

Abstract
The tandem ylide-formation/[2,3]-sigmatropic rearrangement between donor/acceptor rhodium-carbenoids and chiral allyl alcohols is a convergent C—C bond forming process, which generates two vicinal stereogenic centers. Any of the four possible stereoisomers can be selectively synthesized by appropriate combination of the chiral catalyst Rh2(DOSP)4 and the chiral alcohol.
1. INTRODUCTION
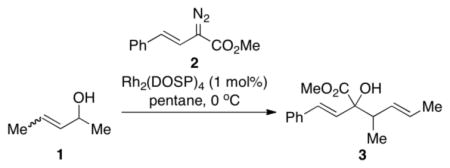
Chiral allylic alcohols are readily available and have been widely used as versatile building blocks in organic synthesis.1 Recently we discovered an unexpected reaction between rhodium carbenoids and allylic alcohols.2 Normally alcohols react with carbenoids to form O—H insertion products.3–5 However, we found that the reaction between donor/acceptor-substituted carbenoids and racemic allylic alcohols bearing a 3,3-dimethyl functionality resulted in an enantioselective [2,3]-sigmatropic rearrangement.2a,6 Homoallylic alcohols containing a single stereogenic center were formed in which the enantioselectivity was governed by the chirality of the catalyst rather than the chirality of the starting alcohol. As the resulting products can be used in extended domino sequences,2b we became interested in broadening the substrate scope and generality of the reaction. In particular, we wished to explore the possibility of generating products containing vicinal stereocenters in a stereoselective manner (Scheme 1). In this paper, we demonstrate that all four of the possible stereoisomers of the products can be selectively and predictably generated by using the appropriate combination of chiral allylic alcohol and chiral catalyst. The allylic stereocenter of the products is controlled by the chirality of the allylic alcohol and the alkene geometry, whereas the homoallylic stereocenter is dictated by the chirality of the catalyst.
Scheme 1.

Rhodium(II)-catalyzed [2,3]-sigmatropic rearrangement of allyl alcohols
2. RESULTS AND DISCUSSION
We began our investigations by studying the reaction of the stereoisomers of 3-penten-2-ol (1) with styryldiazoacetate 2, catalyzed by either Rh2(R-DOSP)4 or Rh2(S-DOSP)4 (Table 1). The reactions of the four possible combinations of (E)-1 and Rh2(DOSP)4 revealed that all the stereoisomers of the products 3 could be obtained in a stereoselective manner (>9:1 dr and >99% ee7) (entries 1–4). A comparison of entries 1 and 2 (and 3 and 4) demonstrated that the stereo-center at C3 of the product was governed by the configuration of the allyl alcohol. In contrast, a comparison of entries 1 and 3 (and 2 and 4) demonstrated that the chiral catalyst controled the configuration at C2. The reactions of (S, Z)-1 with Rh2(R-DOSP)4 and Rh2(S-DOSP)4 were also examined (entries 5 and 6). Significant matched and miss-matched interactions between the chiral entities were displayed in these reactions.8 The Rh2(R-DOSP)4-catalyzed reaction of (S,Z)-1 with 2 was very efficient, generating (2S,3R)-3 in 69% yield and with 94:6 dr and >99% ee (entry 5). The stereochemical configuration of the product was the same as that of the product derived from the Rh2(R-DOSP)4-catalyzed reaction of (R,E)-1 (entry 4). However, the Rh2(R-DOSP)4-catalyzed reaction of (S,Z)-1 with 2 was a missmatched reaction. In this case a 3:1 mixture of diastereomers was produced in low overall yield (35% for the major diastereomer) (entry 6).
Table 1.
Stereocontrolling elements of the tandem ylide formation/[2,3]-sigmatropic rearrangement
| ||||||
|---|---|---|---|---|---|---|
| entrya | Substrate | Rh2(DOSP)4 | Product | Yield, %b | drc | ee, %d |
| 1 |
S,E-1 |
S |
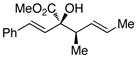 2R,3R-3 |
70 | 92:8 | >99 |
| 2 |
R,E-1 |
S |
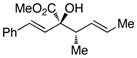 2R,3S-3 |
64 | 91:9 | >99 |
| 3 |
S,E-1 |
R |
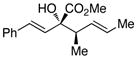 2S,3R-3 |
54 | 92:8 | >99 |
| 4 |
R,E-1 |
R |
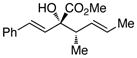 2S,3S-3 |
78 | 95:5 | >99 |
| 5 |
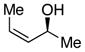 S,Z-1 |
R |
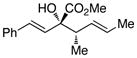 2S,3S-3 |
69 | 94:6 | >99 |
| 5 |
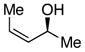 S,Z-1 |
S |
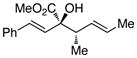 2R,3S-3 |
35 | 75:25 | >99 |
Reaction conditions: To a pentane solution of the allyl alcohol (1 equiv) and Rh2(S-DOSP)4 (0.01 equiv)at 0 °C under an atmosphere of Ar was added a solution of the diazo compound (1.1 equiv) in pentane solution over 1.5 h. The reaction was stirred for a further 1 h at 0 °C and then concentrated under reduced pressure.
