

Abstract
A3 adenosine receptor (A3AR) ligands have been modified to optimize their interaction with the A3AR. Most of these modifications have been made to the N6 and C2 positions of adenine as well as the ribose moiety, and using a combination of these substitutions leads to the most efficacious, selective, and potent ligands. A3AR agonists such as IB-MECA and Cl-IB-MECA are now advancing into Phase II clinical trials for treatments targeting diseases such as cancer, arthritis, and psoriasis. Also, a wide number of compounds exerting high potency and selectivity in antagonizing the human (h)A3AR have been discovered. These molecules are generally characterized by a notable structural diversity, taking into account that aromatic nitrogen-containing monocyclic (thiazoles and thiadiazoles), bicyclic (isoquinoline, quinozalines, (aza)adenines), tricyclic systems (pyrazoloquinolines, triazoloquinoxalines, pyrazolotriazolopyrimidines, triazolopurines, tricyclic xanthines) and nucleoside derivatives have been identified as potent and selective A3AR antagonists. Probably due to the “enigmatic” physiological role of A3AR, whose activation may produce opposite effects (for example, concerning tissue protection in inflammatory and cancer cells) and may produce effects that are species dependent, only a few molecules have reached preclinical investigation. Indeed, the most advanced A3AR antagonists remain in preclinical testing. Among the antagonists described above, compound OT-7999 is expected to enter clinical trials for the treatment of glaucoma, while several thiazole derivatives are in development as antiallergic, antiasthmatic and/or antiinflammatory drugs.
Keywords: A3 adenosine receptor, A3 adenosine receptor agonist, A3 adenosine receptor antagonist, Purines, Structure activity relationship, Nucleoside, G protein-coupled receptor, Neoceptor
1 Introduction
The four subtypes of adenosine receptors (ARs), designated A1, A2A, A2B, and A3, are all seven-transmembrane spanning (7TM) receptors that couple to G proteins. The A3AR inhibits adenylate cyclase through coupling to Gi. A3AR activation may lead to an activation of the phospholipase C pathway through the β, γ subunit. The A3AR is found at a high receptor density in the lungs, liver, and in immune cells such as neutrophils and macrophages, as well as at lower densities in the heart and brain (Fredholm et al. 2001) The A3AR is expressed in neurons in the brain (Lopes et al. 2003; Yaar et al. 2002).
ARs in general, and the A3AR in particular, are involved in many of the body’s cytoprotective functions. Recently, agents that act at the A3AR have been targeted for pharmaceutical development based on their anti-inflammatory, anticancer, and cardioprotective effects. For example, activation of the cardiac A3AR preconditions cardiac myocytes against ischemic damage (Strickler et al. 1996; Tracey et al. 2003) and protects against apoptosis. Selective A3AR agonists have been shown to protect cardiac muscle in various ischemic models and are protective against the cardiotoxic effects of the anticancer drug doxorubicin (Shneyvais et al. 2001). A3AR antagonists are of interest as potential antiglaucoma agents (Yang et al. 2005) and as anticancer agents (Gessi et al. 2008).
Agonist ligands for the ARs, including the A3AR, are almost exclusively nucleoside derivatives. The search for antagonists of the A3AR in the early 1990s initially encountered an unanticipated difficulty: the lack of an obvious lead structure. Previously, efforts to develop antagonist ligands for the A1 and A2A ARs focused on xanthine derivatives. However, at the A3AR, the prototypical AR antagonists (i.e., the xanthines) are typically much weaker in binding than at the other AR subtypes. This observation stimulated the screening of structurally diverse heterocyclic molecules as potential antagonists (Moro et al. 2006). Chemically diverse leads were discovered in this process that were subsequently optimized to achieve high antagonist selectivity for the A3AR.
While the A3AR may be activated by orthosteric agonists that are competitive with adenosine, the action of nucleosides at this receptor may also be enhanced by allosteric modulators. Several heterocyclic classes of positive allosteric modulators of the A3AR, including 1H-imidazo-[4,5-c]quinolines such as LUF6000 (N-(3,4-dichloro-phenyl)-2-cyclohexyl-1H-imidazo[4,5-c]quinolin-4-amine) and pyridinylisoquinolines, have been reported (Gao et al. 2005; Göblyös et al. 2006).
The structure–activity relationships (SARs) of nucleoside derivatives in binding to the A3AR and other ARs have been extensively studied, leading to the development of both selective agonists and, more recently, antagonists. Most of the useful modifications of adenosine 1 (Fig. 1) to achieve high A3AR affinity and selectivity have been made at the N6 or C2 positions of adenine or on the ribose group of adenosine. The systematic probing of SAR of both adenosine derivatives and nonpurine antagonists is frequently guided by molecular modeling (Kim et al. 2003), in which the receptor protein is modeled based on structural homology to the light receptor, rhodopsin. The effects of substitution at various sites (i.e., on the nucleobase and ribose moiety) on both the affinity and relative efficacy of nucleoside derivatives at the A3AR have been extensively probed (Gao et al. 2003, 2004). The approach initially taken to identify agonists for the newly cloned A3AR was to screen known AR ligands in binding assays. The agonist NECA 2 (adenosine 5′ -N-ethyluronamide) was found to be highly potent but nonselective for this receptor (Zhou et al. 1992). The structural features that promoted A3AR potency were combined, leading to the first selective A3 agonist, IB–MECA (N6-(3-iodobenzyl)-5′ -N-methylcarboxamidoadenosine), developed in 1993 at the National Institutes of Health (Jacobson et al. 1993). This potent A3AR agonist IB–MECA 3 and its more selective 2-chloro analog, Cl–IB–MECA 4 (2-chloro-N6-(3-iodobenzyl)-5′ -N-methylcarboxamidoadenosine), are used widely as pharmacological tools. A related derivative 5 is widely used as an iodinated radioligand for the A3AR. IB–MECA and Cl–IB–MECA have entered clinical trials for the treatment of rheumatoid arthritis and cancer (Baharav et al. 2005; Ohana et al. 2001).
Fig. 1.

Structures of adenosine and widely used agonist probes of the A3AR
One problem encountered in refining selective A3AR ligands into pharmaceutically useful agents has been the species dependence of binding. This difference in affinity reflects the difference in sequence between the rodent and the human receptors, with only a 74% sequence identity between the rat (r) and human (h) A3ARs (Fredholm et al. 2001). The species-dependence of A3AR affinity is particularly pronounced for agonists that contain small alkyl N6 substituents and for various heterocyclic antagonists, both of which are more potent in binding to the human than to the rat A3AR (Yang et al. 2005). The first report of a cloned receptor sequence to be later identified as an A3AR was that of the rat (Meyerhof et al. 1991; Zhou et al. 1992), and this species was initially used for screening purposes. Nevertheless, many of the nucleoside analogs that were shown to be rat A3AR agonists, including Cl–IB–MECA and IB–MECA, were later found to be moderately selective for the human A3AR after it was cloned (Jacobson and Gao, 2006; Salvatore et al. 1993).
The ligand recognition within the putative binding site of the ARs has also been probed through extensive mutagenesis to confirm the predictions concerning ligand recognition made using molecular modeling (Kim et al. 2003). The hydrophobic environment surrounding the purine ring of AR agonists, as found in the putative A2AAR model, is defined mainly by residues of TM5 and TM6 (Kim et al. 2003). This region is very similar to the putative binding region of hydrophobic heterocyclic (e.g., triazolopyrimidine) antagonists. An exocyclic amino group is common to both adenosine agonists and to typical heterocyclic antagonists, and this amine is generally required to donate a hydrogen bond to the receptor protein. Amino acid residues involved in the ligand recognition in the putative A2A and A3 AR binding sites have been reviewed (Kim et al. 2003).
2 A3AR Agonists
The subtype selectivity of adenosine derivatives as AR agonists has been probed extensively, principally through modification of the N6-amine moiety (where large hydrophobic groups tend to produce A1AR and A3AR selectivity, Table 1) and the C2 position (where large hydrophobic groups tend to produce A2AAR selectivity, but have also been shown to enhance A3AR selectivity, Table 2). The ribose moiety is less amenable than the adenine moiety to the addition of steric bulk, although substitution of the 5′ -CH2OH moiety with certain amides, ethers, or other hydrophilic groups has resulted in enhancement of A3AR selectivity.
Table 1.
