) were raised on a 12 h light/dark cycle with water and food ad libitum and crossed to obtain embryonic day (E)16 embryos. All the animal experiments were conducted in accordance with the guidelines of the National Institute for Physiological Science's Animal Care and Use Committee. The embryos used for primary neuronal cultures were genotyped during the process of cell culturing and for most experiments only the cells from γ2−/− embryos were plated. Two legs were isolated from each embryo, one was kept at –80°C for subsequent confirmation of the genotype. Another leg was used for quick genotyping using a method modified from McClive & Sinclair (2001). Briefly, legs were washed twice in distilled water and once in DB-K (Digestion Buffer without Proteinase-K) (50 mm KCl, 19 mm Tris-HCl; pH = 8.3, 0.1 mg ml−1 gelatin, 0.45% NP40, 0.45% Tween-20), then incubated in 50 μl DB (Digestion Buffer) (DB-K + 60 U ml−1 proteinase-K) at 55°C for 5 min. Proteinase-K was inactivated by incubation at 95°C for 10 min followed by a brief vortexing. The undigested tissue was sedimented by a centrifugation for 20 s at 14,000 g. One microlitre of the supernatant was used in a 31.5 μl PCR reaction (1x Takara PCR reaction buffer, 0.2 mm dNTPs, 250 nm primers, 0.79 U Takara-Taq), applying the following programme: 1 s at 98°C, 30 s at 60°C, 2 min at 72°C, repeated 28 times. The PCR primers were: CATCTCCATCGCTAAGAATGTTCGGGAAGT (γ2−/+), ATGCTCCAGACTGCCTTGGGAAAAGC (γ2−) and GCTGACAAAATAATGCAGGGTGCCATACTC (γ2+). The PCR products were subjected to an electrophoresis at high current (300 mA, 4°C) for ∼12 min. The genotype of the embryos selected for the primary culture was further confirmed later by a conventional DNA isolation (DNeasy Blood and Tissue Isolating Kit, Qiagen), followed by a similar PCR and conventional gel electrophoresis.
Primary cortical neuronal cultures
Primary cultures of cortical neurons were prepared from mouse embryos (E16). Pregnant mice were killed by cervical dislocation. The embryos were immediately dissected and placed in ice-cold CMF-HBSS (Gibco). The brains were removed from individual embryos and the neocortex was isolated and cut into small pieces. The tissue was collected into a 15 ml tube and sedimented by low-speed centrifugation (800 g, 1 min). The supernatant was replaced with 2 ml 0.25% trypsin (Gibco) and 4 mg ml−1 DNase I in CMF-HBSS and incubated at 37°C for 15 min, with gentle agitation every 3 min. Enzymatic dissociation was stopped by adding 0.8 ml FCS and cells were physically dissociated by triturating with a pipette. After that, 5 ml of MEM+ (Minimum Essential Media) solution was added (5% FCS, 2% B-27 supplement (Gibco), 50 mm l-glutamate (Gibco) and 92.75% MEM (200 mg l−1 CaCl2, 400 mg l−1 KCl, 200 mg l−1 MgSO4, 6.8 g l−1 NaCl, 2.2 g l−1 NaHCO3, 158 mg l−1 NaH2PO4.2H2O, 7 g l−1d-glucose, 0.5 mg l−1 choline chloride, 0.5 mg l−1d-calcium pantothenate, 0.5 mg l−1 folic acid, 0.5 mg l−1 i-inositol, 0.9 mg l−1 nicotinamide, 0.5 mg l−1 pyridoxal hydrochloride, 0.5 mg l−1 thiamine hydrochloride, 0.04 mg l−1 riboflavine, 1x MEM amino acid solution, 100 U l−1 penicillin–100 μg l−1 streptomycin, 15 mm Hepes (pH = 7.25)), briefly mixed and centrifuged for 10 min at 180 g at 4°C. The pellet was re-suspended in 3 ml MEM+, filtered through a cell strainer (100 μm, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and diluted in MEM+ to a final concentration of 9 × 104 cells ml−1. Cells from γ2−/− and γ2+/+ embryos (1.8 × 104 cells/dish) were seeded onto poly-l-lysine-coated glass-bottomed 35 mm cell culture dishes (Iwaki, Tokyo, Japan) and cultured at 37°C in a humidified 5% CO2 atmosphere. For electrophysiological experiments, cells were seeded onto 35 mm cell culture dishes containing poly-l-lysine-coated coverslips (Matsunami, Osaka, Japan). After 4 h of incubation 1–2.5 μl of lentiviral vector was added to the cultures. All chemicals and reagents were obtained from Sigma, unless otherwise stated.
Immunohistochemistry on neuronal cultures
Immunoreactions were performed on cells cultured for 19–27 DIV. Cells were fixed with 10% paraformaldehyde for 30 min at 37°C, washed in PBS, and blocked with PBS containing 0.1% Triton X-100 and 5% normal goat serum (PBST-NGS). The cells were then incubated at 4°C in PBST-NGS containing the primary antibodies for 16 h. The following primary antibodies were used: rabbit anti-γ2 subunit (1:500; gift from Prof. Werner Sieghart), mouse anti-β2/3 subunit (Millipore Corporation, Billerica, MA, USA; MAB341), mouse anti-AU1 (1:5000; Covance Inc., Princeton, NJ, USA; MMS-130R), mouse anti-Flag M2 (1:500–2500; Sigma F3165), mouse anti-HA (1:500; Abcam Inc., Cambridge, MA, USA; 12CA5, ab16918) and mouse anti-Strep-tag II (1:500; IBA GmbH, Gottingen, Germany, 21507001). After washing, the cells were incubated in secondary antibodies: Alexa488-conjugated goat anti-mouse IgG (Invitrogen, A11029), or Alexa594-conjugated goat anti-rabbit IgG (Invitrogen, A11037), or Alexa488-conjugated bungarotoxin (1:500; Invitrogen B13422). Finally, the cells were washed in PBS and were mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA).
In vitro whole-cell patch-clamp recording of mIPSCs from cultured cortical neurons
Dissociated cells were prepared as described above, cultured for 19–33 DIV and used for electrophysiological examination of mIPSCs. Cells cultured on coverslips were transferred to the recording chamber and superfused continuously at room temperature with a solution containing (in mm): 119.0 NaCl, 2.5 KCl, 2.0 CaCl2, 1.3 MgCl2, 1.0 NaH2PO4, 26.2 NaHCO3, and 11.0 glucose (saturated with 95% O2 and 5% CO2). Cells were visualized using a ×60 water-immersion objective on a BX51WI upright microscope (Olympus, Tokyo, Japan). Whole-cell recordings were made with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), signals were filtered at 2 kHz, digitized at 10 kHz with Digidata 1440A interface and collected using pCLAMP 10 acquisition software (Molecular Devices). The pipette solution contained (in mm): 60 CsCH3SO3, 60 CsCl, 10 Hepes, 10 EGTA, 4 MgCl2, 4 ATP, 0.5 GTP and were titrated to pH 7.2 with CsOH. Recorded cells were voltage clamped at −68 mV, and as the equilibrium potential for Cl− with the above solutions was calculated as −16 mV, IPSCs appeared as inward currents. TTX (0.5 μm), CNQX (5 μm) and dl-AP5 (50 μm) (all from Tocris Bioscience, Bristol, UK) were added to the superfusate to block action potentials, AMPA receptors, and NMDA receptors, respectively. Zolpidem (1 μm) (Sigma) and SR95531 (30 μm) (Tocris Bioscience) were used. Data analysis was performed using AxoGraph X (AxoGraph Scientific, Sydney, Australia), and statistical analysis was performed using Microsoft Excel (Microsoft, Redmond, WA, USA) and StatView (SAS Institute, Cary, NC, USA). Time from 20% to 80% of the peak amplitude was measured as rise time, and time to decay to 1/e times the peak amplitude was taken as decay time.
