

Abstract
The physiological role of the P2Y6 nucleotide receptor may involve cardiovascular, immune and digestive functions based on the receptor tissue distribution, and selective antagonists for this receptor are lacking. We have synthesized a series of symmetric aryl diisothiocyanate derivatives and examined their ability to inhibit phospholipase C (PLC) activity induced by activation of five subtypes of recombinant P2Y receptors. Several derivatives were more potent at inhibiting action of UDP at both human and rat P2Y6 receptors expressed in 1321N1 human astrocytes than activation of human P2Y1, P2Y2, P2Y4 and P2Y11 receptors. The inhibition by diisothiocyanate derivatives of 1,2-diphenylethane (MRS2567) and 1,4-di-(phenylthioureido) butane (MRS2578) was concentration-dependent and insurmountable, with IC50 values of 126 ± 15 nM and 37 ± 16 nM (human) and 101 ± 27 nM (rat), respectively. A derivative of 1,4-phenylendiisothiocyanate (MRS2575) inhibited only human but not rat P2Y6 receptor activity. MRS2567 and MRS2578 at 10 μM did not affect the UTP (100 nM)-induced responses of cells expressing P2Y2 and P2Y4 receptors, nor did they affect the 2-methylthio-ADP (30 nM)-induced responses at the P2Y1 receptor or the ATP (10 μM)-induced responses at the P2Y11 receptor. Other antagonists displayed mixed selectivities. The selective antagonists MRS2567, MRS2575 and MRS2578 (1 μM) completely blocked the protection by UDP of cells undergoing TNFα-induced apoptosis. Thus, we have identified potent, insurmountable antagonists of P2Y6 receptors that are selective within the family of PLC-coupled P2Y receptors.
Keywords: P2Y6 nucleotide receptor, GPCR, Pyrimidines, Purines, Isothiocyanate, Apoptosis
1. Introduction
In addition to their intracellular functions in signaling and genetic coding, nucleotides have an important role as extracellular signaling molecules. The P2 receptors consist of two superfamilies: 8 G protein-coupled 7TM receptors (P2Y) and 7 ligand-gated ion channels (P2X) [1–3]. The P2Y1,2,4,6,11 receptors are coupled preferentially to the activation of phospholipase C (PLC), via Gq/11 proteins. The P2Y12,13,14 receptors are coupled preferentially to the inhibition of adenylate cyclase. The agonist preference of these eight receptors differs greatly and includes both adenine and uracil nucleotides [4]. The pyrimidine-selective receptors include: P2Y4, P2Y6 and P2Y14 receptors, which are activated by UTP, UDP and UDP-glucose, respectively. P2Y2 receptors are activated by both ATP and UTP. The remaining subtypes are activated by adenine nucleotides.
The physiological role of the P2Y6 receptor may involve cardiovascular, immune, and digestive functions, based on the receptor tissue distribution [4–8]. P2Y6 receptors are associated with vasoconstriction [9]. Recently, we have reported that activation by UDP of P2Y6 receptors inhibited apoptosis induced by TNFα through a mechanism dependent on PKC and Erk [10]. Also, it was reported that extracellular nucleotides stimulated contractions of human cerebral arteries primarily by activation of the P2Y6 receptor [11], suggesting antagonists of the P2Y6 receptor may be useful in the treatment of vasospasm.
Selective antagonists for the P2Y6 receptor have not previously been reported [4]. The lack of selective receptor probes for the P2Y6 and other subtypes of nucleotide receptors has impeded the ability to examine their pharmacology. DIDS (1) and its dihydro derivative, H2DIDS (2) (Fig. 1) have been shown to inhibit the PLC activity induced by UDP at rat P2Y6 receptors [12]. It appears that both isothiocyanate groups are needed for this activity, since SITS (3) was inactive. In this study we have explored the structure–activity relationship (SAR) of aryl diisothiocyanates at P2Y receptors. We tested these compounds as antagonists of human P2Y1, P2Y2, P2Y4 and P2Y6 receptors stably expressed in 1321N1 astrocytoma cells and the human P2Y11 receptor stably expressed in CHO cells.
Fig. 1.

Structures of diisothiocyanates tested previously as inhibitors of P2Y receptors.