Isolated yield of the major diastereomer.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by chiral HPLC.
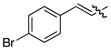
The tandem ylide formation/[2,3]-sigmatropic rearrangement was examined with a series of donor/acceptor-substituted diazoacetates with a variety of aryl and alkenyl substituents. In all cases, the major diastereomer was produced with very high asymmetric induction (>99% ee), but the diastereoselectivity was variable. In the case of the aryldiazoacetates, 4a and 4b, the diastereo-selectivity was ≥9:1 (Table 2, entries 1–2). The p-bromostyryl derivative 4c (entry 3) was comparable to the unsubstituted phenyl system (Table 1, entry 1). The butenyl- and propenyl-substituted diazo compounds (4d and 4e, respectively) underwent the rhodium-catalyzed transformation with high levels of asymmetric induction (entries 4 and 5). These results are consistent with previous examples of the high enantioselectivity exhibited in the Rh2(S-DOSP)4 catalyzed reactions of diazoacetates 4a-e.2 In entry 6, the unsubstituted vinyldiazoacetate 4f was obtained in modest yield (43%) and with poor diastereoselectivity (79:21 dr) It is well established that Rh2(S-DOSP)4–catalyzed cyclopropanations with 4f proceed with moderate enantiocontrol9 and the moderate diastereoselectivity observed in entry 6 is consistent with a low level of stereocontrol by Rh2(S-DOSP)4 in this case.
Table 2.
Reaction of (S,E)-2 with 4a-f
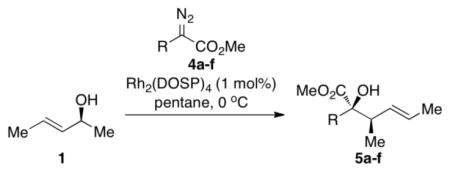
| |||||
|---|---|---|---|---|---|
| entry | comp’d | R | yield, %b | drc | eed |
| 1 | a |
|
56 | 94 : 6 | >99 |
| 2 | b |
|
66 | 90 : 10 | >99 |
| 3 | c |
|
69 | >95 : 5 | >99 |
| 4 | d |
|
60 | >95 : 5 | >99 |
| 5 | e |
|
55 | >95 : 5 | >99 |
| 6 | f |
|
43 | 79 : 21 | >99 |
Same reaction conditions as described in Table 1.
Isolated yield of the major diastereomer.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by chiral HPLC.
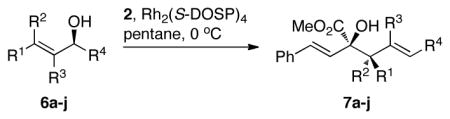
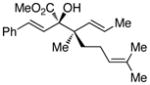
The tolerance of the reaction to various substituents on the alcohols was then studied, and these results are summarized in Table 3. In general, extended aliphatic and aryl substituents at the C3 position of the alcohol 6 were well tolerated (entries 1–2) including the 3,3-disubstituted substrate (entry 3). This substrate exemplified the utility of the metal-carbenoid transformation, facilitating the high yielding preparation of a product bearing two contiguous quaternary stereogenic centers with high levels of enantioselectvity and diastereoselectivity. Allyl alcohols with relatively bulky substituents, such as isopropyl and trimethylsilyl (6d and 6e) also afforded the corresponding rearrangement products, but the yields were modest (60% and 42%, respectively). An array of alcohols bearing C2-substitution (6f-h) were evaluated, and they were also amenable to this transformation (entries 6–8). It was expected that in the metal-bound oxonium-ylide intermediate formed any functionality at C2 would be oriented away from the catalyst and thus, would have little consequence on the reactivity. Finally, the effect of various functional groups at the carbinol position was explored in entries 9–11 and in all cases the desired products were formed. Of particular significance is the reaction of the mono-benzyl-protected 1,2-diol 6k, which was capable of selective reaction at the allylic alcohol over the benzyl ether functionality.