Binding affinities of monosubstituted adenosine derivatives (N6-substituted) at the human A3AR expressed in CHO cells and at A1 and A2A ARs, and maximal A3AR agonist effect
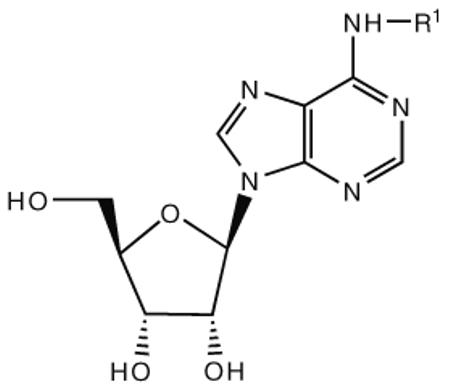
| Compound | Substitution R1 = |
pKi at A1ARa |
pKi at A2AARa |
pKi at A3ARa |
%Activation, A3ARa |
|---|---|---|---|---|---|
| 6 | CH3 | 7.22b | <5b | 8.03 | 96 |
| 7 | CH2CH3 | 8.31b | 5.05b | 8.34 | 102 |
| 8 | OCH3 | 6.65b | <5b | 7.55 | 107 |
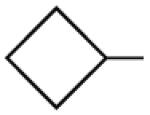
| 9 |
|
9.15b | 5.76b | 8.19 | 100 |
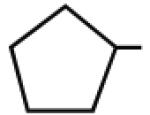
| 10 |
|
9.35b | 6.34b | 7.14 | 97 |
| 11 |
|
9.05b | 6.28b | 7.14 | 76 |
| 12 |
|
8.48b | 6.18b | 7.83 | 102 |
| 13 |
|
6.76b | 6.55b | 7.38 | 55 |
| 14 |
|
7.35 | <5b | 8.36 | 80 |
| 15 |
|
7.89, 7.89b | 7.17, 7.17b | 8.68, 6.62b | 84 |
| 16 |
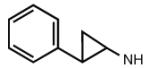
|
6.91 | 5.60 | 9.06 | 101 |
| 17 |
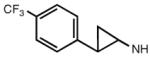
|
6.98 | 5.60 | 8.72 | 101 |
| 18 |
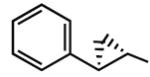
|
7.82b | 5.52b | 9.20 | 100 |
| 19 |
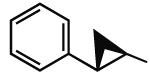
|
7.93b | 5.60b | 7.62 | 87 |
| 20 |
|
8.15 | 6.24 | 7.04 | 100 |
| 21 |
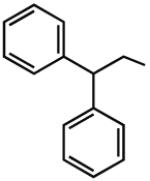
|
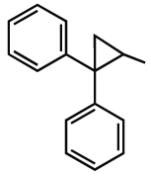
7.30 | 6.29 | 8.41 | 0 |
| 22 |
|
6.31 | <5 | 5.48 | 87 |
| 23 |
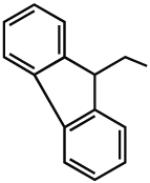
|
7.85 | 6.84 | 9.04 | 99 |
A3AR binding experiments were performed with membranes prepared from adherent CHO cells stably transfected with cDNA encoding the human A3AR, using as radioligand [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I–AB–MECA; 2000 Ci/mmol) at a final concentration of 0.5 nM, in Tris·HCl buffer (50 mM, pH 8.0) containing 10mM MgCl2, 1mM EDTA. Nonspecific binding was determined using 10 μM Cl–IB–MECA. The mixtures were incubated at 25°C for 60 min. Maximal A3AR agonist effect is the inhibition of forskolin-stimulated adenylate cyclase at 10 μM using a reference value for Cl–IB–MECA of 100%. (Gao et al. 2003; Tchilibon et al. 2004).
In rat brain (Gao et al. 2003; Tchilibon et al. 2004).
Table 2.
Binding affinities of monosubstituted adenosine derivatives (2-ether-substituted) at the human A3AR expressed in CHO cells and at A1 and A2A ARs, and maximal A3AR agonist effect
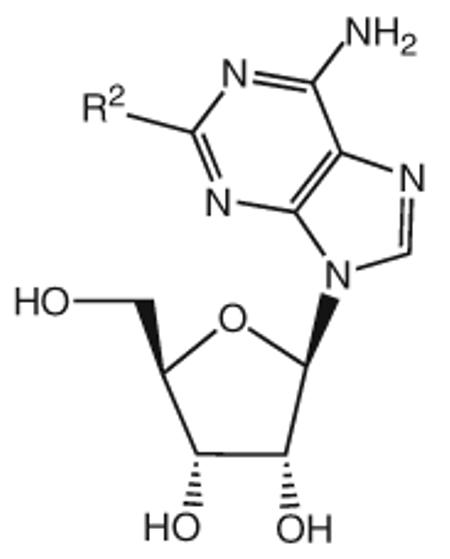
| Compound | Substitution R1 = |
pKi at A1ARa |
pKi at A2AARa |
pKi at A3ARa |
%Activation, A3ARa |
|---|---|---|---|---|---|
| 29 | Cl | 8.12 | 6.20 | 7.06 | 100 |
| 30 |
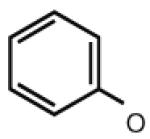
|
5.29 | 7.36 | 6.44 | 32 |
| 31 |
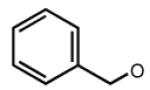
|
6.19 | 6.23 | 6.93 | 17 |
| 32 |
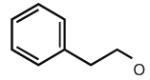
|
6.66 | 8.03 | 7.27 | 71 |
| 33 |
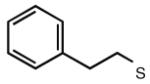
|
5.43 | 6.23 | 5.71 | ND |
| 34 |
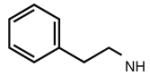
|
6.28 | 7.21 | 6.51 | 72 |
| 35 |
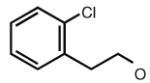
|
6.66 | 7.75 | 6.85 | 1 |
| 36 |
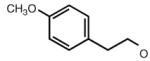
|
6.54 | 7.19 | 6.98 | 91 |
| 37 |
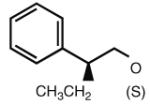
|
5.32 | 7.57 | 6.76 | 0 |
| 38 |
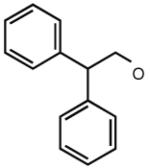
|
<5 | 6.51 | 7.27 | 0 |
A3AR binding experiments were performed with membranes prepared from adherent CHO cells stably transfected with cDNA encoding the human A3AR, using as radioligand [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I–AB–MECA; 2000 Ci/mmol) at a final concentration of 0.5 nM, in Tris·HCl buffer (50 mM, pH 8.0) containing 10mM MgCl2, 1mM EDTA. Nonspecific binding was determined using 10 μM Cl–IB–MECA. The mixtures were incubated at 25°C for 60 min. ND, not determined. Maximal A3AR agonist effect is inhibition of forskolin-stimulated adenylate cyclase at 10 μMusing a reference value for Cl–IB–MECA of 100% (Gao et al. 2004)
The binding of a nucleoside to the A3AR and its activation of the receptor are separate processes that appear to have distinct structural requirements. There is no general correlation between the affinity of a given nucleoside derivative in binding to the A3AR and its ability to fully vs. partially activate the receptor (Table 1). Specific functionality on the nucleoside structure that lowers efficacy relative to that of a full agonist (e.g., NECA) has been identified. For example, N6-benzyl and certain 2-position substituents on the adenine moiety reduce the relative efficacy at the A3AR. 2-Chloro alone does not reduce A3AR efficacy, but, in combination with a substituted N6-benzyl moiety, it leads to a further reduction (Gao et al. 2002). Other N6 substitutions have been studied using the same criteria. For example, the relative efficacy of N6-(2-phenylethyl) derivatives is extremely sensitive to substitution of the phenyl ring and the β-methylene carbon (Tchilibon et al. 2004).
Modifications of the ribose moiety have also been explored for effects on both A3AR binding affinity and efficacy (Gao et al. 2004; van Tilburg et al. 2002). SAR studies also indicate that flexibility in the ribose 5′ region is a prerequisite for A3AR activation, in concert with a proposed required rotation of TM6 (Kim et al. 2006). Thus, with proper manipulation of groups at the N6 and/or ribose moieties, a high-affinity agonist may be converted into a selective A3AR antagonist. Conversely, agonist function may be maintained fully with proper derivatization of the ribose moiety. A flexible 5′ -uronamide moiety is particularly well suited to maintaining efficacy, and even overcomes the reduction of efficacy induced by various adenine substituents at the N6 and C2 positions.
2.1 Substitution of the Adenine Moiety of Adenine Nucleosides
2.1.1 N6 Position
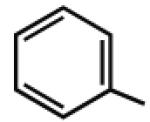
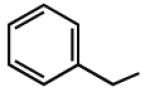
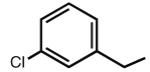
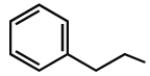
Multiple studies have been undertaken to optimize the N6 position of adenosine in order to design selective A3AR agonists (Table 1, 6–23). Addition of small groups such as methyl (6) and oxymethyl (8) to the N6 amine gave at least a tenfold increase in potency over adenosine and increased the selectivity of the ligand for the human A3AR over other human ARs (Volpini et al. 2007). However, increasing the alkyl chain length to an ethyl group (7) increased the affinity of the ligand for both the A3 and A1 ARs, thus, decreasing the selectivity (Gao et al. 2003). Larger alkyl chains were not well tolerated at the N6 position, and increased branching of the chain caused a decrease in A3AR affinity and efficacy. Various cycloalkyl groups were also appended to the N6-amino group. Adding an N6-cyclobutyl (9) or cyclopentyl (10) ring resulted in agonists that had greater affinity to the A1AR than the A3AR (Gao et al. 2003) but were full agonists at the A3AR. Analogs bearing larger N6-cycloalkyl rings such as 11 were only partially efficacious as A3AR agonists. When a benzyl ring was attached to the N6 amine (13), the compound was three- to four-fold selective in binding to the human A3AR in comparison to the A1 and A2A ARs, but only displayed a 55% relative efficacy at the A3AR. N6-Phenyladenosine (12) was fully efficacious as an A3AR agonist. N6-(2-Phenylethyl)adenosine (15) was the most potent in binding to the A3AR among a series of arylalkyl-substituted homologs. However, the N6-benzyl and the N6-phenyl substituents provided greater selectivity than 2-phenylethyl for the A3AR. Generally, halogen substitution at the 3 position of the N6-benzyl ring caused an increase in A3AR affinity and selectivity. For example, N6-(3-chlorobenzyl)adenosine (14) showed a tenfold selectivity for the A3AR and a nanomolar affinity. Halogen substitution at other positions of the ring frequently decreased the A3AR affinity.