2. Materials and methods
2.1. Chemical synthesis
2.1.1. General procedure for the synthesis of 4 and 5
A mixture of the diamine (12 or 13, 1.0 mmol), sodium bicarbonate (4.0 mmol), water (10 ml) and chloroform (30 ml) was stirred at room temperature. After 10 min thiophosgene (4 mmol, Aldrich Chemical Co.) was added and the mixture stirred for 2 h at room temperature. The phases were separated, the organic layer was dried (Na2SO4) and evaporated. The residue was purified by silica gel chromatography eluting with ethyl acetate-petroleum ether (5:95) to furnish the product as a solid (yield 65–70%). trans-1,2-Di-(4-isothiocyanatophenyl)ethylene (4). 1H NMR (CDCl3): δ 7.14 (s, 2H), 7.34 (t, J = 12 Hz, 4H), 7.58 (d, J = 9 Hz, 4H). FAB-MS 295.1 (M + 1), 1,2-di-(4-isothiocyanatophenyl)ethane (5). 1H NMR (CDCl3): δ 2.87 (s, 4H), 6.92–7.22 (m, 8H). FAB-MS 297.1 (M + 1).
2.1.2. General procedure for the synthesis of 6–11
Either 1,3- or 1,4-phenylenediisothiocyanate (14 or 15, 5 mmol) [13] was dissolved in dry acetonitrile (20 ml). To the above solution was added alkyl diamine (1 mmol) in acetonitrile (10 ml), and the resulting reaction mixture was stirred at room temperature for 1 h. Solvent was removed by evaporation, and the residue was purified by silica gel chromatography eluting with methanol–chloroform (5:95) to furnish as a solid (yield 55–60%). 1,2-Di-[(4-isothiocyanatophenyl)-thioureido] ethane (6). 1H NMR (DMSO-d6): δ 3.61–3.78 (m, 4H), 7.51 (d, J = 8.7 Hz, 4H), 7.56 (d, J = 7.2 Hz, 4H), 8.00 (brs, 2H), 9.76 (brs, 2H). FAB-MS 445.1 (M + 1). 1,3-Di-[(4-isothiocyanatophenyl)-thioureido] propane (7). 1H NMR (DMSO-d6): δ 1.62–1.83 (m, 2H), 3.38–3.60 (m, 4H), 7.42 (d, J = 12 Hz, 4H), 7.52 (d, J = 12 Hz, 4H), 7.95 (brs, 2H), 9.65 (brs, 2H). FAB-MS 459.1 (M + 1). 1,4-Di-[(4-isothiocyanato phenyl)-thioureido] butane (8). 1H NMR (DMSO-d6): δ 1.51 (brs, 4H), 3.42 (brs, 4H), 7.20–7.58 (m, 8H), 7.85 (brs, 2H), 9.62 (brs, 2H). FAB-MS 473.1 (M + 1).
1,2-Di-[(3-isothiocyanato phenyl)-thioureido]ethane (9). 1H NMR (DMSO-d6): δ 3.71 (brs, 4H), 7.08–7.20 (m, 2H), 7.28–7.42 (brs, 4H), 7.61 (brs, 2H), 8.03 (brs, 2H), 9.72 (brs, 2H). FAB-MS 445.1 (M + 1). 1,3-Di-[(3-isothiocyanato-phenyl)-thioureido] propane (10). 1H NMR (DMSO-d6): δ 1.76–2.01 (m, 2H), 3.32 (brs, 4H), 6.95–7.45 (m, 6H), 7.55 (brs, 2H), 8.00 (brs, 2H), 9.65 (brs, 2H). FAB-MS 459.1 (M + 1). 1,4-Di-[(3-isothiocyanato phenyl)-thioureido] butane (11). 1H NMR (DMSO-d6): δ 1.58 (m, 4H), 3.45 (brs, 4H), 7.04–7.20 (m, 2H), 7.25–7.40 (m, 4H), 7.65 (m, 2H), 7.95 (brs, 1H), 9.62 (brs, 2H). FAB-MS 473.1 (M + 1).
2.2. Cell culture and membrane preparation
Human 1321N1 astrocytoma cells stably transfected with the hP2Y1–6 receptors and CHO cells stably transfected with the human P2Y11 receptors [14,15] were grown at 37 °C in a humidified incubator with 5% CO2/95% air in Dulbecco’s modified Eagle’s medium (JRH Biosciences, Inc.) supplemented with 10% fetal bovine serum (FBS), 100 Units/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. The cells were grown to ~60% confluence for the experiments.