Lentiviral injections
Male GABAARγ277Ilox mice between 1 and 7 months old were anaesthetized with ketamine:piplophen:xylazine (62.5:6.25:12.5 μg (g body weight)−1) and ∼0.6 μl lentivirus solution was stereotaxically injected into the neocortex at 0.1 μl min−1 flow rate. The animals were kept in ABSL-2 containment for at least 2 weeks before further use. All procedures were carried out in accordance with the ethical guidelines of the Institute of Experimental Medicine of the Hungarian Academy of Sciences, which is in line with the European Union regulations on animal experimentation.
Immunofluorescence reactions on brain sections
Two to ten weeks after the lentiviral injections adult male GABAARγ277Ilox mice (1–7 months old; bred in the Institute of Experimental Medicine's Animal facility; C57BL/6) were deeply anaesthetized with ketamine before transcardial perfusion with ice-cold fixative. Mice were perfused with fixatives containing 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB, pH = 7.3) or in 2% paraformaldehyde in 0.1 m sodium acetate (pH = 6) buffer for 15 min. Coronal sections, 60 μm thick, were cut from the forebrain. Sections were then blocked in 10% normal goat serum (NGS) made up in Tris-buffered saline (TBS, pH = 7.4), followed by incubations in primary antibodies diluted in TBS containing 2% NGS and 0.1% Triton X-100. The following primary antibodies were used: rabbit anti-AU1 (1:1000; Abcam), mouse anti-AU1 (1:500; Covance), mouse anti-Cre (1:5000; Millipore), rabbit anti-Cre (1:1000; Covance), mouse anti-Kv2.1 (1:1000; Millipore), mouse anti-gephyrin (1:1000; SYSY, Göttingen, Germany), rabbit anti-green fluorescent protein (anti-GFP) (1:1000; Invitrogen), guinea pig anti-γ2 (1:1000; gift from Prof. J.-M. Fritschy) and rabbit anti-γ2 (1:1000; SYSY). The following secondary antibodies were used to visualize the immunoreactions: Alexa488-conjugated goat anti-rabbit and anti-mouse (1:1000; Invitrogen), Cy3-conjugated goat anti-rabbit, anti-mouse and donkey anti-guinea pig (1:1000, Jackson ImmunoResearch Europe Ltd, Newmarket, UK) IgGs.
Image acquisition
Immunofluorescence labelling was visualized by an inverted confocal scanning microscope (FV300, Olympus Co., Tokyo, Japan) using a ×40 (NA = 0.75) objective or with an upright confocal scanning laser microscope (FV1000, Olympus) using a ×60 objective (NA = 1.35). Sequential acquisition of multiple channels was used for multi-colour images to avoid spectral crosstalk between channels.
SDS-digested freeze-fracture replica-labelling (SDS-FRL)
For SDS-FRL five adult (1–7 months old) male GABAARγ277Ilox mice were injected with either a mixture of Cre-recombinase and eGFP or a mixture of Cre-2A-AU1γ277F and eGFP-expressing lentiviruses. Two to ten weeks after the injections mice were perfused with a fixative containing 2% paraformaldehyde in 0.1 m PB for 15 min. Coronal sections, 80 μm thick, were cut from the neocortex. Small blocks containing the injected area indicated by the endogenous eGFP fluorescence were cut out in such a way that the injection site dominated the centre of the block surrounded by a small non-injected area. Blocks were high pressure frozen and replicas were prepared as described earlier (Lorincz & Nusser, 2010). Tissue debris was digested from the replicas with gentle stirring in a TBS solution containing 2.5% SDS and 20% sucrose (pH = 8.3) at 80°C overnight. The replicas were blocked with 5% bovine serum albumin (BSA) in TBS for one hour. This was followed by an overnight incubation in TBS containing 1% BSA and the following primary antibodies: rabbit anti-AU1 (1:300; Abcam), rabbit anti-neuroligin-2 (1:1000; SYSY), mouse anti-β3 (1:1000; NeuroMab, UC Davis, CA, USA), guinea pig anti-γ2 (1:600; gift from Prof. J.-M. Fritschy) IgGs. Replicas were then incubated in TBS containing 5% BSA and the following secondary antibodies: goat anti-rabbit IgGs coupled to 10 nm gold particles, goat anti-mouse IgGs coupled to 15 nm gold particles, and goat anti-guinea pig IgGs coupled to 15 nm gold particles (all in 1:100; all from British Biocell International, Cardiff, UK). Replicas were rinsed in TBS and distilled water before they were picked up on copper parallel bar grids and examined with a transmission electron microscope (JEM-1011, JEOL Ltd, Tokyo, Japan) equipped with a high-resolution bottom-mounted CCD camera (Cantega G2, Olympus Soft Imaging Solution GmbH, Munster, Germany).
Acute slice preparation
Two weeks after virus injection, GABAARγ277Ilox mice were deeply anaesthetized with isoflurane in accordance with the ethical guidelines of the Institute of Experimental Medicine Protection of Research Subjects Committee. After decapitation, the brain was removed and placed into ice-cold artificial cerebrospinal fluid (ACSF) containing (in mm): 230 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 24 NaHCO3, 4 MgCl2 and 0.5 CaCl2. Coronal slices from the cerebral cortex were cut at 250 μm thickness with a Vibratome (Leica VT1200S; Leica Microsystems) and were stored in ACSF containing (in mm): 126 NaCl, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 24 NaHCO3, 2 MgCl2 and 2 CaCl2. All extracellular solutions were bubbled continuously with 95% O2 and 5% CO2, resulting in a pH of 7.4. After a 30 min recovery period at 33°C, slices were further incubated at room temperature until they were transferred to the recording chamber.
Electrophysiological recordings from acute slices and data analysis
Somatic whole-cell recordings were performed at 26.0 ± 0.9°C using IR-DIC on an Olympus BX51WI microscope with a ×40 water immersion objective. Recordings were carried out using a mixed potassium gluconate- and KCl-based intracellular solution (containing in mm: 65 potassium gluconate, 70 KCl, 2.5 NaCl, 1.5 MgCl2, 0.025 EGTA, 10 Hepes, 2 Mg-ATP, 0.4 Mg-GTP, 10 creatinine phosphate, and 8 biocytin; pH = 7.33; 270–290 mosmol l−1). Voltage-clamp recordings of mIPSCs at a holding potential of –70 mV were carried out in the presence of 1 μm TTX (Alomone Labs, Jerusalem, Israel) to block voltage-gated sodium channels and 3 mm kynurenic acid to inhibit ionotropic glutamate receptors. For animals injected with the lenti- or adeno-associated viruses containing Cre-2A-AU1γ277F the effects of 1 μm zolpidem was also tested. For paired recordings, TTX was omitted from the ACSF, and the same intracellular solution was used for both cells. Cells were held in whole-cell current-clamp mode, and a sequence of hyper- and depolarizing current injections were used to determine firing parameters. The putative postsynaptic pyramidal cell was then recorded in voltage-clamp mode whilst brief (1–3 ms) depolarizing currents were injected into the putative presynaptic fast-spiking basket cell every 3 s. Recordings were performed with a MultiClamp 700B amplifier (Molecular Devices). Data were digitized on-line at 20 kHz and filtered at 3 kHz with a low-pass Bessel filter. Individual mIPSCs were detected and analysed off-line with EVAN 1.5 (Nusser et al. 2001). Access resistance (Ra) was subject to 70% compensation and was continuously monitored. If Ra changed >20% during the recording, the cell was discarded from the analysis. All recordings were rejected if the uncompensated Ra became >20 MΩ.
Visualization of the recorded cells
After recordings, slices were fixed in 2% paraformaldehyde with 15% picric acid in 0.1 m PB for 24 h and then extensively washed with PB and TBS before re-sectioning at 60 μm thickness. Sections were blocked in TBS with 10% NGS for 1 h, and then incubated overnight in mouse anti-Cre primary antibody (Millipore) diluted 1:5000 in TBS containing 0.1% Triton X-100 and 2% NGS. Sections were then washed in TBS and incubated in TBS containing Cy5-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) diluted 1:500 and streptavidin-conjugated Cy3 diluted 1:1000 (Jackson ImmunoResearch) to visualize the recorded cell. Slices were then washed in TBS, mounted and viewed using an Olympus FV1000 confocal microscope.