Addition of certain larger N6 substituents also increased the potency and affinity of the ligands at the A3AR. For instance, N6-(trans-2-phenylcyclopropropyl)adeno sine (16) was a full agonist with high selectivity and a subnanomolar potency (Gao et al. 2003). Further variations on this substituent were prepared, and the importance of conformational factors in the relative efficacy was demonstrated. The addition of one bond to bridge the phenyl rings could change an antagonist into an agonist. Thus, while N6-(2,2-diphenylethyl)adenosine (21) was an antagonist at the A3AR, adding a bond between the phenyl groups to create N6-(9-fluorenylmethyl)adenosine (23) restored the efficacy. This compound also had a subnanomolar A3AR affinity but was less selective than N6-(trans-2-phenylcyclopropyl)adenosine (Tchilibon et al. 2004). The most selective compound of the series was N6-(trans-2-(3-trifluoromethyl)phenyl)-1-cyclopropyl adenosine (17), which had a 100-fold selectivity at the A3AR in comparison to the A1AR.
Various labs have combined sterically bulky N6 groups with a 5′ -uronamide moiety on the ribose group to make potent, selective A3AR agonists. The first A3AR-selective compounds combined a 5′ -N-alkyluronamide with an N6-benzyl group (Gallo-Rodriguez et al. 1994; Jacobson et al. 1993; van Galen et al. 1994). One of the most common A3 agonists, Cl–IB–MECA (Fig. 1), has an N6-iodobenzyl group, a 2-chloro group, and a 5′ -methylcarboxamido group. This compound has a Ki of 0.33 nM at the rat A3AR, but Ki values of only 2,500 and 1,400 nM at rat A1 and A2A ARs, respectively (Kim et al. 1994a). At the human ARs, the binding affinities of Cl–IB–MECA are (nM): A1AR 220, A2AAR 5400, and A3AR 1.4. Thus, Cl–IB–MECA is more selective for the rat A3AR than the human A3AR (Melman et al. 2008a). The 125I form of I–AB–MECA (N6-(4-amino-3-iodobenzyl)-5′ -N-methylcarboxamidoadenosine, 5, Fig. 1) is commonly used as a high-affinity radioligand for characterizing binding to the A3AR of various species.
Baraldi et al. (1998) prepared a series of N6-substituted-aminosulfonylphenyl derivatives of NECA (e.g., compound 24). Among these compounds, the most favorable substituents of the sulfonamido group for increasing affinity at the A3AR were small alkyl groups, such as ethyl or allyl moieties, and disubstitution of thesulfonamido group. The A3AR selectivity was increased by the addition of a saturated heterocyclic ring, such as piperidine or morpholine, to the sulfonamido moiety.
Finally, A3 selective fluorescent probes have also been made by attaching 7-nitrobenzofurazan fluorophores to NECA derivatives using an alkyl spacer (e.g., compound 25). These compounds displayed 500-fold selectivity at the A3AR and bound in the low nanomolar range (Cordeaux et al. 2008).
2.1.2 Adenine 2 Position
Many modifications at the 2 position of adenosine (Table 2, 29–38) tend to increase A2AAR potency, but some additions have been found to contribute to A3AR selectivity. Adding a simple 2-chloro group (29) increased the A3AR affinity in comparison to adenosine, but it also significantly increased the potency at the A1AR (Gao et al. 2004). Generally, 2-ether modifications decreased A3AR affinity, with certain exceptions. For example, adding a 2-i-pentyloxy moiety increased A3AR affinity threefold, and the compound was slightly selective. 2-Benzyloxy substitution (31) decreased the efficacy to 17% of the full agonist Cl–IB–MECA. 2-Phenylethyloxy substitution (32) often increased affinity at both the A3 and A2A ARs, but many such analogs displayed a decreased efficacy as A3AR agonists. Other 2-ethers, such as 2-(2,2-diphenylethyloxy)adenosine (38), were A3 AR antagonists, in curious parallel to the effect of the same group when placed at the N6 position (Tchilibon et al. 2004).
Many other substitutions at the 2 position of adenosine were combined with previously introduced substitutions at the N6 position of adenosine. For instance, adding a 2-cyano group to N6-(3-iodobenzyl)adenosine created an A3AR antagonist, but when the 2-cyano group was added to N6-methyladenosine, the compound was a full agonist that was 30-fold selective for the human A3AR in comparison to the A1AR (Ohno et al. 2004). However, when other small modifications were made at the 2 position of N6-methyladenosine, such as an amino or a trifluoromethyl group, there was a decrease in selectivity and affinity toward the A3AR. Elzein et al. (2004) synthesized a series of 2-pyrazolyl-N6-substituted adenosine derivatives that were very potent and selective for the A3AR. Cosyn et al. (2006b) found that several 2-triazol-1-yl substitutions of N6-methyladenosine increased affinity at the A3AR. However, in order to maintain efficacy, a 5′ -ethyluronamide was necessary. 9-(5-Ethylcarbamoyl-β-D-ribofuranosyl)-N6-methyl-2-(4-pyridin-2-yl-1,2,3-triazol-1-yl)adenine 26 (LC257, Fig. 2) was a full agonist with a Ki of 1.8 nM at the A3AR and a minimum of 900-fold selectivity over other ARs.
Fig. 2.

Structures of a novel, multiply substituted A3AR agonists
Additions at the 2 position of NECA often increased potency and/or selectivity. For instance, 2-(3-hydroxy-3-phenyl)propyn-1-yl-NECA 27 (PHPNECA) (Fig. 2) exhibited a subnanomolar affinity at the A3 receptor (Volpini et al. 2002). Also, Zhu et al. (2006) made a series of N6-ethyl-2-alkynyl-NECA derivatives which had sub- to low nanomolar affinities and were very selective in comparison to the A2A and A2B ARs, with some selectivity over the A1AR. The most potent compound in that series (28) had a (p-(methoxy)phenyl)alkynyl substituent at the 2 position.
2.2 Ribose Modifications
2.2.1 Modification of Ribose Hydroxyl Groups
Many modifications have been made to the ribose ring. As mentioned above, the 5′ -N-alkyluronamide modification has been particularly fruitful. Gallo-Rodriguez et al. (1994) initially found that adding a 5′ -N-methyluronamide group to N6-benzyl derivatives increased the binding affinity at all three ARs examined and resulted in several of the compounds gaining selectivity for the A3AR. They also found that adding a 5′ -N-ethyluronamide more than doubled the potency of several N6-benzyl derivatives of adenosine. Other modifications at the ribose 5′ position, such as alkylthioethers (van Tilburg et al. 2002) have been found to modulate affinity and efficacy at the A3AR.
Both the 2′ - and the 3′ - hydroxyl groups contribute to the binding process, since replacing either of these groups in Cl–IB–MECA with a fluoro group caused a significant drop in both affinity and efficacy (Gao et al. 2004). A less drastic decrease in binding and efficacy was seen when the 3′ -hydroxyl of the adenosine analogs was replaced with an amino group (DeNinno et al. 2003). When a methylene spacer was added between the 3′ -amino and ribose groups, there was a total loss of affinity (Van Rompaey et al. 2005). Also, 3′ -deoxy-3′ -acetylamino analogs were weak at the A3AR. However, DeNinno et al. (2003, 2006) found that the 3′ -amino substitution was tolerated and gave high selectivity when the 5′ and N6 positions of adenosine were also appropriately modified in compounds 39 and 40 (Fig. 3). Replacement of the 3′ -hydroxyl with an azido group generally abolished A3AR activation. The 2′ -hydroxyl group appeared to be more important than the 3′ -hydroxyl group, because when it was replaced with the fluoro group there was no binding or activation of the A3AR (Gao et al. 2004).
Fig. 3.