For membrane preparation, human astrocytoma cells expressing human P2Y1 receptors were grown to approximately 80% confluence and then harvested. The cells were homogenized and suspended and then centrifuged at 100 × g for 5 min at room temperature. The pellet was resuspended in 50 mM tris(hydroxymethyl)aminomethane (Tris) hydrochloride buffer (pH 7.4). The suspension was homogenized with a polytron homogenizer (Brinkmann) for 10 s and was then recentrifuged at 20,000 × g for 20 min at 4 °C. The resultant pellets were resuspended in Tris buffer (pH 7.4), and the suspension was stored at −80 °C until the binding experiments. The protein concentration was measured with the Bradford assay [16].
2.3. Determination of inositol phosphates
The quantity of inositol phosphates was measured by a modification of the method of Kim et al. [17] and Gao et al. [18]. Agonists were dissolved as stock solutions in Tris buffer (pH 7.4), and antagonists were dissolved in DMSO (5 mM) and stored at −20 °C. The antagonists were not stable to storage in aqueous medium. The P2Y1–6-1321N1 and P2Y11-CHO cells were grown to confluence in 6-well plates in the presence of myo-[3H]inositol (2 μCi/ml) for 24 h. Cells were then treated for 30 min at 37 °C with antagonists or buffer in the presence of 20 mM LiCl, followed by another 30 min incubation at 37 °C with the appropriate agonist. Agonists used were: P2Y1, 2-MeSADP; hP2Y2, UTP; hP2Y4, UTP; hP2Y6, UDP; hP2Y11, ATP. The reaction was terminated upon aspiration of the medium and addition of cold formic acid (20 mM). After 30 min, supernatants were neutralized with NH4OH, and applied to Bio-Rad Dowex AG1-X8 anion exchange columns. All of the columns were then washed with water followed by a 60 mM sodium formate solution containing 5 mM sodium tetraborate. Total inositol phosphates were eluted with 1 M ammonium formate containing 0.1 M formic acid, and radioactivity were measured using a liquid scintillation counter.
2.4. Radioligand binding assays
P2Y1 receptor binding experiments were performed as previously described [19]. Briefly, membranes (40 μg protein) from astrocytoma cells stably expressing human P2Y1 receptors were incubated with [3H]MRS2279 (8 nM) for 30 min at 4 °C in a total assay volume of 200 μl. For adenosine A1 receptor binding, an agonist radioligand [3H]R-PIA (2.0 nM) was incubated with membranes (40 μg protein/tube) from CHO cells stably expressing human adenosine A1 receptors for 60 min at 25 °C [20]. Radiolabeled ligand concentrations used in all assays approximated the Kd values of the receptor. Binding reactions were terminated by filtration through Whatman GF/B glass-fiber filters under reduced pressure with a MT-24 cell harvester (Brandel), and radioactivity was determined with a 1414 liquid scintillation counter (Wallac, Win Spectral, Perkin Elmer Life Sciences).
2.5. Induction of apoptosis
TNFα was used to induce apoptosis. Medium containing 5 ng/ml cycloheximide was added to the cells grown to ~60% confluence. Cycloheximide was included in all experiments concerning TNFα-induced apoptosis. The cells were treated with both UDP and TNFα for 4 h. Antagonists (1 μM) were added to the incubation medium 20 min prior to addition of UDP (100 nM) and TNFα (20 ng/ml). After 4 h the media was changed and was left for 16 h. The media contained 5 ng/ml cycloheximide during the entire incubation. Cell death was observed 16 h later.
2.6. Degree of cell death
After treatment, cells were washed once with PBS and were stained with propidium iodide (PI) solution (final concentration; 2 μg/ml). The numbers of unstained (live) and stained (dead) cells were measured with a Cytoflour 4000 (Perseptive Biosystems) fluorescence plate reader, with excitation at 485 nm and emission at 645 nm.
2.7. Reagents
myo-[3H]Inositol (20 Ci/mmol) was obtained from American Radiolabeled Chemicals. Dowex AG 1-X8 resin was purchased from Bio-Rad. DMEM and FBS were from Life Technologies. [3H]MRS2279 was from Perkin-Elmer, and [3H]R-PIA was from Amersham. All other reagents were purchased from Sigma Chemical Co.
2.8. Statistical analysis
Pharmacological parameters were analyzed by Graph-PAD Prism software. Data were expressed as mean ± S.E.
3. Results
3.1. Chemistry
Symmetric aryl diisothiocyanate derivatives (4–11, Table 1) were synthesized as shown in Fig. 1. Diaryl-4,4′-diisothiocyanates (4 and 5) were prepared in good yields from their corresponding amines 12 and 13, respectively, by a standard procedure using thiophosgene and sodium bicarbonate [13]. The symmetric diisothiocyanates (6–11) were synthesized in good yield by a different route, i.e. from the interaction of alkyl diamines and large excess of either 1,3- or 1,4-phenylenediisothiocyanates (Fig. 2).