Results
Generation and testing different immuno- and affinity-tagged GABAA receptor γ2 subunits
Thorough verification of the unaltered subcellular distribution and synaptic density of the virally introduced γ277F requires it to be identified and distinguished from the genomically coded γ277I. A powerful way of doing this is the genetic introduction of either a small peptide sequence (tag) with high antigenicity (immuno-tagging) or high affinity to small molecules (affinity tagging) or reporter proteins with fluorescent properties (Kittler et al. 2000; Terpe, 2003; Giepmans et al. 2006). However, the tagging entails potential problems; the most obvious one is that the introduced tag itself affects the function and/or subcellular location of the target protein (Previtali et al. 2000; Limon et al. 2007).
First, we aimed to develop a widely applicable method for the efficient generation and testing of multiple tags and tag insertion sites. To facilitate the insertion of different affinity tags into different positions on the target protein we developed a DNA insertion method based on the work of Tomic et al. (1990). Our method combines the unrestricted freedom of choice of insertion positions used in the PCR-based mutagenesis techniques with the robustness and accuracy of directional ligation at restriction endonuclease (RE)-generated overhanging DNA ends. Type IIs REs (Szybalski et al. 1991) generate protruding DNA termini that differ from their recognition sequence by cutting at a defined distance from their recognition sites. A replaceable insert can be inserted into any position on a plasmid by inverse PCR (Ochman et al. 1988) and the insert can be completely removed later by a type IIs RE if the recognition sequence of the enzyme is present on the insert. The enzymatic cleavage leaves behind a at the insertion point surrounded by single-stranded regions of different lengths, determined by the type of the IIs RE used. The generated single-stranded ends can be used for ligation with synthetic oligo-nucleotides, holding complementary ends (Fig. 1).
We inserted five immuno- (Flag, 3XFlag (3XF), StrepII (SII), haemagglutinin (HA), and AU1) and one chemo-affinity (α-bungarotoxin binding site; BBS) tags between the 4th and 5th amino acid residues (referred to as N-terminal insertion) and at the C-terminus of the mature GABAAR γ2 subunit. The tagged γ2 subunit genes were used to replace the GFP gene in the pFUGW (Lois et al. 2002) lentiviral plasmid for generating lentiviral particles. Once lentiviruses containing N- and C-terminal-tagged γ2 subunits were generated, we focused on the following questions. How efficiently can the tagged γ2 subunits be detected? Does tagging affect the precise subcellular location of the subunit? Does tagging alter the kinetic and pharmacological properties of the receptors? To address these questions, we have applied light microscopic (LM) immunofluorescent reactions and patch-clamp electrophysiology in embryonic cortical cultures of control and GABAAR γ2−/− mice and performed LM immunofluorescent and electron microscopic freeze-fracture replica immunogold labelling of the tagged γ2 subunit and in vitro patch-clamp electrophysiology on brain slices of GABAARγ277Ilox mice.
Rescue of synaptic GABAARs with lentiviral transduction of wild-type γ2 subunits in γ2−/− cultured cortical cells
Viral transduction of wild-type neurons with tagged γ2 subunits has the disadvantage that the introduced subunit has to compete with the wild-type γ2 subunit naturally expressed by the cells. Such a competition could result in unforeseen alterations in the assembly, trafficking and localization of the tagged subunit. Furthermore, the tagged subunit might be incorporated only into a small fraction of the receptors, precluding its efficient detection and functional scrutiny. To overcome this difficulty, we carried out viral transduction of neurons in which the endogenous γ2 subunit is knocked out (γ2−/−; Günther et al. 1995). Unfortunately such deletion is lethal; therefore we were restricted to the use of primary neuronal cultures generated from γ2−/− mice at embryonic day 16. First we used an anti-γ2 and an anti-β2/3 subunit antibody to determine the detectability and subcellular distribution of these subunits in γ2+/+ cultures (∼21 DIV). As shown in Fig. 2A, our antibodies revealed a diffuse labelling throughout the somato-dendritic compartments of the cultured neurons with many strong immunoreactive clusters similar to that reported previously (Craig et al. 1994; Essricht et al. 1998). Next, we assessed the distribution of these subunits in cultured cells obtained from γ2−/− mice (Fig. 2B). Immunolabelling for the γ2 subunit completely disappeared in these cells, demonstrating both the lack of γ2 subunit proteins as well as the specificity of our anti-γ2 labelling. A dramatic change in the distribution of β2/3 subunits was also observed similar to that reported earlier for another GABAAR subunit (α2; Essricht et al. 1998). The diffuse, non-clustered distribution of the β subunits is consistent with the essential role of the γ2 subunit for the synaptic clustering of the receptors. Next, we asked whether the lack of synaptic enrichment of the GABAARs could be rescued by viral transduction of the γ2−/− cells with the γ277F subunit. Application of viruses to the cultured cells resulted in a partial and random transduction among the cells; some cells showed strong immunoreactivity for the γ2 subunit, whereas others were just as negative as γ2−/− cells in non-transduced dishes (Fig. 2C). The immunopositive cells showed a prominent clustering of both the γ2 and the β2/3 subunits, similar to that seen in γ2+/+ cells. In summary, these experiments demonstrate that the lentiviral transduction of γ2−/− cells with the wild-type γ2 subunit can rescue the synaptic distribution of γ2 and β2/3 subunits.
Figure 2. GABAA receptor clustering is rescued in γ2−/− cultures following lentiviral introduction of wild-type γ2 subunit.
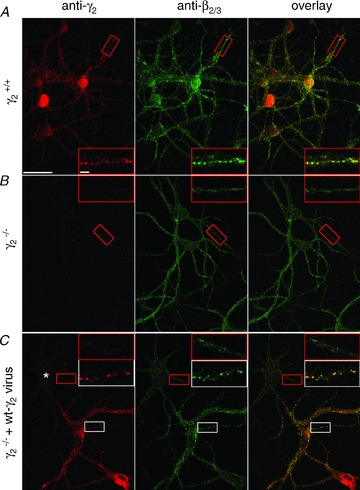
A–C, immunolocalization of the γ2 and β2/3 subunits in primary neuronal cell cultures derived from wild-type (γ2+/+; A), knock out (γ2−/−; B and C) cultures without (A and B) and following infection with wild-type γ2-expressing lentiviruses (C). A, the γ2 and β2/3 subunits colocalize and form dendritic clusters in cultured neurons. B, in γ2−/− cultures, immunolabelling for the γ2 subunit completely disappears, whereas that for the β2/3 subunits became diffuse, without forming dendritic clusters. C, a subset of cells in γ2−/− cultures are infected with wild-type γ2 subunit-expressing lentivirus and show intense punctate labelling for both the γ2 and β2/3 subunits. Note that β2/3 labelling remained diffuse in an uninfected cell (asterisk). Boxed regions are enlarged in insets. Scale bars: main panels: 50 μm; insets: 5 μm.