Table 3.
Scope of the allyl alcohol 6a
| ||||||
|---|---|---|---|---|---|---|
| entry | comp’d | 6 | 7 | yield, %b | drc | ee, %d |
| 1 | a |
|
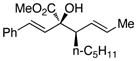
|
83 | >95 : 5 | >99 |
| 2 | b |
|
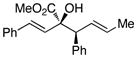
|
71 | >95 : 5 | >99 |
| 3 | c |
|
|
82 | >95 : 5 | >99 |
| 4 | d |
|
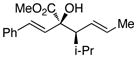
|
60 | >95 : 5 | >99 |
| 5 | e |
|
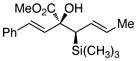
|
42 | >95 : 5 | 99 |
| 6 | f |
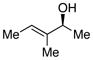
|
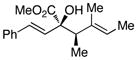
|
61 | >95 : 5 | >99 |
| 7 | g |
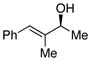
|
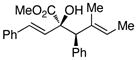
|
68 | >95 : 5 | >99 |
| 8 | h |
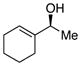
|
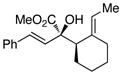
|
77 | >95 : 5 | >99 |
| 9 | i |
|
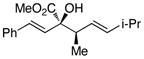
|
75 | >95 : 5 | >99 |
| 10 | j |
|
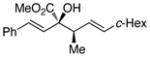
|
86 | >95 : 5 | >99 |
| 11 | k |
|
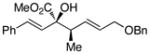
|
70 | >95 : 5 | >99 |
Same reaction conditions as described in Table 1.
Isolated yield of the major diastereomer.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by chiral HPLC.
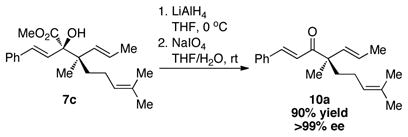
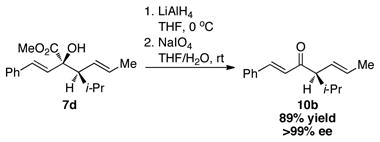
The synthetic utility of the rhodium-catalyzed sigmatropic rearrangement with the chiral alcohols lies in the ability to generate two adjacent stereogenic centers in a controlled and predictable manner. A distinctive feature of the transformation is the generation of a quaternary hydroxyl carbonyl moiety bearing a vicinal stereocenter, which is a structural feature embedded in a number of natural products.10 We also decided to demonstrate the broader synthetic potential of the reaction by illustrating a two-step conversion of the products to enones, containing a chiral center α to the carbonyl (equations 1 and 2). Enones containing quaternary (10a) and tertiary (10b) stereocenters α to the carbonyl were readily prepared in excellent yields. A particularly appealing feature of this approach to chiral enones is the likelihood that a chiral catalyst would not be required because the stereogenic center α to the carbonyl is controlled by the chirality of the starting alcohol.
 |
(1) |
 |
(2) |
Due to the uniformly high levels of asymmetric induction for the tandem ylide-formation/[2,3]-sigmatropic rearrangement, we sought a general transition-state model which would rationalize the observed stereochemical results.6 It has been well established that the Rh2(S-DOSP)4-catalyzed reactions of vinyldiazoacetates results in attack at the Re face of the vinylcarbenoid.11 The [2,3]-sigmatropic rearrangement would be expected to proceed through an envelope-like transition state, in which A1,3-strain is minimized.12 A reasonable model, which takes into account the established stereochemical understanding of these reactions is shown in Figure 1. Re face attack of the carbenoid by (S,E)-1 would generate an intermediate that would preferentially undergo a 2,3-sigmatropic rearrangement through TS-A, in which the A1,3 strain is minimized. This transition state would lead to the formation of the observed (2R,3R) isomer. Likewise, the reaction of (R,E)-1 would proceed through TS-B, which would generate the (2R,3S) isomer. The Re face attack on the carbenoid controls the stereochemistry at C2 in the product and at least in the case of (E)-1, the carbenoid-induced stereogenic center does not have a significant influence on the stereochemistry of the [2,3]-sigmatropic rearrangement.
Figure 1.

Transition-state analysis for the formation of 3.