Table 1.
Inhibition by aryl diisothiocyanate derivatives of the activation of PLC induced by P2Y receptor agonists
| Compound (MRS number) |
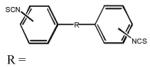
|
Position of NCS | Percent inhibitiona |
||||
|---|---|---|---|---|---|---|---|
| hP2Y1 | hP2Y2 | hP2Y4 | hP2Y6 | hP2Y11 | |||
| 4 (2568) | trans-CH=CH– | 4 | <5 | <5 | <5 | <5 | <5 |
| 5 (2567) | –CH2–CH2– | 4 | <5 | <5 | <5 | 95 ± 7 | <5 |
| 6 (2564) | –NHCSNH(CH2)2NHCSNH– | 4 | <5 | <5 | <5 | 77 ± 4 | 62 ± 4 |
| 7 (2575) | –NHCSNH(CH2)3NHCSNH– | 4 | <5 | <5 | <5 | 96 ± 6 | <5 |
| 8 (2576) | –NHCSNH(CH2)4NHCSNH– | 4 | 90 ± 5 | 92 ± 4 | 77 ± 4 | 78 ± 2 | <5 |
| 9 (2570) | –NHCSNH(CH2)2NHCSNH– | 3 | 80 ± 4 | 40 | <5 | 71 ± 10 | <5 |
| 10 (2577) | –NHCSNH(CH2)3NHCSNH– | 3 | <5 | <5 | 98 ± 2 | 86 ±6 | <5 |
| 11 (2578) | –NHCSNH(CH2)4NHCSNH– | 3 | 20 ± 3 | <5 | <5 | 100 ± 6 | <5 |
Concentration of antagonists was 10 μM. Specific subtypes of human P2Y receptors were expressed in 1321N astrocytoma cells (hP2Y1, hP2Y2, hP2Y4, and hP2Y6) or in CHO cells (hP2Y11) [13]. Agonists used were: P2Y1, 2-MeSADP (30 nM); hP2Y2, UTP (100 nM); hP2Y4, UTP (100 nM); hP2Y6, UDP (300 nM); hP2Y11, ATP (10 μM). The concentrations of agonists used were roughly equivalent to their EC50 values. The potency (IC50 values) of MRS2567, MRS2575 and MRS2578 at both human and rat P2Y6 receptors were listed in the text.
Fig. 2.

Synthetic routes for the preparation of diisothiocyanate derivatives.
3.2. Inhibition of agonist-induced production of inositol phosphates by aryl diisothiocyanate derivatives
The ability of the diisothiocyanate derivatives to inhibit agonist-induced accumulation of inositol phosphates in 1321N1 human astrocytes expressing P2Y6 receptors and other P2Y subtypes was studied. Fig. 3 indicates that the concentration–response curve for UDP acting at the P2Y6 receptor (EC50 230 ± 83 nM) was inhibited by the compounds 5 and 11, but in an insurmountable manner. Table 1 indicates that similar effects were obtained for other compounds in the series and at other subtypes of P2Y receptors. Typically, the effect was inhibited with the diisothiocyanate in a micromolar concentration range. Full concentration–response curves for the inhibition of the effects of UDP at P2Y6 receptors were measured for compounds 5 (MRS2567) and 11 (MRS2578) (Fig. 4), derivatives of 1,4- and 1,3-phenylendiisothiocyanate, respectively. Results indicated that the production of inositol phosphates in human P2Y6-transfected 1321N1 astrocytoma cells in response to 300 nM UDP was inhibited in a concentration-dependent manner with IC50 values of 126 ± 15 nM and 37 ± 16 nM, respectively. Also, these two selective inhibitors of the hP2Y6 receptor activation had approximately identical effects on rat P2Y6 receptors. MRS2567 and MRS2578 inhibited the response to 300 nM UDP at the rat P2Y6 receptor expressed in 1321N1 cells in a concentration-dependent manner, with IC50 values of 101 ± 27 nM and 98 ± 11 nM, respectively. MRS2567 and MRS2578 at 10 μM did not affect the UTP (100 nM)-induced responses of cells expressing human P2Y2 or P2Y4 receptors, nor did they affect the 2-MeSADP (30 nM)-induced responses of cells expressing the P2Y1 receptor. In addition, they did not affect the ATP (10 μM)-induced responses of cells expressing the P2Y11 receptor. Thus, MRS2567 and MRS2578 selectively blocked P2Y6 receptor activity versus activity at P2Y1, P2Y2, P2Y4 or P2Y11 receptors. However, MRS2577 blocked activity at both P2Y4 and P2Y6 receptors, without affecting other subtypes.