Detectability and subcellular distribution of tagged GABAAR γ2 subunits
Once the rescue of synaptic clustering of GABAARs by the viral introduction of γ2 subunits was verified, we turned to the six N- and six C-terminal-tagged γ2 subunit-containing lentiviral constructs and tested the detectability and the subcellular distribution of the tagged subunits in γ2−/− cultured cells. As a general strategy, we used double immunofluorescent labelling with anti-γ2 and anti-tag antibodies. Strong, mainly cytoplasmic immunosignal for the γ2 subunit was detected in most transduced cells irrespective of the tag and tagging location (cf. left panels on Fig. 3 and Supplemental Figs 1 and 2 (available online only)). In spite of this, the detection of the different tags showed enormous variability. For example, the Flag and BBS visualization resulted in the weakest signal and this was insertion site independent (Supplemental Figs 1 and 2). The N-terminal insertion of SII gave a weaker signal than the same tag at the C-terminal position (Supplemental Fig. 1), whereas for the AU1 tag the N-terminal tagging position resulted in a much stronger immunolabelling than the same tag at the C-terminal position (cf. Fig. 3B and Supplemental Fig. 2). The HA (Supplemental Fig. 2) and 3XF (Supplemental Fig. 1) tags could be visualized with relatively high efficiency, but the pattern of the labelling was different from that seen for the γ2 subunit in wild-type cultures and in γ2−/− cultures transduced with γ2 subunits. A uniform and strong intracellular labelling was observed rather than clear synaptic plasma membrane clustering of the receptors, indicating that the insertion of these tags could have changed the subcellular distribution of the tagged γ2 subunits. Out of the 12 different conditions, only the N-terminal AU1-tagged γ2 subunit (AU1γ277F) provided a subcellular labelling pattern similar to that found in wild-type cultures. A clustered immunolabelling pattern was obtained with an anti-γ2 subunit antibody, which showed an almost perfect co-localization with the β2/3 subunits (Fig. 3A). The immunosignal obtained with the anti-AU1 antibody possessed a very high signal compared to the noise, consistent with the results of a previous study (Shevtsova et al. 2006). The images were dominated by clear, very intense synaptic clusters and low intensity extrasynaptic dendritic labelling (Fig. 3B).
Figure 3. Tagging the γ2 subunit with the AU1 tag at an N-terminal position does not alter its subcellular distribution, but allows its efficient detection.
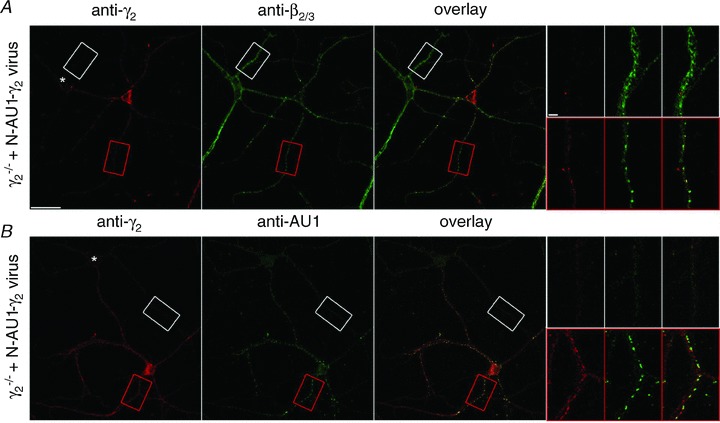
Cultured γ2−/− cortical neurons were infected with AU1γ277F subunit- expressing lentivirus. Infected cells were double immunolabelled with either anti-γ2 and anti-β2/3 subunit antibodies (A) or anti-γ2 and anti-AU1 antibodies (B). Higher magnification views of dendrites from the boxed areas are shown in the insets. Immunoreactive clusters for the γ2 and β2/3 subunits colocalize in the infected dendrites (red boxes), but not in the uninfected cells (asterisk, white boxes). Note the very strong and punctate labelling obtained with the anti-AU1 antibody. Scale bars, main panels: 50 μm; insets: 5 μm.
Unaltered kinetic and pharmacological properties of AU1-tagged γ2 subunits in cultures
The amplitude, rise and decay kinetics of IPSCs depend on the number, density and kinetic properties of postsynaptic GABAARs and on the spatio-temporal profile of GABA in the synaptic cleft. Thus, the detailed analysis of the amplitude and kinetics of GABAAR-mediated IPSCs is an excellent tool to deduce these properties of the receptors. In the next set of experiments, we carried out whole-cell voltage-clamp recordings of IPSCs evoked by spontaneous vesicular release of GABA. In the presence of a voltage-gated Na+ channel blocker (0.5 μm TTX), NMDA (50 μm AP5) and non-NMDA (5 μm CNQX) receptor antagonists, spontaneously occurring inward currents were recorded from wild-type cultured cortical cells (Fig. 4A). A high Cl−-containing intracellular solution facilitated the detection of mIPSCs and resulted in inward current when recorded at –68 mV. The currents had fast rise times and exponential decays and an average amplitude of 54.1 ± 7.7 pA. In γ2−/− cells, mIPSCs could be readily detected (Fig. 4B), in agreement with a previous study (Essricht et al. 1998), and had a similar rise time, peak amplitude, but a significantly slower decay time (35.7 ± 3.8 ms vs. 16.6 ± 0.9 ms) compared to γ2+/+ cells (Fig. 4B and F). When the γ2−/− cells were transduced with either wild-type γ2 (Fig. 4C) or AU1γ277F (Fig. 4D) subunits, the amplitude and kinetics of mIPSCs were fully rescued and were indistinguishable from those recorded from control cells. Because the γ2 subunit confers BZ sensitivity to most GABAARs, we applied 1 μm zolpidem, a BZ site agonist, and examined its effect on the amplitude and kinetics of the mIPSCs. As shown in Fig. 4E, zolpidem increased the amplitude and prolonged the decay of mIPSCs in control cells, but had no effect on γ2−/− mIPSCs, consistent with the lack of γ2 subunits and its essential role in conferring BZ sensitivity. The effect of zolpidem on mIPSC amplitudes and decays was fully rescued by the viral introduction of γ277F or AU1γ277F subunits. In summary, our physiological and pharmacological experiments demonstrate the lack of unwanted effects following insertion of the AU1 tag into the N-terminal region of the γ2 subunit.
Figure 4. Electrophysiological verification of the unaltered properties of the N-terminal AU1-tagged γ2 subunits.
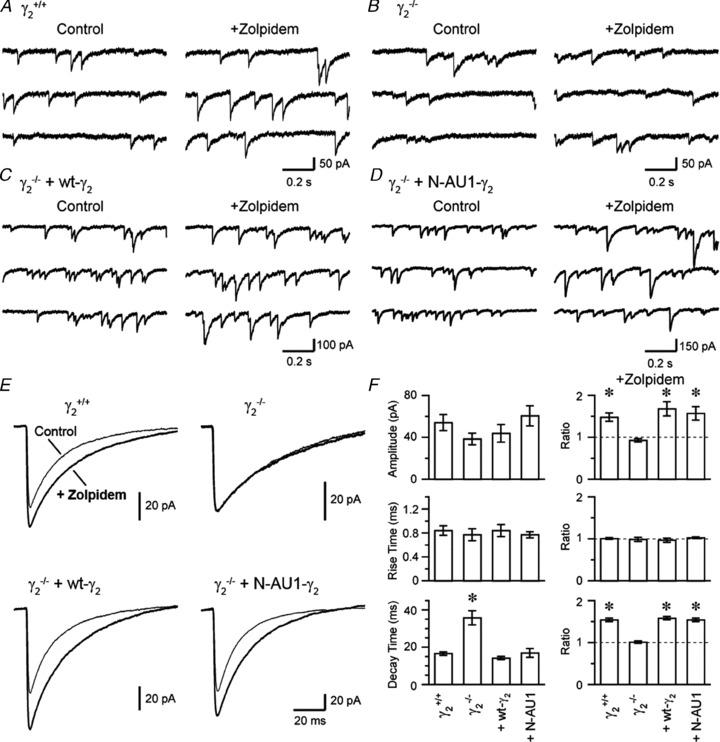
A–D, continuous 3 s whole-cell voltage-clamp recordings are shown from γ2+/+ (A), γ2−/− (B), γ2−/−+γ277F (C) and γ2−/−+AU1γ277F (D) cortical cultured neurons in the presence of 0.5 μm TTX, 5 μm CNQX and 50 μm AP5 (Control) and after addition of 1 μm zolpidem (+Zolpidem). E, superimposed averaged mIPSCs are shown in the presence and absence of zolpidem in γ2+/+ (top left), γ2−/− (top right) cultures without viral transduction and in γ2−/− cultures transduced with wild-type γ2 subunit (bottom left) and γ2−/− cultures transduced with AU1γ277F subunit (bottom right). F, summary of the kinetics and the effect of zolpidem on GABAergic mIPSCs recorded under different conditions. The ratios of the amplitude, the rise time and the decay time before and after zolpidem application are shown on the right. Compared to mIPSCs recorded in γ2+/+ cultured cells, mIPSCs in γ2−/− cultured cells had similar amplitudes and rise times, but had ∼2 times longer decay times. In addition, zolpidem affected the amplitude and the decay time of the wild-type mIPSCs, but was ineffective in modulating γ2−/− mIPSCs. The kinetic properties and the benzodiazepine sensitivity of the γ2−/− cultures are restored following the lentiviral transduction with the γ277F subunit and the AU1γ277F subunit. The * on the left columns indicates significant differences (P < 0.05) using ANOVA and Fisher's PLSD post hoc test and the * on the right column indicates significant difference (P < 0.05) from control following zolpidem application using a paired t test.