Structures of ribose ring-modified selective A3AR agonist probes
2.2.2 Modification of the Pentose Ring
4′ -Thio derivatives were usually equipotent or slightly more potent at ARs than their oxygen equivalents (Jeong et al. 2006a). Many 4′ -thio derivatives of adenosine have been found to be full agonists. For example, LJ-529 41 (2-chloro-N6-(3-iodobenzyl)-4′ -thioadenosine-5′ -methyluronamide) (Fig. 3) is a highly potent ligand (Ki = 0.38 nM against 125I]–AB–MECA binding to the human A3AR expressed in CHO cells). In the same 4′-thio-modified series, a wide variety of ribose 5′ -alkyluronamides have shown that there is tolerance for groups larger than N-ethyl (Jeong et al. 2006a). For example, compounds 42 and 43 were full agonists with Ki values of 3.6 and 18 nM at the hA3AR, respectively. The nature of the N-alkyl or N-arylalkyl group can modulate affinity and efficacy at the A3AR (Jeong et al. 2008).
However, when the thio modification was combined with shifting the adenine moiety of Cl–IB–MECA from the 1′ to the 4′ position of the ribose ring, the compound was curiously transformed into a potent antagonist (Gao et al. 2004).
Ring-constrained nucleosides have been used to define conformational preferences at the A3AR. Medicinal chemists frequently utilize the approach of conformationally constraining otherwise flexible molecules to probe the “active” conformation(s) and to increase ligand affinity by overcoming the energy barriers needed to attain this preferred conformation. Nucleoside analogs containing novel rigid ring systems in place of the ribose ring have been explored as ligands for the ARs. The focus on conformational factors of the ribose or ribose-like moiety allows the introduction of general modifications that lead to enhanced potency and selectivity at certain subtypes of these receptors. One ring system selected for this purpose is the methanocarba (bicyclo[3.1.0]hexane) ring system, which has been incorporated in either of two isomeric forms that adopt either a North (N) or South (S) envelope conformation (Jacobson et al. 2000; Marquez et al. 1996). These ribose modifications were combined with known enhancing modifications at other positions on the molecule to explore the resulting SARs. (N)-Methanocarba-adenosine was favored in binding at the A3AR by 150-fold over the (S) conformation and by 2.5-fold over adenosine. Doubly modified nucleoside derivatives containing the (N)-methanocarba ring system have confirmed that this conformation of the pseudoribose ring is highly preferred over the (S) conformation for agonists at the A3AR in general.
Introducing an (N)-methanocarba modification to adenosine 5′ -ethyluronamide increased the human A3AR binding affinity by sixfold. This modification also demonstrated that the ring oxygen is not required for binding or activation of the receptor (Lee et al. 2001).
Highly selective ring-constrained agonists of the A3AR have been designed and synthesized based on the (N)-methanocarba ring system (Fig. 3). This led to the introduction of MRS3558 44 ((1′ R,2′ R,3′ S,4′ R,5′ S)-4-{2-chloro-6-[(3-iodophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo-[3.1.0]-hexane-2,3-diol) as a full agonist with subnanomolar potency at the A3AR and its congeners (e.g., 45 and 46) as full agonists with nanomolar potency at the A3AR (Tchilibon et al. 2005). The SAR of MRS3558 and related congeners as A3AR agonists (Melman et al. 2008a) was recently explored in detail. The utility of MRS3558 in treating lung injury was shown in a model of ischemia reperfusion lung injury (Matot et al. 2006). In this series of (N)-methanocarba nucleosides, a 5′ -uronamide moiety is needed in order to achieve full efficacy at the A3AR. The corresponding 5′ -alcohol is an antagonist of the A3AR. The 5′ -uronamide moiety overcomes the loss of efficacy associated with substitution of the N6 and ribose ring moieties. Thus, in the (N)-methanocarba series, as in the ribose series, a freely rotating 5′ -uronamide that is able to make and break multiple hydrogen bonds provides a necessary degree of flexibility during the receptor activation step.
2.3 Nonadenine Nucleosides and Nonnucleosides as A3AR Agonists
Occasionally, nonadenine nucleotides are also found to activate the A3AR. For instance, xanthines such as caffeine are generally found to act as antagonists, but N-methyl-1,3-dibutylxanthine 7-β-D-ribofuronamide 48 (DBXRM) acted as a moderately selective A3AR agonist (Kim et al. 1994b).
A series of atypical, nonnucleoside agonist ligands that activated various ARs were reported (Chang et al. 2005). In addition to compounds in this family of pyridine-3,5-dicarbonitriles that were selective agonists of the A1AR, various members of this series substantially activated the A3AR.
2.4 Further Optimization of A3AR Agonists Using Multiple Modifications
Interestingly, certain modifications (such as a 5′ -alkylamide or an N6-methyl group) can restore efficacy to previously modified compounds. For instance, adding a 2-chloro group to N6-cyclopentyladenosine creates an A3AR antagonist (Gao et al. 2002), but activation is restored by the 5′ -methylcarboxamide and 4-thio substitutions. This is particularly interesting since 4′ -thioadenosine is also an A3AR antagonist, and 2-chloro-4-thioadenosine is only a partial agonist (Jeong et al. 2006b).
A series of (N)-methanocarba-2,N6-disubstituted adenine nucleosides were made by Tchilibon et al. (2004), who found that adding the (N)-methanocarba, 2-chloro, and 5′ -methyluronamido groups significantly improved the selectivity and efficacy of several compounds. For instance, N6-(2,2-diphenylethyl)adenosine was an A3AR antagonist with 12-fold and 130-fold selectivity over A1 and A2A ARs, respectively. However, by adding the above substitutions, the compound became a full agonist with a Ki of 0.69 nM and a selectivity of close to 2,000-fold over A1 and A2A AR (Tchilibon et al. 2004). Also, the 2-cyano derivative of N6-methyl adenosine was a full agonist whereas the 2-cyano derivative of N6-(2-phenylcyclopropyl) adenosine was an A3AR antagonist (Ohno et al. 2004).
Adding several substitutions may also improve selectivity for the A3AR. Adding an N6-methyl group and 2-chloro group to 4′ -thioadenosine-5′ -methyluronamide created a compound with a Ki of 0.28 nM and a nearly 5,000-fold selectivity for the A3AR (Jeong et al. 2006a). A series of these compounds was made by varying the N6 and 5′ groups. While none of these derivatives could match the potency and selectivity of the original compound, it was found that 4′ -thioadenosine derivatives were often more potent than their oxy counterparts. The most potent compound was 9-(3-amino-3-deoxy-5-methylcarbamoyl-β-D-ribofuranosyl)-2-amino-N6-methylpurine. Another highly substituted yet extremely potent N6-methyl derivative is 2-chloro-N6-methyl-4-thioadenosine-5-methyluronamide, which has a Ki of 0.28 nM (Jeong et al. 2006a). N6-Methylation also seems to improve human A3AR selectivity, as N6-methyl-2-(2-phenylethyl)-adenosine is much more selective than 2-(2-phenylethyl)-NECA (Volpini et al. 2002). While large 2-position substitutions are not always tolerated, (2R,3S,4R)-tetra hydro-2-(hydroxymethyl)-5-(6-(methylamino)-2-(4-pyridin-2-yl)-1H-pyrazol-1-yl) -9H-purin-9-yl) furan-3,4-diol had a Ki of 2 nM and was extremely selective (Van Rompaey et al. 2005).
Recently, new potent and A3-selective N6,2-disubstituted adenosine derivatives have been reported. Volpini et al. (2007) made a series of N6-methoxy-2-alkyladenosine derivatives, of which N6-methoxy-2-p-acetylphenylethylMECA was the most potent and selective. This compound had a Ki of 2.5 nM at the human A3AR and selectivities of 21,000 and 4,200 against A1 and A2A ARs, respectively. Recently, a series of water-soluble A3AR agonists were synthesized (DeNinno et al. 2006). Of these compounds, (2S,3S,4R,5R)-3-amino-5-{6-[5-chloro-2-(2-oxo-2-piperazin-1-yl-ethoxy)-benzylamino]-purin-9-yl}-4-hydroxy-tetrahydro-furan-2-carboxylic acid methylamide was the most potent/selective derivative, with a Ki of 10 nM. Van Rompaey et al. (2005) found that adding additional substitutions to 3-amino-3-deoxyadenosine increased the potency, but these compounds were only partial agonists. 9-[3-Amino-3-deoxy-5-(methylcarbamoyl)-β-D-ribofuranosyl]-N6-(5-chloro2-methoxybenzyl)adenine had a Ki of 27 nM and was extremely selective for the A3AR, but had an efficacy of only 51%. Cosyn et al. (2006a) made a series of 3′ -amino-3′ -deoxy congeners that were highly selective for the A3AR.
The selectivity at the mouse A3AR of analogs containing the (N)-methanocarba ring system was reduced due to an increased tolerance of this ring system at the mouse A1AR (Melman et al. 2008a). Substitution of the 2-chloro atom with iodo or hydrophobic alkynyl groups tended to increase the A3AR selectivity (up to 430-fold) in mouse and preserve it in human. Extended and chemically functionalized alkynyl chains attached at the C2 position of the purine moiety preserved A3AR selectivity more effectively than similar chains attached at the 3 position of the N6-benzyl group. For example, the carboxylic acid congener MRS5151 47 (Fig. 3) is a highly potent agonist (Ki 2.38 nM at hA3AR) and is selective in binding at human (6,260-fold) and mouse (431-fold) A3ARs in comparison to A1ARs in the same species.