Fig. 3.

Inositol phosphate production in human P2Y6-transfected 1321N1 human astrocytes. After labeling with myo-[3H]inositol (1 μCi/106 cells) for 24 h, the cells were treated for 30 min at 37 °C with antagonists in the presence of LiCl, followed by addition of the agonist, UDP, for another 30 min. The quantity of inositol phosphates was analyzed after extraction though Dowex AG 1-X8 columns (see Section 2). Data shown are from the combined results of three independent experiments in triplicate. The EC50 value is listed in Section 3.
Fig. 4.
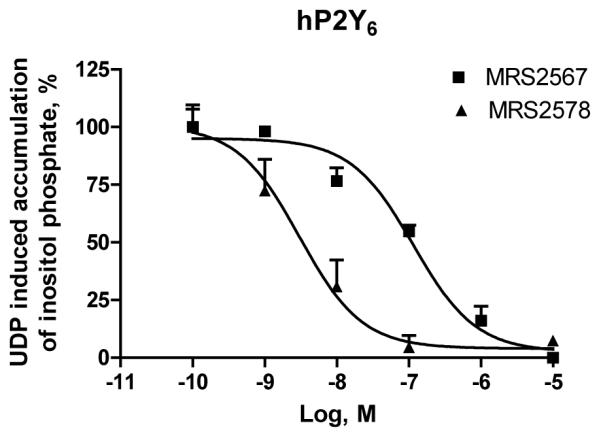
Concentration dependence of inhibition by compounds 5 and 11 of inositol phosphate production induced by UDP (300 nM) in human P2Y6-receptor transfected 1321N1 human astrocytes. After labeling with myo-[3H]inositol (1 μCi/106 cells) for 24 h, the cells were treated for 30 min at 37 °C with the antagonist or buffer in the presence of LiCl, followed by addition of agonist, UDP, for another 30 min. The quantity of inositol phosphates was analyzed after extraction though Dowex AG 1-X8 columns (see Section 2). Data shown represent the mean ± S.E.M. of three independent experiments in duplicate. IC50 values are listed in Section 3.
It was interesting that another derivative of 1,4-phenylendiisothiocyanate (MRS2575) inhibited only human P2Y6 receptor activity. This compound at 10 μM concentration had no effect on rat P2Y6 or human P2Y1, P2Y2, P2Y4 and P2Y11 receptors (Table 1). This antagonist inhibited in an insurmountable manner UDP activity at the P2Y6 receptor. Inhibition of human P2Y6 receptor activity by MRS2575 occurred in a concentration-dependent manner with an IC50 value of 155 ± 49 nM (Fig. 5A and B).
Fig. 5.

(A) Inositol phosphate production in human P2Y6-transfected 1321N1 human astrocytes. (B) Concentration dependence of inhibition by compound MRS2575 (7) of inositol phosphate production induced by UDP (300 nM) in human P2Y6-receptor transfected 1321N1 human astrocytes. After labeling with myo-[3H]inositol (1 μCi/106 cells) for 24 h, the cells were treated for 30 min at 37 °C with the antagonist or buffer in the presence of LiCl, followed by addition of agonist, UDP, for another 30 min. The quantity of inositol phosphates was analyzed after extraction though Dowex AG 1-X8 columns (see Section 2). Data shown represent the mean ± S.E.M. of three independent experiments in triplicate.
3.3. Aryl diisothiocyanate derivatives in receptor binding experiments
Binding experiments at the human A1 AR expressed in CHO cell membranes indicated that compounds MRS2567 (5), MRS2575 (7) and MRS2578 (11) at 10 μM concentration displayed no significant inhibition of radioligand binding (data not shown).
The diisothiocyanate derivatives MRS2576 (8) and MRS2570 (9) inhibited agonist effects at hP2Y1 receptors as well as at P2Y6 receptors, and consequently we examined the ability to interfere with radioligand binding at the former subtype. The high affinity antagonist [3H]MRS2279 was used as a P2Y1 receptor radioligand in binding to membranes from astrocytoma cells stably expressing human P2Y1 receptors. Specific binding of [3H]MRS2279 was defined in these experiments using the antagonist MRS2179 (10 μM). Essentially no inhibition of binding of [3H]MRS2279 was detected in the presence of 10 μM MRS2576 and MRS2570 (data not shown), both of which antagonized the P2Y1 receptor-mediated effects of 2-MeSADP.