3. CONCLUSION
In summary, the tandem ylide-formation/[2,3]-sigmatropic rearrangement between donor/acceptor rhodium-carbenoids and chiral allyl alcohols is a convergent C—C bond forming process, which generates two vicinal stereogenic centers. Any of the four possible stereoisomers can be selectively synthesized by appropriate combination of the chiral catalyst Rh2(DOSP)4 and the chiral alcohol. Only traces of O—H insertion products are observed in these reactions, which further illustrates the difference in reactivity of donor/acceptor carbenoids compared to conventional carbenoids, lacking a donor group.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (GM099142). We thank Dr. Ken Hardcastle (Emory University) for the X-ray crystallographic structural determination.
Footnotes
Supporting Information. Synthetic details and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Hodgson DM, Humphreys PG. Science of Synthesis. 2008;36:583–665. [Google Scholar]; (b) Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394–1401. doi: 10.1021/ar7001123. [DOI] [PubMed] [Google Scholar]; (c) Jeso V, Micalizio GC. J Am Chem Soc. 2010;132:11422–11424. doi: 10.1021/ja104782u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kerrigan MH, Jeon SJ, Chen YK, Salvi L, Carroll PJ, Walsh PJ. J Am Chem Soc. 2009;131:8434–8445. doi: 10.1021/ja809821x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Light-burn TE, De Paolis OA, Cheng KH, Tan KL. Org Lett. 2011;13:2686–2689. doi: 10.1021/ol200782d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xie Y, Floreancig PE. Chem Sci. 2011;2:2423–2427. [Google Scholar]; (g) Yamashita Y, Gopalarathnam A, Hartwig JF. J Am Chem Soc. 2007;129:7508–7509. doi: 10.1021/ja0730718. [DOI] [PubMed] [Google Scholar]
- 2.(a) Li Z, Davies HML. J Am Chem Soc. 2010;132:396–401. doi: 10.1021/ja9075293. [DOI] [PubMed] [Google Scholar]; (b) Parr BT, Li Z, Davies HML. Chem Sci. 2011;2:2378–2382. doi: 10.1039/C1SC00434D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reviews: Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylide. Wiley; New York: 1998. pp. 433–436.Miller DJ, Moody CJ. Tetrahedron. 1995;51:10811–10843.
- 4.For representative recent examples of O—H insertion, see: Bartrum HE, Blakemore DC, Moody CJ, Hayes CJ. Chem-Eur J. 2011;17:9586–9589. doi: 10.1002/chem.201101590.Chamni S, HeQ-LDang Y, Bhat S, Liu JO, Romo D. Chem Biol. 2011;6:1175–1181. doi: 10.1021/cb2002686.Peddibhotla S, Dang Y, Liu JO, Romo D. J Am Chem Soc. 2007;129:12222–12231. doi: 10.1021/ja0733686.Maier TC, Fu GC. J Am Chem Soc. 2006;128:4594–4595. doi: 10.1021/ja0607739.Chen C, Zhu SF, Liu B, Wang L-X, Zhou Q-L. J Am Chem Soc. 2007;129:12616–12617. doi: 10.1021/ja074729k.Zhu SF, Chen C, Zhou QL. Angew Chem Int Ed. 2008;47:932–934. doi: 10.1002/anie.200704651.Zhu SF, Song XG, Li Y, Cai Y, Zhou QL. J Am Chem Soc. 2010;132:16374–16376. doi: 10.1021/ja1078464.Wood JL, Moniz GA, Pflum DA, Stoltz BM, Holubec AA, Dietrich HJ. J Am Chem Soc. 1999;121:1748–1749.Wood JL, Moniz GA. Org Lett. 1999;1:371–374. doi: 10.1021/ol990697x.
- 5.For a computational study about the difference between copper(I) and dirhodium(II) complex in catalytic asymmetric OH insertion reactions, see: Liang Y, Zhou H, Yu Z-X. J Am Chem Soc. 2009;131:17783–17785. doi: 10.1021/ja9086566.