Cre-recombinase-mediated removal of GABAAR γ2 subunits from GABAARγ277Ilox cortical cells
Next, we carried out in vivo experiments to remove the GABAAR γ2 subunits from cortical cells. By transducing neocortical pyramidal cells of GABAARγ277Ilox mice following stereotaxic injection of Cre-recombinase-containing lentiviruses we tested the subcellular distribution of GABAAR subunits. Figure 5A demonstrates that the local injection of Crerecombinase-containing lentiviruses into the neocortex results in a large reduction in the immunolabelling for the γ2 subunit. A similar injection into the contralateral side with lentiviruses expressing eGFP caused no change in the γ2 immunofluorescent labelling. Examining the LM immunofluorescent reaction at high magnifications revealed that the γ2 subunit was missing only from the Cre-expressing neurons (Fig. 5B). In these cells, perisomatic plasma membranes were completely devoid of γ2 subunit immunolabelling. To verify further the lack of synaptic labelling of the γ2 subunit, we performed SDS-digested freeze-fracture replica labelling (SDS-FRL) (Fujimoto, 1995; Masugi-Tokita & Shigemoto, 2007; Lorincz & Nusser, 2010) on cortical tissue obtained from the injection sites. Because SDS treatment in this method washes away all the soluble proteins, we were unable to detect the virally introduced Cre-recombinase. We knew from the LM immunofluorescent reactions, however, that not all, but only a fraction of the cells were virally transduced in the middle of the injection area. Thus, we predicted that by examining a large number of cell bodies within the injection area at the electron microscopic level, we would randomly encounter Cre-negative as well as Cre-positive cells. Indeed, we found the exoplasmic plasma membrane faces (E-faces) of cell bodies without any (Fig. 5D) and with many γ2 (Fig. 5F) immunopositive synapses. Since our anti-γ2 antibody reacts with extracellular epitopes of the subunit, labelling for the γ2 subunit appears on the E-face, which lacks clear structural features for demarcating GABAergic synapses (Kasugai et al. 2010). In order to unequivocally identify the GABAergic synapses on E-face structures, all experiments were performed with the ‘mirror replica method’ (Hagiwara et al. 2005). With this method, replicas are generated from both matching sides of the fractured tissue surfaces, allowing the examination of the corresponding E-faces and protoplasmic or P-faces of exactly the same membranes. On the P-face, immunolabelling for neuroligin-2 and GABAAR β3 subunit (Fig. 5C and E) together with clusters of intramembrane particles clearly demarcated GABAergic synapses (Kasugai et al. 2010), allowing the direct examination of the same synapses on the corresponding E-face with respect to their immunoreactivity for the γ2 subunit. On cell bodies that contained γ2 subunit-immunopositive GABAergic synapses, all examined synapses were immunopositive for the γ2 subunit. In contrast, if a soma had synapses that were immunonegative for the γ2 subunit, all of its synapses were immunonegative. These results provide compelling evidence for the total lack of GABAAR γ2 subunits in the perisomatic GABAergic postsynaptic plasma membranes of neocortical neurons following viral transduction with Cre-recombinase.
Figure 5. In vivo lentiviral expression of Cre-recombinase disrupts the expression of the GABAAR γ2 subunit in neocortical neurons of GABAARγ277Ilox mice.
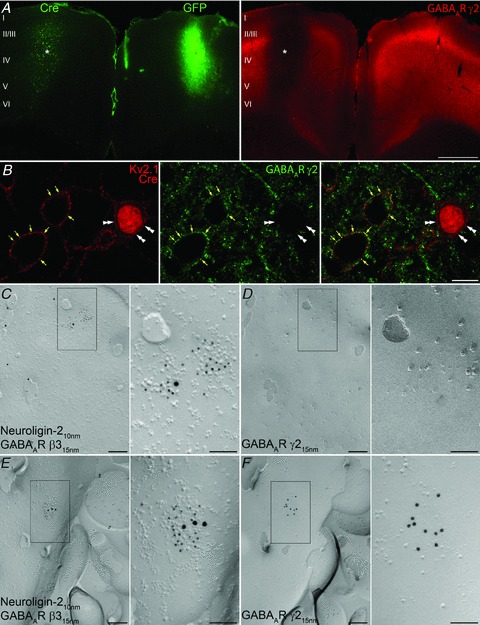
A, double immunofluorescent reaction reveals the absence of GABAAR γ2 immunoreactivity (right panel, asterisk) in the neocortex expressing Cre-recombinase (left panel, asterisk), whereas immunoreactivity for the γ2 subunit is not altered in the contralateral neocortex transduced with eGFP, which was detected by its fluorescence. B, punctate immunolabelling for the γ2 subunit is associated with the plasma membrane (yellow arrows) of non-transduced, control neocortical neurons outlined by a Kv2.1 subunit immunolabelling, but is not present in the plasma membrane (double arrowheads) of a Cre-recombinase-expressing neuron. C and D, P- (C) and E-faces (D) of the same somatic plasma membrane of a transduced cell are shown using the ‘mirror replica technique’. Enlarged views of the boxed areas in C and D are shown on the right. Immunogold particles labelling neuroligin-2 (10 nm gold) and GABAAR β3 (15 nm gold) are accumulated over the postsynaptic membrane of a GABAergic synapse marked by the loose cluster of intramembrane particles, whereas immmunogold labelling for the GABAAR γ2 (15 nm gold) subunit is absent from the corresponding E-face of the same postsynaptic membrane. E and F, P- (E) and E-faces (F) of the somatic plasma membrane of a control, non-transduced cell body are shown using the ‘mirror replica technique’. Immmunogold particles for the γ2 subunit (15 nm gold) are concentrated in the E-face of the postsynaptic membrane, also labelled for neuroligin-2 (10 nm gold) and GABAAR β3 (15 nm gold) on the corresponding P-face. Scale bars: 500 μm (A); 10 μm (B); 200 nm (C, D, E and F); 100 nm (insets).
Efficient detectability, unaltered synaptic localization and kinetic properties of the AU1-tagged γ2 subunit-containing GABAARs
Next, we tested the detectability and subcellular distribution of virally introduced AU1-tagged GABAAR γ2 subunits in vivo. For this, we generated lenti- and adeno-associated viruses containing Cre-recombinase and AU1γ277F, separated by a 2A-peptide-like sequence (Donnelly et al. 2001; Cre-2A-AU1γ277F). Following the stereotaxic injection of these viruses into the neocortex of GABAARγ277Ilox mice, LM immunofluorescent reactions demonstrated the strong expression of Cre-recombinase and AU1 around the injection site (Fig. 6A). The plasma membranes of all neurons with nuclear labelling for Cre-recombinase were strongly decorated by intense, punctate immunofluorescent labelling for AU1 (Fig. 6B). Such AU1 clusters co-localized with the GABAergic synapse marker gephyrin (Fig. 6C), indicating that the AU1-tagged γ2 subunits took up proper synaptic locations. For a direct demonstration of this, we carried out SDS-FRL in the injection area and immunolabelled mirror replicas with neuroligin-2 and the GABAAR β3 subunit on P-faces (Fig. 6D and F) and the γ2 subunit and AU1 on the corresponding E-faces (Fig. 6E and G). Somatic plasma membranes of all neurons contained large numbers of neuroligin-2- and β3-immunopositive synapses (Fig. 6D and F), which were all immunopositive for the γ2 subunit as well. Only some of the cells contained synapses that were labelled for AU1 (Fig. 6E). However, all synapses in such cells contained a large number of gold particles labelling the AU1 epitope, demonstrating a high efficiency of the AU1 immunodetection using SDS-FRL and correct synaptic targeting of the AU1-tagged γ2 subunit.