3.4. Inhibition of the anti-apoptotic effects of a P2Y6 receptor agonist
The study of P2Y receptors has been a challenge, largely due to the paucity of pharmacological tools for identification of receptor subtypes. Since truly selective antagonists for most of the subtypes have not yet been developed, the characterization of P2Y receptors has mainly consisted of monitoring agonist responses, such as activation of PLC. One functional effect of P2Y6 nucleotide receptor activation is its cellular protective role. Activation of the rat P2Y6 receptor expressed in 1321N1 astrocytoma cells by UDP has been shown to protect the cells from undergoing apoptosis induced by TNFα [10,21]. The P2Y6 receptor is a GPCR that is coupled to PLC, and the subsequent activation of protein kinase C (PKC) by the receptor has been shown to relate to this protective effect. We studied the present set of P2Y6 receptor antagonists in a model of the anti-apoptotic effects of P2 receptor agonists.
We have extended previous results with the rat P2Y6 receptor [10,21] to the human P2Y6 receptor. The activation of the human P2Y6 receptor by UDP (100 nM) in the presence of TNFα (20 ng/ml) and cycloheximide (5 ng/ml), which sensitizes the TNFα response, significantly reduced cell death (Fig. 6). Four antagonists of P2Y6 receptors were used in the experiment, including the selective aryl diisothiocyanate derivatives MRS2567 (5), MRS2575 (7) and MRS2578 (11) and the non-selective derivative MRS2564 (6) (1 μM). Measurements of intracellular PI staining, a measure of cell death, indicated that MRS2567, MRS2575 and MRS2578 completely blocked the protection by UDP (100 nM). Therefore, it was demonstrated using antagonists that P2Y6 receptor activation by UDP played a crucial role in preventing 1321N1 astrocytoma cells from undergoing TNFα-induced apoptosis.
Fig. 6.
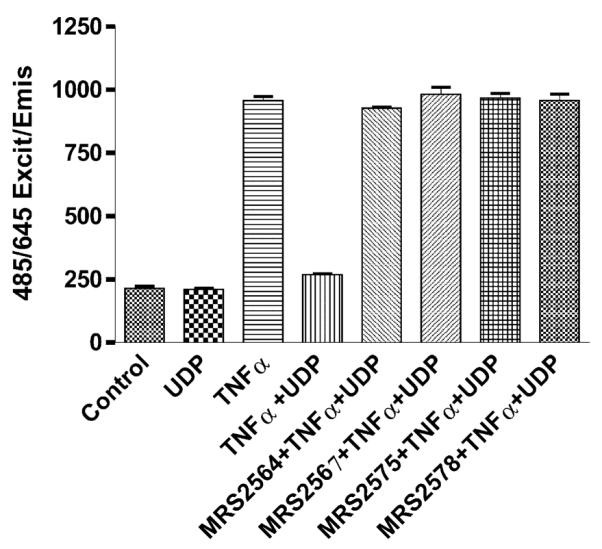
Effects of P2Y6 receptor antagonists on cell death induced by the treatment of hP2Y6 receptor-expressing 1321N1 astrocytoma cells with TNFα. Cells were treated with both UDP and TNFα for 4 h. Antagonists (1 μM) were added to the incubation medium 20 min prior to addition of UDP (100 nM) and TNFα (20 ng/ml). The media contained 5 ng/ml cycloheximide during the entire incubation. After 16 h of continuous treatment (as described in Section 2) the cell culture medium was replaced by medium containing PI (2 μg/ml). Since PI transverses only leaky or lysed cell membranes, DNA of dead cells can be stained and fluorescence emission measured, while living cells will not be stained. Fluorescence was measured using the Cytofluor 4000 microplate reader (excitation 485 nm/emission 645 nm), giving a direct number of dead cells. Results represent the mean value of two experiments, each performed in triplicate.