3 A3AR Antagonists
Initial attempts at obtaining potent and highly selective A3AR antagonists focused on wide pharmacological screening of different heterocyclic compounds (Jacobson et al. 1995; Ji et al. 1996; Siddiqi et al. 1995). One of the first nonxanthine heterocyclic derivatives (Fig. 4) found to be selective for the human A3AR (Ki 0.65 nM) was MRS1220 (N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzeneacetamide) 49, which was based on appropriate acylation of the exocyclic amino group of this class of known AR antagonists (Kim et al. 1996). During subsequent evaluations, different classes of nonxanthine nitrogen-containing molecules were identified as potent A3AR antagonists: flavonoids, 1,4-dihydropyridines and pyridines, triazoloquinazolines, isoquinolines and quinazolines (Baraldi et al. 2003a; Mullër et al. 2003). The 1,4-dihydropyridine (DHP) derivative MRS1191 (1,4-dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyridinedicarboxylic acid, 3-ethyl 5-(phenylmethyl) ester) 51 was structurally optimized for binding to the A3AR (Ki 31 nM) from library screening that identified various DHP calcium channel blockers as weak A3AR antagonists (Jacobson et al. 1997). The pyridine derivative MRS1523 (5-propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate) 50 was the first heterocyclic A3AR antagonist to display considerable potency and selectivity for the rat A3AR (Ki 113 nM), as well as the human (18.9 nM) and mouse A3AR (Li et al. 1998). In this section, the recent advancements in this field have been summarized, with particular attention paid to the most important reports of the last five years.
Fig. 4.

Structures of heterocyclic derivatives that are widely used as selective human A3AR antagonists
3.1 Recent Developments in Nonpurine Heterocycles
3.1.1 Thiazole and Thiadiazole
IJzerman and coworkers investigated a series of 3-(2-pyridinyl)-isoquinoline derivatives for their affinity at the A3AR (Van Muijlwijk-Koezen et al. 2000). The effect of an additional nitrogen atom was valued by synthesizing bioisosteric quinazoline derivatives. The compounds VUF 8504 (4-methoxy-N-(3-(2-pyridinyl)-1-isoquinoli nyl)benzamide, 52) and VUF 5574, (N-(2-methoxyphenyl)-N′ -(2-(3-pyridyl)quina zolin-4-yl)urea, 53) (Fig. 4) display considerable A3AR affinity and appreciable selectivity versus A1 and A2A AR subtypes.
The bicyclic system of isoquinoline and quinazoline has been replaced by several monocyclic rings (Van Muijlwijk-Koezen et al. 2001). Some thiazole and thiadiazole derivatives were shown to be most promising candidates for the identification of new A3AR ligands.
The derivative N-[3-(4-methoxy-phenyl)-[1,2,4]thiadiazol-5-yl]-acetamide (54, Fig. 5) has been claimed to be the most potent A3AR antagonist of the series, exhibiting a Ki value of 0.79 nM at hA3AR and antagonistic properties in a cAMP functional assay (Jung et al. 2004). A series of potent and selective A3AR antagonists have been obtained via an optimization study of compound 55 that revealed that a 5-(pyridine-4-yl) moiety on the 2-aminothiazole ring was optimal for enhanced receptor potency and selectivity (Press et al. 2004). Of particular note, N-[4-(3,4,5-trimethoxyphenyl)-5-pyridin-4-ylthiazol-2-yl]-acetamide 56 showed subnanomolar affinity at the human A3AR as a competitive antagonist of [125I]I–AB–MECA, binding with 1,000-fold selectivity versus the other ARs.
Fig. 5.

Thiazole and thiadiazole derivatives as human A3AR antagonists
Binding affinity data on thiazole and thiadiazole derivatives at the hA3AR have been subjected to QSAR analysis (Bhattacharya et al. 2005). This study disclosed the importance of the molecular electrostatic potential surface (Wang–Ford charges) in relation to atoms C2, C5, C7, X8 and S9 (Fig. 5), the last two playing the most important roles. Furthermore, the A3AR binding affinity increases with decreasing lipophilicity of the compounds and in the presence of short alkyl chains—methyl (Me) or ethyl (Et)—at the R position.
3.1.2 Pyrazoloquinolines
The binding affinities at bovine A1 and A2A ARs and at human cloned A3ARs of some 2-arylpyrazolo[3,4-c]quinolin-4-ones along with their corresponding 4-amines and 4-substituted-amino derivatives were reported by Colotta et al. (2000). The 4-benzoylamido derivative 57 (Fig. 6) displayed one of the best binding profiles of the series of A3AR antagonists. The same group recently reported an extension of the SAR study of this class of compounds (Colotta et al. 2007) which high-lighted that bulky and lipophilic acyl–amino groups at the 4 position seemed able to promote hA3AR potency and selectivity. Selected compounds of these series were tested in an in vitro rat model of cerebral ischemia and prevented the irreversible failure of synaptic activity induced by oxygen and glucose deficiency in the hippocampus, thus confirming that potent and selective A3AR antagonists may substantially increase the tissue resistance to ischemic damage.
Fig. 6.

Pyrazoloquinoline derivatives as human A3AR antagonists
The synthesis and the affinity profile at ARs of a series of 2-phenyl-2,5-dihydro-pyrazolo[4,3-c]quinolin-4-ones, conceived as structural isomers of the parent 2-arylpyrazolo[3,4-c]quinoline derivatives, have also been reported (Baraldi et al. 2005a). Some of the synthesized compounds showed A3AR affinities in the nanomolar range and good selectivities, as evaluated in radioligand binding assays at hARs. In particular, substitution at the 4 position of the 2-phenyl ring with methyl, methoxy, or chlorine and the presence of a 4-oxo functionality gave good activity and selectivity (58).
3.1.3 Triazoloquinoxalines
Triazolo[4,3-a]quinoxalines
Interesting studies performed in the last decade by Colotta and coworkers highlighted that the 1,2,4-triazolo[4,3-a]quinoxalin-1-one moiety is an attractive scaffold for obtaining potent and selective hA3AR antagonists (Colotta et al. 2004; Lenzi et al. 2006). Intensive efforts in the chemical synthesis of compounds based on the systematic substitution of the 2, 4 and 6 positions of the tricyclic template, along with molecular modeling investigations performed to rationalize the experimental SAR findings, led to the identification of optimal structural requirements for A3AR affinity and selectivity. In particular, the introduction into the triazoloquinoxaline moiety of a 4-oxo (59) or a 4-N-amido (60, Fig. 7) function affords selective and/or potent A3AR antagonists, indicating that a C = O group (either extranuclear or nuclear) is necessary for A3AR affinity. This suggested that the probable engagement of this site of the molecule is a hydrogen bond with the A3AR binding site. Hindering and lipophilic acyl–amino moieties at the 4 position showed enhanced A3AR affinity (60). Substitution of the 4 position of the 2-phenyl ring with a methoxy or a nitro group and 6-nitro substitution, as well as the combination of these substituents, afforded nanomolar A3AR affinity and better A3AR selectivity. 1-Oxo, 6-nitro, and 4-amino groups have been proposed to be involved in hydrogen bonds that anchor the antagonists to the binding site.
Fig. 7.

Triazoloquinoxaline derivatives as A3AR antagonists
Triazolo[1,5-a]quinoxalines
Some 2-aryl-8-chloro-1,2,4-triazolo[1,5-a]quinoxaline derivatives have been synthesized and tested in radioligand binding assays at bovine (b) A1 and bA2AARs and at hA1 and hA3ARs (Catarzi et al. 2005a, b). The SAR of these compounds are in agreement with those of previously reported for 2-aryl-1,2,4-triazolo[4,3-a]quinoxalines and 2-arylpyrazolo[3,4/4,3-c]quinolines, thus suggesting a similar AR-binding mode. These studies provided some interesting compounds; among them, 2-(4-methoxyphenyl)-1,2,4-triazolo[1,5-a]quinoxalin-4-one (61, Fig. 7) is the most potent and selective hA3AR antagonist of this series.
3.1.4 Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines
The first example of an AR antagonist containing the pyrazolo-triazolo-pyrimidine scaffold (Cacciari et al. 2007) was reported by Gatta and coworkers (Gatta et al. 1993).