Figure 6. In vivo lentiviral replacement of γ277I with AU1γ277F in neocortical neurons.
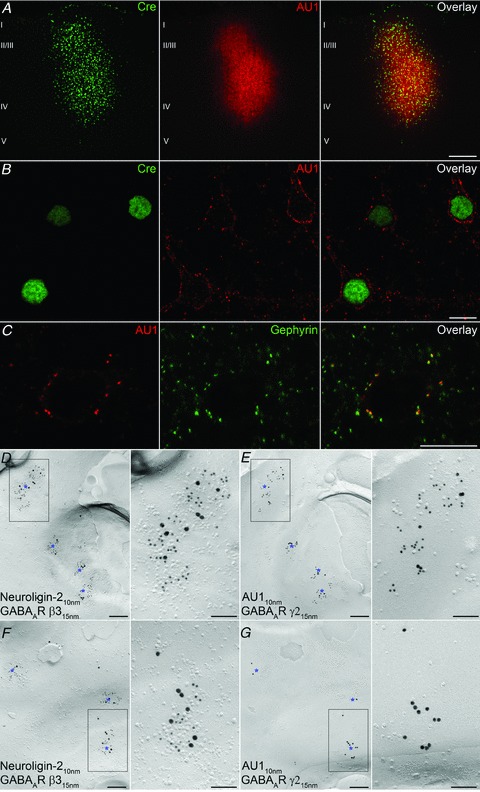
A, double immunofluorescent reactions demonstrate the co-expression of Cre-recombinase and AU1 in neocortical neurons transduced with a lentivirus containing Cre-2A-AU1γ277F. B, higher magnification view showing the nuclear expression of Cre-recombinase and plasma membrane-associated AU1 labelling in the transduced neurons. C, co-localization of AU1 and gephyrin in the perisomatic synapses of a transduced neuron. D and E, P- (D) and E-faces (E) of a somatic plasma membrane of a transduced cell are shown using the ‘mirror replica technique’. Higher magnification images of the boxed areas are shown on the right. Immunogold particles for the AU1 (10 nm gold) and GABAAR γ2 subunit (15 nm gold) are concentrated in the E-face of the postsynaptic membrane also labelled for neuroligin-2 (10 nm gold) and GABAAR β3 (15 nm gold) on the corresponding P-face. F and G, P- (F) and E-faces (G) of a somatic plasma membrane of a non-transduced cell are shown using the ‘mirror replica technique’. Immunogold particles for the γ2 subunit (15 nm gold), but not that of the AU1 (10 nm gold) are concentrated in the E-face of the postsynaptic membrane. The same synapses are also labelled for neuroligin-2 (10 nm gold) and β3 subunit (15 nm gold) on the corresponding P-face. Asterisks indicate GABAergic synapses. Scale bars: 200 μm (A); 10 μm (B and C); 200 nm (D, E, F and G); 100 nm (insets).
In our final sets of experiments, we performed whole-cell voltage-clamp recordings from acute cortical slices 2–7 weeks after the injection of Cre-2A-AU1γ277F lenti- and adeno-associated viruses. In all cells, fast rising, exponentially decaying spontaneous inward currents were recorded in the presence of TTX (1 μm) and kynurenic acid (3 mm; Fig. 7A and B), which were blocked by the GABAAR antagonist SR95531. When the recorded cells were grouped based on their immunonegativity or positivity for Cre-recombinase determined post hoc, there was no significant difference in the frequency (3.6 ± 2.4 Hz, n = 8 Cre– cells vs. 4.1 ± 2.7 Hz, n = 5 Cre+ cells), amplitude (11.2 ± 3.0 pA vs. 11.1 ± 4.0 pA), 10–90% rise time (0.49 ± 0.11 ms vs. 0.55 ± 0.13 ms) and weighted decay time constant (τw; 6.3 ± 2.3 ms vs. 6.7 ± 0.8 ms) of mIPSCs between the two cell populations (Fig. 7C). However, when the effect of 1 μm zolpidem was tested on the decay of mIPSCs, there was no significant change (P = 0.25, paired t test) in Cre-negative cells (before drug: 6.3 ± 2.3 ms vs. drug: 7.3 ± 4.0 ms), but a significant (P < 0.05, paired t test) slowing of the decay was observed in Cre-positive cells (before drug: 6.7 ± 0.8 ms vs. drug: 11.8 ± 2.8 ms). We then specifically investigated the restoration of zolpidem sensitivity of the perisomatic synaptic receptors activated by fast spiking GABAergic basket cells. For this, we performed double whole-cell recordings in the absence of TTX from a presynaptic fast-spiking basket cell and a postsynaptic pyramidal cell (PC; Fig. 7D). Brief depolarizing current injections into the presynaptic cell reliably evoked fast rising, exponentially decaying IPSCs, the decay of which was prolonged by 1 μm zolpidem (Fig. 7E). Post hoc analysis of the recorded cells revealed that the postsynaptic PC was immunopositive for Cre-recombinase (Fig. 7F). These results demonstrate the unaltered synaptic localization of GABAARs and similar kinetics of postsynaptic currents following viral swapping of γ277I with AU1γ277F, and the restoration of zolpidem sensitivity of the synaptic currents.
Figure 7. Virus-mediated replacement of γ277I with AU1γ277F restores IPSC sensitivity to zolpidem in Cre-immunopositive layer 2/3 pyramidal cells.
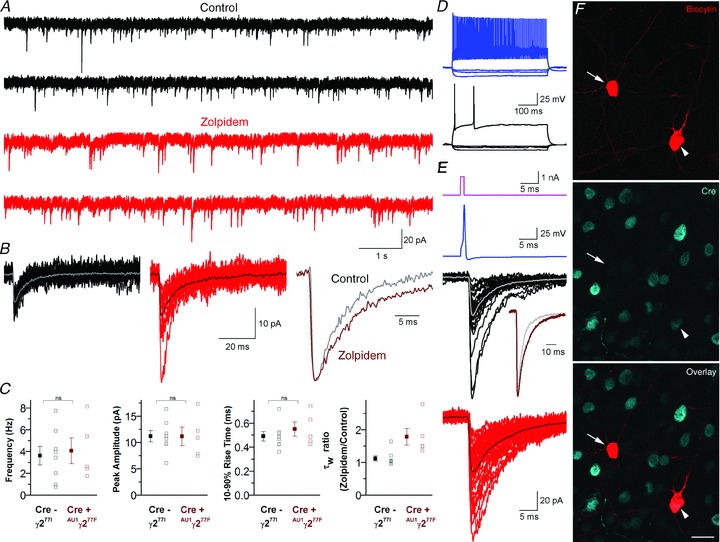
A, continuous current recordings from a Cre-positive layer 2/3 pyramidal cell in ACSF (upper, black) and 1 μm zolpidem (lower, red). B, superimposed consecutive mIPSCs in control (left, black, average in grey) and 1 μm zolpidem (centre, red, average in dark red) from the cell shown in A. Peak-scaled, peak-aligned average traces are shown on the right. C, plots of the population mean ± SEM and individual cell mean values of mIPSC frequency, peak amplitude, 10–90% rise time and the weighted decay time constant (τw) ratio (zolpidem/control) from 8 Cre-negative (grey) and 5 Cre-positive (dark red) cells. D, voltage responses of a fast-spiking basket cell (upper, blue) and a layer 2/3 pyramidal cell (lower, black) to 500 ms hyperpolarizing and depolarizing current injections. E, brief current pulses (top, magenta) were used to trigger single action potentials in the basket cell (upper, blue), evoking IPSCs in the simultaneously recorded pyramidal cell (lower, individual sweeps in black, average in grey). After wash in of 1 μm zolpidem (bottom, individual sweeps in red, average in dark red), peak amplitude and τw values were increased. The change in kinetics can be clearly seen from the superimposed peak-scaled, peak-aligned average traces (inset in lower panel). F, single confocal section showing post hoc visualization of biocytin (red) and immunofluorescent detection of Cre-recombinase (cyan) in the recorded basket cell (Cre-negative, arrow) and pyramidal cell (Cre-positive, arrowhead) shown in panels D and E. Scale bar: 20 μm.