4. Discussion
The present study demonstrated that MRS2567 and MRS2578 block agonist effects at both human and rat P2Y6 receptors, and MRS2575 selectively blocks effects at human P2Y6 but not rat P2Y6 receptors. This is the first report of selective antagonists for P2Y6 receptors, albeit insurmountable. Previous studies have shown that DIDS and H2DIDS block P2Y6 receptors at concentrations of 10–100 μM and are clearly less potent than MRS2567, MRS2578 and MRS2575 [12]. Compounds MRS2567 and MRS2578 can block UDP-stimulated activity at concentrations less than 1 μM (for human P2Y6 at IC50 values 126 ± 15 nM and 37 ± 16 nM, for rat P2Y6 at IC50 values 101 ± 27 nM and 98 ± 11 nM, respectively). Interestingly, MRS2575 is a selective antagonist for human P2Y6 receptors with an IC50 value of 155 ± 49 nM, while it has no effect on rat P2Y6 receptors. Since other compounds in this series inhibited other P2Y receptor subtypes, a panel of such diisothiocyanates may be useful in characterizing a given pharmacological response to extracellular nucleotides. For example, the following mixed selectivities were observed: MRS 2564 (P2Y6, P2Y11), MRS 2576 (P2Y1, P2Y2, P2Y4, P2Y6), and MRS 2577 (P2Y4, P2Y6).
It is likely that the isothiocyanate groups are involved in the antagonistic effects at P2Y6 receptors. All of the derivatives 4–11 evaluated as P2Y receptor antagonists included two isothiocyanate groups. Isothiocyanate groups are potent electrophiles that are sufficiently stable in aqueous medium for biochemical experiments, yet may be chemically reactive towards certain nucleophilic groups on biopolymers [22], such as Lys and Cys side chains. The possible covalent reaction of these antagonists with the P2Y6 receptor protein has not been demonstrated. Future studies will be needed to establish the location(s) of cross-linking, if any, with the receptor. The degree of inhibition and specificity of the antagonists depended on the chain length and position of ring substitution. Thus, these inhibitory effects follow a specific SAR pattern and are not the result of general reactivity of the electrophilic group. The selectivity of these derivatives was further indicated by their inability to block binding at the human A1AR, which is know to be sensitive to other electrophilic reagents [13].
The inability of these aryl diisothiocyanate derivatives to produce a parallel rightward shift of the concentration–response curves suggests that they are insurmountable P2YR antagonists [23]. Moreover, the failure of selected derivatives to displace radioligand binding at the human P2Y1R suggests that they are non-competitive antagonists at the P2Y1 subtype. Since there is no radioligand for P2Y6 receptors, it was not possible to carry out similar binding experiments at this subtype. An additional functional experiment using the antagonists has supported the finding [10,21] that activation of the human P2Y6 receptor by the agonist UDP prevented TNFα-induced apoptosis, since selective antagonists MRS2575, MRS2567, and MRS2578 at a concentration of 1 μM completely blocked agonist-induced activation of the P2Y6 receptor.
In summary, we have identified potent, insurmountable antagonists of P2Y6 receptors that are selective within the family of PLC-coupled P2Y receptors. These selective antagonists could be used as pharmacological tools for defining the role of P2Y6 and other P2Y receptors.
Acknowledgments
L.K. Mamedova and B.V. Joshi thank Gilead Sciences (Foster City, CA) for financial support. We thank Prof. Robert Nicholas and Prof. T. Kendall Harden of the University of North Carolina (Chapel Hill, NC, USA) for providing the P2Y6-receptor-transfected cells. We thank Dr. Neli Melman (NIDDK) for technical assistance.
Abbreviations
- CHO
Chinese hamster ovary
- DIDS
4,4′-diisothiocya-natostilbene-2,2′-disulfonic acid disodium salt
- MRS2279
2-chloro-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate
- PIA
N6-phenylisopropyladenosine
- PLC
phospholipase C
- TNFα
tumor necrosis factor α
- Tris
tris(hydroxymethyl)aminomethane
References
- [1].Barnard EA, Burnstock G, Webb TE. G-protein coupled receptors for ATP and other nucleotides: a new receptor family. Trends Pharmacol Sci. 1994;15:67–70. doi: 10.1016/0165-6147(94)90280-1. [DOI] [PubMed] [Google Scholar]
- [2].North RA. A family of ion channels with two hydrophobic segments. Curr Opin Cell Biol. 1996;8:474–83. doi: 10.1016/s0955-0674(96)80023-8. [DOI] [PubMed] [Google Scholar]