A wide number of compounds (MRE series) originated from the structure– activity optimization work based on systematic substitution at the C2, C5, C9, N7, and N8 positions (Baraldi et al. 2002a; 2003b; 2006). The N7-substituted derivatives were found to bind principally to the hA2AAR (Baraldi et al. 2002b), while the most potent and selective hA3AR antagonists in this series were derived from the combination of a small alkyl chain at the N8-pyrazole position with a (substituted)phenylcarbamoyl chain at the N5 position (Baraldi et al. 2003a). The compound designated MRE-3008-F20, (5-N-(4-methoxyphenylcarbamoyl)amino-8-propyl-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine, 62) (Fig. 8), one of several high-affinity antagonists of this series, is a highly potent A3AR ligand (Ki = 0.29 nM against [125I]I–AB–MECA binding to human AR receptors expressed in HEK293 cells) with good selectivity over the other hARs. It showed antagonist activity in a functional assay blocking the effect of IB–MECA on cAMP production in CHO cells with an IC50 value of 4.5 nM. [3H] MRE 3008-F20 shows a Kd value of 0.82 ± 0.08 nM and a Bmax value of 297 ± 28 fmol mg−1 protein (Varani et al. 2000).
Fig. 8.

A3AR antagonists based on a pyrazolo-triazolo-pyrimidine scaffold
An important problem with the pyrazolo-triazolo-pyrimidine series was the low water solubilities typically observed, which could limit their use as pharmacological and diagnostic ligands. The bioisosteric replacement of the phenyl ring of the 5-phenylcarbamoyl moiety with a 4-pyridyl moiety (Maconi et al. 2002) provided high water solubility while enhancing hA3AR affinity. Compound MRE-3005-F20, (5-N-(4-methoxyphenylcarbamoyl)amino-8-ethyl-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine, 63) and the corresponding HCl salt, which showed very high affinities and good selectivities at the hA3 receptor subtype, with Ki values in the picomolar range (40 and 10 pM, respectively), can be considered ideal candidates for pharmacological and clinical investigations of the hA3AR subtype. Receptor modeling ascribed this increase in affinity, compared to neutral arylcarbamate derivatives, to strong electrostatic interactions between the pyridinium moiety and the side chain carbonyl oxygen atoms of Asn274 and Asn278, both located on TM7. Additional studies suggested that involvement of the residue Tyr254 in a hydrogen bond with the pyridyl ring was responsible for both enhanced receptor affinity and selectivity (Tafi et al. 2006). The replacement of the N5-pyridine moiety with several N5-heteroaryl rings produced a general loss of affinity and selectivity at the hA3AR (Pastorin et al. 2006).
In order to rationally design and synthesize hA3AR antagonists with improved binding and/or absorption, distribution, metabolism, and excretion (ADME) profiles, and as suitable clinical candidates, different molecular modeling investigations have been carried out in the last years. Particular attention has been paid to the pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine family, the most potent class of A3AR antagonists ever reported (Tafi et al. 2006). A combined target-based (highthroughput molecular docking) and ligand-based (CoMFA) (comparative molecular field analysis) drug design approach has recently been performed by Moro and coworkers (Moro et al. 2005), which defined a novel “Y-shaped” binding motif for pyrazolo-triazolo-pyrimidines and rationally delineated some key ligand–receptor interactions for this class of molecules as follows: (1) steric control around the 3 and 4 positions of the N5-phenyl ring justifies the decrease in affinity of 3- or 4-substituted-phenyl derivatives; (2) an important π–π interaction takes place between the 2-furyl ring and two phenylalanine residues of the binding site; (3) a hydrophobic pocket, bordered by two hydrophilic amino acids, surrounds the N8 interaction area; and (4) strong hydrogen bonding is possible between a residue of Asn and the N4 of the triazolo ring.
3.1.5 Various Heterocycles
In the last few years, other classes of heterocyclic compounds have been identified as A3AR antagonists, but large structural dissimilarities meant that none of these could be classified into particular family groups. The quinoxaline derivative 64 (Fig. 9) deserves to be mentioned here, not only because of its good binding profile as an A3AR antagonist, but also (especially) due to the novelty of the strategy applied to its design, which was based on a 3D database-searching approach (Novellino et al. 2005). There is increasing evidence of the importance of 2D/3D database searching as a valuable tool to discover novel lead compounds for the A3AR and for other G-protein-coupled receptors (GPCRs) (Costanzi et al. 2008).
Fig. 9.
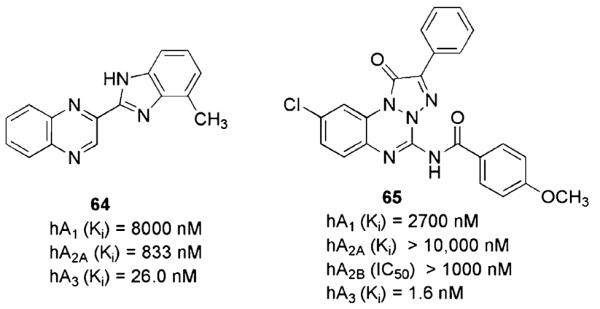
A3AR antagonists based on quinoxaline and triazolobenzotriazinone scaffolds
The structural manipulation of a series of phenyltriazolobenzotriazindiones, previously described as ligands at the central benzodiazepine receptor, led Da Settimo and coworkers to the identification of a series of aminophenyltriazolobenzotriazinones. Among these, compound 65, a result of a systematic lead optimization, stands out for its remarkable potency and selectivity at the A3AR (Ki values at the A1, A2A, A3ARs of 2,700 > 10,000, 1.6 nM, respectively, and IC50 value from cAMP assay at the A2B > 1,000 nM) (Da Settimo et al. 2007). Interestingly, the triazolobenzotriazinone nucleus is isomeric with that of the triazoloquinoxalinone series described above (compounds 59–61, Fig. 7).
3.2 Purine Derivatives
3.2.1 Adenines
The first class of A3AR-selective antagonists with a bicyclic structure strictly correlated to the adenine nucleus was claimed in 2005 by Biagi and coworkers (Biagi et al. 2005). The authors described the synthesis of a series of N6-ureidosubstituted-2-phenyl-9-benzyl-8-azaadenines whose adenine-like structure was responsible for the antagonist activity and whose phenylcarbamoyl group ensures selectivity at the A3AR. The structure–activity relationship studies were performed based on the systematic optimization of substituents at the 2, 6 and 9 positions of the bicyclic scaffold, and led to the desired enhancement of A1/A3 selectivity (compound 66, Fig. 10).
Fig. 10.

A3AR antagonists based on nonnucleoside adenine scaffolds
Basing on the finding that the known differentiation agent “reversine” (2-(4-morpholinoanilino)-N6-cyclohexyladenine) exerted a moderate antagonist activity at the hA3AR (Ki value of 0.66 μM), Jacobson and coworkers developed a series of reversine analogs, focusing their attention on the substitution pattern at the 2 and N6 positions of the adenine scaffold (Perreira et al. 2005). One of most interesting compounds in terms of hA3AR affinity and selectivity, MRS3777, (2-(phenyloxy)-N6-cyclohexyladenine, 67), combines the N6-cyclohexyl moiety of reversine with a 2-phenyloxy group. A few derivatives tested in binding assays to the rat A3AR seemed to reflect the species dependence of the affinity typical of most known nonnucleoside A3AR antagonists, and were shown to be inactive at 10 μM in this species.
3.2.2 Triazolopurines
Okamura et al. (Okamura et al. 2002, 2004a) recently reported the study of a new series of 1,2,4-triazolo[5,1-i]purines. This research group highlighted the structural similarity between the new class of compounds and the triazoloquinazoline derivatives and consequently evaluated the corresponding A3AR affinities. These investigations led to potent and selective hA3AR ligands, the most potent of which are reported in Fig. 11 (68,69). In particular, 5-n-butyl-8-(4-n-propoxyphenyl)-3H-[1,2,4]triazolo[5,1-i]purine (69) exhibited the best selectivity profile of this series (affinity ratios vs. other AR subtypes > 19,600). Compound (70), 5-n-butyl-8-(4-trifluoromethylphenyl)-3H-[1,2,4]triazolo-[5,1-i]purine (OT-7999), significantly reduced intraocular pressure in cynomolgus monkeys at 2–4 h following topical application (500 mcg) (Okamura et al. 2004b).
Fig. 11.

A3AR antagonists based on triazolopurine scaffolds
3.2.3 Tricyclic Xanthines
Natural antagonists for ARs such as caffeine and theophylline show, in general, low affinity for the A3AR subtype (Baraldi et al. 2003a; van Galen et al. 1994). In a recent study, the approach based on the ring annelation of xanthine derivatives for the development of AR antagonists was considered in depth (Drabczyńska et al. 2003).