Discussion
In the present work, we have developed a fast, efficient cloning strategy for site-independent insertion of different immuno- and affinity tags to target proteins. First, we have examined six N- and six C-terminal-tagged GABAAR γ2 subunits and found that only a single construct fulfilled all the requirements; namely high detection efficiency, genuine wild-type-like subcellular distribution and the rescue of mIPSCs in γ2−/− cultures, while conferring normal amplitude, kinetics and BZ pharmacology to the GABAARs. We then generated both lenti- and adeno-associated viruses, which contained both the Cre-recombinase and the N-terminal AU1-tagged wild-type γ2 subunit, separated by a 2A-peptide-like sequence. These viruses allowed the brain region-specific swapping of γ277I with AU1γ277F. The resulting GABAARs had normal synaptic enrichment, unaltered kinetic properties and restored sensitivity to zolpidem.
Most of the recently developed pharmacogenetic approaches rely on the genetic introduction of an exogenous protein to the targeted mammalian neuron. This is true for the Drosophila allatostatin receptor (Lechner et al. 2002; Tan et al. 2006), the C. elegans ivermectin-sensitive Cl− channels (Slimko et al. 2002), the mutated human muscarinic receptor (Armbruster et al. 2007; Ferguson et al. 2011) and the α7 nicotinic and glycine receptor chimera (Magnus et al. 2011). The so-called ‘designer receptor’ approach holds great promise, allowing the selective increase or decrease in the activity of a well-defined neuronal population (Armbruster et al. 2007; Ferguson et al. 2011; Magnus et al. 2011). Here, the ligand-binding sites of known ligand-gated ion channels (e.g. α7 nicotinic) or G-protein-coupled receptors (e.g. human M4 muscarinic) are mutated in order to remove their sensitivity to their endogenous ligands and to confer sensitivity to small synthetic ligands that are otherwise pharmacologically inert. The introduction of such designer receptors either by creating transgenic animals or by using viral vectors makes the targeted cells selectively sensitive to pharmacological manipulations. The application of the appropriate drug activates these receptors, leading to either direct ion flux through the receptors or through G-protein-coupled inward-rectifying K+ channels. There are two potential problems with these methods. One is the unforeseen effects of the genetically coded and mutated receptors on the assembly and subcellular targeting of the endogenous wild-type proteins. How the pentameric assembly of endogenous Gly or nicotinic receptors in the endoplasmic reticulum is affected by the transgenically introduced α7-GlyR is unknown. A potentially even more alarming issue is the partial activation of the mutated receptors with their original endogenous ligands or their unforeseen constitutive activity. These problems would lead to changes in the passive membrane properties and the excitability of the transduced cells in the absence of the designed ligands. As these genetic manipulations take place either throughout the whole life of the animal or over several weeks in the case of viral transduction, such long-standing alterations in the excitability of the target cell population could lead to unforeseen compensatory effects. The pharmacogenetic approach developed by Wulff et al. (2007) is the only available method that does not depend on the introduction of exogenous proteins into the target cell population, conferring a great advantage to this method. However, the generation of two additional mouse lines – one expressing Cre-recombinase and the other expressing the wild-type γ2 subunit – makes the original method of Wulff et al. (2007) cumbersome. Moreover, γ277F subunit expression was driven by the L7 promoter, which may cause altered GABAAR-mediated inhibition during development. Here we demonstrated the development of lenti- and adeno-associated virus-based swapping of mutated γ277I with the AU1-tagged wild-type, zolpidem-sensitive γ2 subunit in cortical pyramidal cells. We also employed a large repertoire of anatomical and in vitro electrophysiological methods to verify the unaltered subcellular/synaptic distribution and unaltered kinetic properties of the virally introduced AU1γ277F subunit.
There is a general agreement that affinity tagging of proteins holds great promise (Giepmans et al. 2006), but could result in potential problems, the lack of which must be rigorously tested before the analysis of function with the tagged protein (Previtali et al. 2000; Limon et al. 2007). For ion channels, such functional tests are often confined to examining the ionic currents flowing through the channels when they are expressed in heterologous expression systems. It is now widely accepted that the function of ion channels is regulated by a large family of interacting proteins, which are only present in their natural subcellular environments (Ehlers et al. 1996; Moss & Smart, 2001; Trimmer & Rhodes, 2004). Thus, it seems rational to suggest that the functional verification of affinity tagging must be carried out in the natural environment of the protein. For most synaptic neurotransmitter receptors, this site is the postsynaptic density opposite to the presynaptic active zone where transmitter molecules are released. Thus the analysis of postsynaptic currents provides the best indication of any alteration of the receptor location, density, ligand-binding and gating properties. In addition, we believe that affinity-tagged proteins should be expressed in cells that do not express genomic, non-tagged copies of the same protein. A simple over-expression approach ends up with a competition between the tagged and non-tagged proteins, the result of which is highly unpredictable. The most foreseeable problem might be that only a small proportion of the proteins in a given location will be the tagged ones, and therefore their altered function might not be detected upon verification. For example, if tagged neurotransmitter receptors are expressed in nerve cells, which endogenously express the same receptor, a scenario might be envisaged whereby only a small fraction of the postsynaptically clustered receptors will be tagged, and therefore their altered conductance, kinetics or pharmacological properties will remain unnoticed when the synaptic currents are examined. This necessitates the use of either conventional knock-out animals/cultured cells or the endogenous gene might be deleted using the Cre-LoxP (or other similar recombination) system in only those cells where the tagged proteins are expressed. In the present study, we used virus-mediated expression of Cre and γ277F, which has a few advantages over the transgenic strategy used by Wulff et al. (2007). First, we can exclude unwanted alterations of the γ2 subunit expression during development that could affect neuronal circuit formation. Second, using multiple kinds of viruses carrying different promoters or elements of alternative conditional recombination systems, it will be possible to express γ277F in multiple cell types of different brain regions.
In the present work, using combined light- and electron microscopic immunolocalizations and in vitro patch-clamp electrophysiology, we have provided a proof of principle for the generation of an immunotagged GABAAR γ2 subunit that has unaltered subcellular distribution, synaptic density and kinetic properties. In γ2−/− cortical cells, the lentiviral expression of wild-type γ2 subunits rescued the kinetic and pharmacological properties of mIPSCs and such physiological and pharmacological rescue was indistinguishable when AU1γ277F was used. Having established the lack of functional effects of N-terminal tagging of the γ2 subunit with the small immunotag AU1 in cultured cells, we turned to in vivo experiments to verify the proper subcellular/synaptic location and the detectability of the tagged receptors with an anti-AU1 antibody. Light microscopic immunofluorescent localization allowed the efficient and highly specific visualization of the AU1γ277F subunit in cortical cells following in vivo stereotaxic injection of lenti- or adeno-asociated viruses. Intense immunofluorescent puncta decorated the Cre-expressing cells without any detectable signal in the surrounding non-transduced cells. Not only was the detection efficiency of the AU1 tag remarkable, but also the extremely low non-specific labelling outside the injection zone. To our great delight, the AU1 tag was also detected very efficiently with the highly sensitive, high resolution SDS-FRL method, opening up new avenues for the applicability of AU1 immunotagging cell surface proteins, including neurotransmitter receptors and ion channels.