- 6.For representative examples of carbenoid reactions involving [2,3]-sigmatropic rearrangement, see: Pirrung MC, Werner JA. J Am Chem Soc. 1986;108:6060–6062. doi: 10.1021/ja00279a076.Roskamp EJ, Johnson CR. J Am Chem Soc. 1986;108:6062–6063. doi: 10.1021/ja00279a077.Clark JS, Hayes ST, Wilson C, Gobbi L. Angew Chem, Int Ed. 2007;46:437–440. doi: 10.1002/anie.200603880.Clark JS, Dossetter AG, Whittingham WG. Tetrahedron Lett. 1996;37:5605–5608.Clark JS, Dossetter AG, Blake AJ, Li WS, Whittingham WG. Chem Commun. 1999:749–750.Clark JS, Wong Y-S. Chem Commun. 2000:1079–1080.Clark JS, Baxter CA, Castro JL. Synthesis. 2005:3398–3404.Doyle MP, Bagheri V, Claxton EE. J Chem Soc, Chem Commun. 1990:46–48.Murphy GK, West FG. Org Lett. 2006;8:4359–4361. doi: 10.1021/ol061772o.Marmsäter FP, Vanecko JA, West FG. Org Lett. 2004;6:1657–1660. doi: 10.1021/ol049493t.Marmsäter FP, West FG. J Am Chem Soc. 2001;123:5144–5145. doi: 10.1021/ja015872v.Ma M, Peng L, Li C, Zhang X, Wang J. J Am Chem Soc. 2005;127:15016–15017. doi: 10.1021/ja055021d.Jaber DM, Burgin RN, Helper M, Zavalij PY, Doyle MP. Org Lett. 2012;14:1676–1679. doi: 10.1021/ol300213u.
- 7.The absolute configurations of five of the [2,3]-sigmatropic rearrangement products were unambiguously determined by X-ray crystallography. The absolute configurations of the remaining products were assigned by analogy. For a full description of the stereochemical assignment of the products, see the supporting information.
- 8.For examples of matchds/missmatched reactions in the rhodium carbenoid field, see: Martin SF, Spaller MR, Liras S, Hartmann B. J Am Chem Soc. 1994;116:4493–4494.Doyle MP, Dyatkin AB, Kalinin AV, Ruppar DA, Martin SF, Spaller MR, Liras S. J Am Chem Soc. 1995;117:11021–11022.Doyle MP, Kalinin AV, Ene DG. J Am Chem Soc. 1996;118:8837–8846.Nadeau E, Ventura DL, Brekan JA, Davies HML. J Org Chem. 2010;75:1927–1939. doi: 10.1021/jo902644f.Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J Am Chem Soc. 2010;132:12422–12425. doi: 10.1021/ja103916t.
- 9.(a) Miller LC, Ndungu JM, Sarpong R. Angew Chem, Int Ed. 2009;48:2398–2402. doi: 10.1002/anie.200806154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J Am Chem Soc. 2010;132:12422–12425. doi: 10.1021/ja103916t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J Am Chem Soc. 2009;131:8329–8332. doi: 10.1021/ja9019484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Mandala SM, Harris GH. Methods Enzymol. 2000;311:335–348. doi: 10.1016/s0076-6879(00)11094-8. [DOI] [PubMed] [Google Scholar]; (b) Kupchan SM, Sigel CW, Matz MJ, Renauld JAS, Haltiwanger RC, Bryan RF. J Am Chem Soc. 1970;92:4476–4477. [Google Scholar]; (c) Kupchan SM, Sigel CW, Matz MJ, Gilmore CJ, Bryan RF. J Am Chem Soc. 1976;98:2295–2300. doi: 10.1021/ja00424a050. [DOI] [PubMed] [Google Scholar]; (d) Purushothaman KK, Chandrasekharan S, Cameron AF, Connolly JD, Labbe C, Maltz A, Rycroft DS. Tetrahedron Lett. 1979;20:979–980. [Google Scholar]; (e) Fakunle CO, Connolly JD, Rycroft DS. J Nat Prod. 1989;52:279–283. [Google Scholar]; (f) Bergstrom JD, Kurtz MM, Rew DJ, Amend AM, Karkas JD, Bostedor RG, Bansal VS, Dufresne C, VanMiddlesworth FL, Hensens OD. Proc Natl Acad Sci USA. 1993;90:80–84. doi: 10.1073/pnas.90.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies HML, Bruzinski PR, Lake DH, Kong N, Fall MJ. J Am Chem Soc. 1996;118:6897–6907. [Google Scholar]
- 12.(a) Mikami K, Azuma KI, Nakai T. Chem Lett. 1983:1379–1382. [Google Scholar]; (b) Mikami K, Azuma KI, Nakai T. Tetrahedron. 1984;40:2303–2308. [Google Scholar]; (c) O’Brien AG. Tetrahedron. 2011;67:9639–9667. [Google Scholar]; (d) Tsai DJS, Midland MM. J Org Chem. 1984;49:1842–1843. [Google Scholar]; (e) Carreira EM, Kvaerno L. Classics in Stereo-selective Synthesis. Wiley-VCH Verlag GmbH & Co; Weinheim: 2009. pp. 532–539. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.