- [3].Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–92. [PubMed] [Google Scholar]
- [4].Jacobson KA, Jarvis MF, Williams M. Purine and pyrimidine (P2) receptors as drug targets. J Med Chem. 2002;45:4057–93. doi: 10.1021/jm020046y. [DOI] [PubMed] [Google Scholar]
- [5].Somers GR, Bradbury R, Trute L, Conigrave A, Venter DJ. Expression of the human P2Y6 nucleotide receptor in normal placenta and gestational trophoblastic disease. Lab Invest. 1999;79:131–9. [PubMed] [Google Scholar]
- [6].Somers GR, Hammet FM, Trute L, Southey MC, Venter DJ. Expression of the P2Y6 purinergic receptor in human T cells infiltrating inflammatory bowel disease. Lab Invest. 1998;78:1375–83. [PubMed] [Google Scholar]
- [7].Hou M, Malmsjö M, Möller S, Pantev E, Bergdahl A, Zhao XH, et al. Increase in cardiac P2X1- and P2Y2-receptor mRNA levels in congestive heart failure. Life Sci. 1999;65:1195–206. doi: 10.1016/s0024-3205(99)00353-7. [DOI] [PubMed] [Google Scholar]
- [8].Bogdanov Y, Rubino A, Burnstock G. Characterisation of subtypes of the P2X and P2Y families of ATP receptors in the foetal human heart. Life Sci. 1998;62:697–703. doi: 10.1016/s0024-3205(97)01168-5. [DOI] [PubMed] [Google Scholar]
- [9].Lewis CJ, Ennion SJ, Evans RJ. P2 purinoceptor-mediated control of rat cerebral (Pial) microvasculature contribution of P2X and P2Y receptors. J Physiol. 2000;527:315–24. doi: 10.1111/j.1469-7793.2000.00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim SG, Soltysiak KA, Gao ZG, Chang TS, Chung E, Jacobson KA. Tumor necrosis factor alpha-induced apoptosis in astrocytes is prevented by the activation of P2Y6, but not P2Y4 nucleotide receptors. Biochem Pharmacol. 2003;65:923–31. doi: 10.1016/s0006-2952(02)01614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nicholas RA, Watt WC, Lazarowski ER, Li Q, Harden K. Uridine nucleotide selectivity of three phospholipase C-activating P2 receptors: identification of a UDP-selective, a UTP-selective and an ATP- and UTP-specific receptor. Mol Pharmacol. 1996;50:224–9. [PubMed] [Google Scholar]
- [12].von Kügelgen I, Jacobson KA. Blockade of P2Y6 receptors by diisothiocyanatostilbene-disulfonic acid (DIDS) and analogues. Naunyn-Schmiedeberg’s Arch Pharmacol. 2001;363:R34. [Google Scholar]
- [13].Stiles GL, Jacobson KA. High affinity acylating antagonists for the A1 adenosine receptor: identification of binding subunit. Mol Pharmacol. 1988;34:724–8. [PMC free article] [PubMed] [Google Scholar]
- [14].Communi D, Govaerts C, Parmentier M, Boeynaems JM. Cloning of a human purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase. J Biol Chem. 1997;272:31969–73. doi: 10.1074/jbc.272.51.31969. [DOI] [PubMed] [Google Scholar]
- [15].Qi AD, Kennedy C, Harden TK, Nicholas RA. Differential coupling of the human P2Y11 receptor to phospholipase C and adenylyl cyclase. Br J Pharmacol. 2001;132:318–26. doi: 10.1038/sj.bjp.0703788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;76:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [17].Kim SG, Ravi G, Hoffmann C, Jung Y-J, Kim M, Chen A, et al. p53-independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative, Cl-IB-MECA. Biochem Pharmacol. 2002;63:871–80. doi: 10.1016/s0006-2952(02)00839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gao ZG, Jeong LS, Moon HR, Kim HO, Choi WJ, Shin DH, et al. Structural determinants of efficacy at A3 adenosine receptors: modification of the ribose moiety. Biochem Pharmacol. 2004;67:893–901. doi: 10.1016/j.bcp.2003.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Waldo GL, Corbitt J, Boyer JL, Ravi G, Kim HS, Ji XD, et al. Quantitation of the P2Y1 receptor with a high affinity radiolabeled antagonist. Mol Pharmacol. 2002;62:1249–57. doi: 10.1124/mol.62.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;6:1675–84. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim SG, Gao ZG, Soltysiak KA, Chang TS, Brodie C, Jacobson KA. P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell Mol Neurobiol. 2003;23:401–18. doi: 10.1023/A:1023696806609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Newman AH, Lueddens HWM, Skolnick P, Rice KC. Novel irreversible ligands specific for “peripheral” type benzodiazepine receptors. J Med Chem. 1987;30:1901–4. doi: 10.1021/jm00393a036. [DOI] [PubMed] [Google Scholar]
- [23].Vauquelin G, Van Liefde I, Vanderheyden P. Models and methods for studying insurmountable antagonism. Trends Pharmacol Sci. 2002;23:514–8. doi: 10.1016/s0165-6147(02)02081-3. [DOI] [PubMed] [Google Scholar]