Acknowledgments
This work was supported by a Fellowship from the Japan Society for the Promotion of Science to M.S.; a grant from Solution Oriented Research for Science and Technology (SORST), Japan Science and Technology Agency to R.S.; grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grants-in-Aid for Scientific Research on Priority Areas – Molecular Brain Science (18022043) to Y.F.; and Grants-in-Aid for Young Scientists (18680903) to K.M.; a Project Grant (090197/Z/09/Z) and an Equipment Grant (083484/Z/07/Z) from the Wellcome Trust; Hungarian National Office for Research and Technology-French National Research Agency TéT Fund (NKTH-Neurogen) to Z.N. Support from these foundations is gratefully acknowledged. We would like to thank Drs Peer Wulff and William Wisden for providing the GABAA receptor γ2-L subunit cDNA and for the GABAARγ277Ilox mice, Dr Bernhard Luscher for the GABAARγ2+/− mice, Profs Werner Sieghart and Jean-Marc Fritschy for the anti-γ2 antibodies and Dr Pavel Osten for plasmids for lentivirus.
Glossary
Abbreviations
- BZ
benzodiazepine
- eGFP
enhanced green fluorescent protein
- GABAAR
type-A GABA receptor
- LM
light microscopic
- PC
pyramidal cell
- RE
restriction endonuclease
- SDS-FRL
sodium dodecyl sulphate-digested freeze-fracture replica-labelling
Author contributions
M.S. and Y.F. generated the lentiviruses, performed the transduction of cells and analysed the data in cultures in Japan. K.M. performed the whole-cell patch-clamp recordings on cultured cells and analysed the data in Japan. M.D.E. carried out patch-clamp recordings in acute slices and analysed his data in Hungary. A.L. performed the immunohistochemical experiments in cortical sections and carried out the SDS-FRL experiments in Hungary. Z.N. and R.S. designed the project and drafted the article. All authors approved the final version of the manuscript.
Supplementary material
S. Fig. 1
S. Fig. 2
S. Fig. 1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway EM. A molecular and genetic arsenal for systems neuroscience. Trends Neurosci. 2005;28:196–201. doi: 10.1016/j.tins.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Craig AM, Blackstone CD, Huganir RL, Banker G. Selective clustering of glutamate and γ-aminobutyric acid receptors opposite terminals releasing the corresponding neurotransmitters. Proc Natl Acad Sci U S A. 1994;91:12373–12377. doi: 10.1073/pnas.91.26.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol. 2001;82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Mammen AL, Lau LF, Huganir RL. Synaptic targeting of glutamate receptors. Curr Opin Cell Biol. 1996;8:484–489. doi: 10.1016/s0955-0674(96)80024-x. [DOI] [PubMed] [Google Scholar]
- Essricht C, Lorez M, Benson JA, Fritschy J-M, Lüscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, Neumaier JF. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K. Freeze-fracture replica electron microscopy combined with SDS digestion for cytochemical labelling of integral membrane proteins. Application to the immunogold labelling of intercellular junctional complexes. J Cell Sci. 1995;108:3443–3449. doi: 10.1242/jcs.108.11.3443. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Günther U, Benson J, Benke D, Fritschy JM, Reyes G, Knoflach F, Crestani F, Aguzzi A, Arigoni M, Lang Y, Bluethmann H, Mohler H, Lüscher B. Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of γ-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara A, Fukazawa Y, Deguchi-Tawarada M, Ohtsuka T, Shigemoto R. Differential distribution of release-related proteins in the hippocampal CA3 area as revealed by freeze-fracture replica labelling. J Comp Neurol. 2005;489:195–216. doi: 10.1002/cne.20633. [DOI] [PubMed] [Google Scholar]
- Kasugai Y, Swinny JD, Roberts JD, Dalezios Y, Fukazawa Y, Sieghart W, Shigemoto R, Somogyi P. Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur J Neurosci. 2010;32:1868–1888. doi: 10.1111/j.1460-9568.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Wang J, Connolly CN, Vicini S, Smart TG, Moss SJ. Analysis of GABAA receptor assembly in mammalian cell lines and hippocampal neurons using γ2 subunit green fluorescent protein chimeras. Mol Cell Neurosci. 2000;16:440–452. doi: 10.1006/mcne.2000.0882. [DOI] [PubMed] [Google Scholar]
- Lechner HA, Lein ES, Callaway EM. A genetic method for selective and quickly reversible silencing of mammalian neurons. J Neurosci. 2002;22:5287–5290. doi: 10.1523/JNEUROSCI.22-13-05287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerchner W, Xiao C, Nashmi R, Slimko EM, van Trigt L, Lester HA, Anderson DJ. Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl− channel. Neuron. 2007;54:35–49. doi: 10.1016/j.neuron.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Limon A, Reyes-Ruiz JM, Eusebi F, Miledi R. Properties of GluR3 receptors tagged with GFP at the amino or carboxyl terminus. Proc Natl Acad Sci U S A. 2007;104:15526–15530. doi: 10.1073/pnas.0706773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;238:906–909. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClive PJ, Sinclair AH. Rapid DNA extraction and PCR-sexing of mouse embryos. Mol Reprod Dev. 2001;60:225–226. doi: 10.1002/mrd.1081. [DOI] [PubMed] [Google Scholar]
- Magnus CJ, Lee PH, Atasoy D, Su HH, Looger LL, Sternson SM. Chemical and genetic engineering of selective ion channel-ligand interactions. Science. 2011;333:1292–1296. doi: 10.1126/science.1206606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masugi-Tokita M, Shigemoto R. High-resolution quantitative visualization of glutamate and GABA receptors at central synapses. Curr Opin Neurobiol. 2007;17:387–393. doi: 10.1016/j.conb.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Naylor D, Mody I. Synapse-specific contribution of the variation of transmitter concentration to the decay of inhibitory postsynaptic currents. Biophys J. 2001;80:1251–1261. doi: 10.1016/S0006-3495(01)76101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previtali SC, Quattrini A, Fasolini M, Panzeri MC, Villa A, Filbin MT, Li W, Chiu SY, Messing A, Wrabetz L, Feltri ML. Epitope-tagged P0 glycoprotein causes Charcot-Marie-Tooth-like neuropathy in transgenic mice. J Cell Biol. 2000;151:1035–1046. doi: 10.1083/jcb.151.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsova Z, Malik JM, Michel U, Scholl U, Bahr M, Kugler S. Evaluation of epitope tags for protein detection after in vivo CNS gene transfer. Eur J Neurosci. 2006;23:1961–1969. doi: 10.1111/j.1460-9568.2006.04725.x. [DOI] [PubMed] [Google Scholar]
- Slimko EM, McKinney S, Anderson DJ, Davidson N, Lester HA. Selective electrical silencing of mammalian neurons in vitro by the use of invertebrate ligand-gated chloride channels. J Neurosci. 2002;22:7373–7379. doi: 10.1523/JNEUROSCI.22-17-07373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szybalski W, Kim SC, Hasan N, Podhajska AJ. Class-IIS restriction enzymes – a review. Gene. 1991;100:13–26. doi: 10.1016/0378-1119(91)90345-c. [DOI] [PubMed] [Google Scholar]
- Tan EM, Yamaguchi Y, Horwitz GD, Gosgnach S, Lein ES, Goulding M, Albright TD, Callaway EM. Selective and quickly reversible inactivation of mammalian neurons in vivo using the Drosophila allatostatin receptor. Neuron. 2006;51:157–170. doi: 10.1016/j.neuron.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- Tomic M, Sunjevaric I, Savtchenko ES, Blumenberg M. A rapid and simple method for introducing specific mutations into any position of DNA leaving all other positions unaltered. Nucleic Acids Res. 1990;18:1656. doi: 10.1093/nar/18.6.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66:477–519. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Wulff P, Goetz T, Leppa E, Linden AM, Renzi M, Swinny JD, Vekovischeva OY, Sieghart W, Somogyi P, Korpi ER, Farrant M, Wisden W. From synapse to behaviour: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat Neurosci. 2007;10:923–929. doi: 10.1038/nn1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.