Some pyrido[2,1-f ]purine-2,4-dione derivatives, which could be considered tricyclic xanthine derivatives, have been reported to exert subnanomolar affinity at the hA3AR (Priego et al. 2002). The most potent compound of this recent series is the 1-benzyl-3-propyl-1H, 3H-pyrido[2,1-f ]purine-2,4-dione derivative (71, Fig. 12), which presents a Ki value of 4.0 ± 0.3 nM at hA3AR. The replacement of the benzyl nucleus at the 1 position with a methyl moiety caused dramatic losses of both affinity and selectivity. The effect of the replacement of the pyridine ring of the pyrido[2,1-f ]purine-2,4-dione core with different five-membered heterocycles was examined. In particular, the synthesis and the SAR profile at the ARs of a series of 1-benzyl-3-propyl-7-aryl/alkyl-1H,6H-pyrrolo[2,1-f ]purine-2,4-dione and 1-benzyl-3-propyl-7-aryl/alkyl-1H,8H-imidazo[2,1-f ]purine-2,4-dione derivatives were recently reported (Baraldi et al. 2005b). Among the examined tricycles, the imidazo[2,1-f ]purine-2,4-dione derivatives were two-to tenfold more potent than the corresponding pyrrolo[2,1-f ]purine-2,4-dione derivatives. The best results were obtained with the introduction of small alkyl chains at the 7 position (1-benzyl-7- methyl-3-propyl-1H,8H-imidazo[2,1-f ]purine-2,4-dione 73, 1-benzyl-7-ethyl-3-propyl-1H,6H-pyrrolo[2,1-f ]purine-2,4-dione 74, Fig. 12). Compound 73 shows a subnanomolar affinity towards the target A3AR, with noteworthy selectivity with respect to the other AR subtypes (Ki (hA3) = 0.8 nM, Ki (hA1/hA3) = 3, 163, Ki (hA2A/hA3) > 6,250, IC50 (hA2B)/Ki (hA3) = 2,570).
Fig. 12.

A3AR antagonists based on tricyclic xanthine scaffolds
The synthesis and biological evaluation of a series of fused xanthine derivatives was investigated by Müller and coworkers (Müller et al. 2002a). In particular, the (R)-4-methyl-8-ethyl-2-phenyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (PSB-11 (76), Fig. 12) exhibited a Ki value of 2.3 nM for the A3AR and good selectivity vs. all other AR subtypes. The radiolabeled derivative of this compound ([3H]PSB-11) exhibited a Kd value of 4.9 nM and a Bmax value of 3,500 fmol mg−1 of protein in human A3AR binding in transfected CHO cells (Müller et al. 2002b). An important innovation of such a series, in comparison with xanthines, is a significant increase in water solubility due to the introduction of a basic nitrogen atom, which can be protonated in physiological conditions. Compound PSB-10, bearing a 2,3,5-trichlorophenyl moiety at the 2 position, showed inverse agonist activity in binding studies in CHO cells expressing recombinant hA3ARs (IC50 = 4 nM) (Ozola et al. 2003). The 2-(4-bromophenyl)-derivative named KF-26777 (77) with subnanomolar affinity at the hA3AR (Ki = 0.2 nM) and high selectivity over A1, A2A and A2B ARs (9,000-, 23,500-, 31,000-fold, respectively) was considered a potential lead molecule for development for the treatment of brain ischemia and inflammatory diseases such as asthma (Saki et al. 2002).
3.3 Nucleoside-Derived A3AR Antagonists
Based on the observation that the relative efficacy of purine nucleosides depends on structural features (see Sect. 2), new subtype-selective nucleoside antagonists of the A3AR have been designed. One of the first such antagonists was the rigid spirolactam MRS1292 (78) (Fig. 13, (2R,3R,4S,5S)-2-[N6-3-iodobenzyl)adenos-9′ -yl]-7-aza-1-oxa-6-oxospiro[4.4]-nonan-4,5-diol) (Gao et al. 2002), which binds potently and selectively to the rat and human A3ARs but does not activate these receptors, and thus acts as an antagonist.
Fig. 13.

A3AR antagonists based on nucleoside scaffolds
Modeling/mutagenesis of ARs has focused on distinct residues related to ligand binding and the relative efficacy of adenosine derivatives, and on a conserved Trp residue (6.48) which is involved in the activation process (termed a “rotamer switch,” Shi et al. 2002). Docking studies of agonists suggest that the activation pathway of the A3AR involves a characteristic anticlockwise rotation of this residue, as viewed from the exofacial side (Kim et al. 2006). The docking of MRS1292 (78) to the A3AR model is not accompanied by rotation of this residue, as occurs with nucleoside agonists, consistent with its action as an antagonist (Kim et al. 2006). Moreover, the affinity and selectivity of MRS1292 occurs across species, unlike most other heterocyclic antagonists for the A3AR reported. This allows its use in nonprimate (e.g., murine) experimental animals used as clinical models. For example, MRS1292 applied directly to the eye in mouse has been shown to be effective in reducing intraocular pressure, which may be predictive of its utility as an antiglaucoma agent (Yang et al. 2005).
The removal of the ability of the 5′ -N-alkyluronamide to donate a hydrogen bond was found to convert agonists into selective antagonists (Gao et al. 2006a). In both the 4′ -oxo and the 4′ -thio series, N-methylation of an N-methylamide (i.e., to form a dimethylamide) resulted in potent and selective A3AR antagonists. Recently, nucleosides that are truncated at the 4′ position were found to act as A3AR antagonists. For example, (2R, 3R, 4S)-2-(2-chloro-6-(3-chlorobenzylamino)-9H -purin-9-yl)tetrahydrothiophene-3,4-diol (LJ-1416, 80) and (2R, 3R, 4S)-2-(2-chloro-6-(3-iodobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (LJ-1251, 81) (Fig. 13) (Jeong et al. 2007) displayed Ki values of 1.66 and 4.16 nM, respectively, at the human A3AR, with > 600-fold selectivity in comparison to the A1AR. LJ-1251 was shown to have neuroprotective properties in an ischemia model in the rat hippocampus (Pugliese et al. 2007). Truncation at the 4′ position of A3AR agonist in the (N)-methanocarba series produces potent and selective A3AR antagonists (Melman et al. 2008b), such as the 3-bromo derivative 1′ R, 2′ R, 3′ S, 4′ R, 5′ S)-4′ -[2-chloro-6-(3-bromobenzylamino)-purine]-2′, 3′ -O-dihydroxybicyclo-[3.1.0]hexane (MRS5147) (83, Fig. 13) (2,900-fold selective for hA3 vs. hA1AR) or its 3-iodo analog, MRS5127 (84) (2,400-fold selective for hA3 vs. hA1AR). MRS5127 (84) displayed a KB (Schild constant) value of 8.9 nM as an antagonist of the human A3AR in a functional assay.
4 Engineering of the A3AR to Avoid Side Effects of Conventional Synthetic Agonists
Although selective agonists of several of the ARs have been known for years, their use as pharmaceutical agents has been impeded by undesirable side effects of exogenously administered adenosine derivatives. In spite of the clinically useful protective properties of adenosine agonists observed in experimental animals, such as protection against ischemic damage and suppression of excessive inflammation, none of the selective synthetic agonists have yet been approved for human therapeutic use. The A2AAR-selective agonist Lexiscan (regadenoson, CV Therapeutics, Palo Alto, CA, USA) (CVT-3146, 1-{9-[(4S, 2R, 3R, 5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-aminopurin-2-yl pyrazol-4-yl)-N-methylcarboxamide) was recently approved for cardiac imaging} in patients. The only other adenosine agonist currently in clinical use is adenosine itself, for the treatment of supraventricular tachycardia and as an aid in cardiac imaging.
Since ARs are widespread in the body, in order to overcome inherent nonselectivity of activating the native ARs using synthetic agonists, we have introduced the concept of neoceptors, by which the putative ligand binding site of a 7TM receptor is re-engineered for activation by synthetic agonists (neoligands) that are built to have a structural complementarity. This is a molecular modeling approach to receptor engineering by which a mutant receptor (neoceptor) is designed for selective activation by a novel synthetic ligand (neoligand) at concentrations that do not activate the native receptor. An amino acid residue of the receptor and a functional group of the ligand moiety thought to be in close proximity can be modified in a complementary fashion so that the two groups exhibit a novel mode of interaction (e.g., reversing the polarity in a salt bridge or introducing unique hydrogen-bonding sites). If a stabilizing interaction exists between these two groups, an increase in affinity is expected at the mutant receptor relative to the native receptor. This strategy is intended for eventual use in gene therapy and may also be useful in mechanistic elucidation, using neoceptor–neoligand pairs that are pharmacologically orthogonal with respect to the native species. Neoceptors have so far been applied successfully to A2A and A3 ARs (Gao et al. 2006b; Jacobson et al. 2001, 2005). Compounds 85–87 (Fig. 14) were found to interact selectively with the H272E mutant hA3AR. All three compounds activated this neoceptor.
Fig. 14.

Compounds that interact selectively with the H272E mutant hA3AR neoceptor
5 Conclusions
A3AR ligands have been modified to optimize their interaction with the A3AR. Most of these modifications have been made to the N6 and 2 positions of adenine as well as the ribose moiety, and using a combination of these substitutions leads to the most efficacious, selective, and potent ligands. A3AR agonists such as IB–MECA and Cl–IB–MECA are now advancing into Phase II clinical trials for treatments targeting diseases such as cancer, arthritis, and psoriasis.
Also, a wide number of compounds exerting high potency and selectivity in antagonizing the hA3AR have been discovered. These molecules are generally characterized by a notable structural diversity, taking into account that aromatic nitrogen-containing monocyclic (thiazoles and thiadiazoles), bicyclic (isoquinoline, quinozalines, (aza)adenines), tricyclic systems (pyrazoloquinolines, triazoloquinoxalines, pyrazolotriazolopyrimidines, triazolopurines, tricyclic xanthines) and nucleoside derivatives have been identified as potent and selective A3AR antagonists. Probably due to the “enigmatic” physiological role of A3AR, whose activation may produce opposite effects (for example, concerning tissue protection in inflammatory and cancer cells) and may produce effects that are species dependent, only a few molecules have reached preclinical investigation. Indeed, the most advanced A3AR antagonists remain in preclinical biological testing. Among the antagonists described above, compound OT-7999 is expected to enter clinical trials for the treatment of glaucoma, while several thiazole derivatives are in development as antiallergic, antiasthmatic and/or anti-inflammatory drugs.
Acknowledgements
This research was supported in part by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA.
Abbreviations
- ADME
Absorption, distribution, metabolism, and excretion
- AR
Adenosine receptor
- b
Bovine
- cAMP
Cyclic adenosine monophosphate
- CHO cells
Chinese hamster ovary cells
- Cl–IB–MECA
2-Chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamido-adenosine
- CoMFA
Comparative molecular field analysis
- CVT-3146
1-{9-[(4S, 2R, 3R, 5R)-3,4-Dihydroxy-5-(hydroxymethyl) oxolan-2-yl]-6-aminopurin-2-yl}pyrazol-4-yl)-N-methylcarboxamide
- DBXRM
7-β-D-Ribofuronamide
- DHP
1,4-Dihydropyridine
- Et
Ethyl
- GPCR
G-protein-coupled receptor
- h
Human
- HEK293 cells
Human embryonic kidney 293 cells
- I–AB–MECA
N6-(4-Amino-3-iodobenzyl)-5′-N-methylcabroxamidoa-denosine
- IB–MECA
N6-(3-Iodobenzyl)-5′-N-methylcarboxamidoadenosine
- KF-26777
2-(4-Bromophenyl)-7,8-dihydro-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one
- LJ-529
2-Chloro-N6-(3-iodobenzyl)-4′-thioadenosine-5′-methyluronamide
- LJ-1251
(2R, 3R, 4S)-2-(2-Chloro-6-(3-iodobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol
- LJ-1416
(2R, 3R, 4S)-2-(2-Chloro-6-(3-chlorobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol
- LUF6000
N-(3,4-Dichloro-phenyl)-2-cyclohexyl-1H-imidazo[4,5-c] quinolin-4-amine
- Me
Methyl
- MRE-3005-F20
5-N-(4-Methoxyphenylcarbamoyl)amino-8-ethyl-2-(2-furyl) pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- MRE-3008-F20
5-N-(4-Methoxyphenylcarbamoyl)amino-8-propyl-2-(2-furyl) pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- MRS1191
1,4-Dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3, 5-pyridinedicarboxylic acid, 3-ethyl 5-(phenylmethyl) ester
- MRS1220
N-[9-Chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzeneacetamide
- MRS1292
(2R, 3R, 4S, 5S)-2-[N6-3-Iodobenzyl)adenos-9′ -yl]-7-aza-1-oxa-6-oxospiro[4.4]-nonan-4,5-diol
- MRS1523
5-Propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate
- MRS3558
(1′ S, 2′ R, 3′ S, 4′ R, 5′ S)-4-{2-Chloro-6-[(3-iodophenylmethyl) amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo-[3.1.0]-hexane-2,3-diol
- MRS3777
2-(Phenyloxy)-N6-cyclohexyladenine
- MRS5127
(1′ R, 2′ R, 3′ S, 4′ R, 5′ S)-4′ -[2-chloro-6-(3-iodobenzylamino)-purine]-2′, 3′ -O-dihydroxybicyclo-[3.1.0]hexane
- MRS5147
(1′ R, 2′ R, 3′ S, 4′ R, 5′ S)-4′ -[2-chloro-6-(3-bromobenzylamino)-purine]-2′, 3′ -O-dihydroxybicyclo-[3.1.0]hexane
- MRS5151
(1′ S, 2′ R, 3′ S, 4′ S, 5′ S)-4′ -[6-(3-chlorobenzylamino)-2-(5-hydroxycarbonyl-1-pentynyl)-9-yl]-2′, 3′-dihydroxybicyclo[3.1.0]hexane-1′ -carboxylic acid N-methylamide
- NECA
adenosine 5′ -N-ethyluronamide
- OT-7999
5-n-Butyl-8-(4-trifluoromethylphenyl)-3H-[1,2,4]triazolo-[5,1-i]purine
- Pr
Propyl
- PSB-10
8-Ethyl-1,4,7,8-tetrahydro-4-methyl-2-(2,3,5-trichlorophenyl)-5H-imidazo[2,1-i]purin-5-one
- PSB-11
(R)-4-Methyl-8-ethyl-2-phenyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one
- QSAR
Quantitative structure–activity relationships
- r
Rat
- SARs
Structure–activity relationships
- TM
Transmembrane domain
- VUF 5574
N-(2-Methoxyphenyl)-N′ -(2-(3-pyridyl)quinazolin-4-yl)urea
- VUF 8504
4-Methoxy-N-(3-(2-pyridinyl)-1-isoquinolinyl)benzamide
References
- Baharav E, Bar-Yehuda S, Madi L, Silberman D, Rath-Wolfson L, Halpren M, Ochaion A, Weinberger A, Fishman P. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–476. [PubMed] [Google Scholar]
- Baraldi PG, Cacciari B, de las Infantas MJ Pineda, Romagnoli R, Spalluto G, Volpini R, Costanzi S, Vittori S, Cristalli G, Melman N, Park K-S, Ji X-d, Jacobson KA. Synthesis and biological activity of a new series of N6-arylcarbamoyl-, 2-(ar)alkynyl-N6-arylcarbamoyl, and N6-carboxamido-derivatives of adenosine-5′ -N-ethyluronamide (NECA) as A1 and A3 adenosine receptor agonists. J Med Chem. 1998;41:3174–3185. doi: 10.1021/jm980147p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi PG, Cacciari B, Borea PA, Varani K, Pastorin G, Da Ros T, Spalluto G. Pyrazolotriazolo-pyrimidine derivatives as adenosine receptor antagonists: a possible template for adenosine receptor subtypes? Curr Pharm Design. 2002a;8:99–110. doi: 10.2174/1381612023392838. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Monopoli A, Ongini E, Varani K, Borea PA. 7-Substituted 5-amino-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as A2A adenosine receptor antagonists: a study on the importance of modifications at the side chain on the activity and solubility. J Med Chem. 2002b;45:115–126. doi: 10.1021/jm010924c. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Tabrizi MA, Fruttarolo F, Bovero A, Avitabile B, Preti D, Romagnoli R, Merighi S, Gessi S, Varani K, Borea PA. Recent developments in the field of A3 adenosine receptor antagonists. Drug Dev Res. 2003a;58:315–329. doi: 10.1016/s0223-5234(03)00042-4. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Fruttarolo F, Tabrizi MA, Preti D, Romagnoli R, El-Kashef H, Moorman A, Varani K, Gessi S, Merighi S, Borea PA. Design, synthesis, and biological evaluation of C9- and C2-substituted pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as new A2A and A3 adenosine receptors antagonists. J Med Chem. 2003b;46:1229–1241. doi: 10.1021/jm021023m. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Tabrizi MA, Preti D, Bovero A, Fruttarolo F, Romagnoli R, Zaid NA, Moorman AR, Varani K, Borea PA. New 2-arylpyrazolo[4,3-c]quinoline derivatives as potent and selective human A3 adenosine receptor antagonists. J Med Chem. 2005a;48:5001–5008. doi: 10.1021/jm050125k. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Preti D, Tabrizi MA, Fruttarolo F, Romagnoli R, Zaid NA, Moorman AR, Merighi S, Varani K, Borea PA. New pyrrolo[2,1-f ]purine-2,4-dione and imidazo[2,1-f ]purine-2,4-dione derivatives as potent and selective human A3 adenosine receptor antagonists. J Med Chem. 2005b;48:4697–4701. doi: 10.1021/jm058008c. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Tabrizi MA, Romagnoli R, El-Kashef H, Preti D, Bovero A, Fruttarolo F, Gordaliza M, Borea PA. Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine template: organic and medicinal chemistry approach. Curr Org Chem. 2006;10:259–275. [Google Scholar]
- Bhattacharya P, Leonard JT, Roy K. Exploring QSAR of thiazole and thiadiazole derivatives as potent and selective human adenosine A3 receptor antagonists using FA and GFA techniques. Bioorg Med Chem. 2005;13:1159–1165. doi: 10.1016/j.bmc.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Biagi G, Bianucci AM, Coi A, Costa B, Fabbrini L, Giorgi I, Livi O, Micco I, Pacchini F, Santini E, Leonardi M, Nofal FA, Salernid OL, Scartonia V. 2,9-Disubstituted-N6-(arylcarbamoyl)-8-azaadenines as new selective A3 adenosine receptor antagonists: synthesis, biochemical and molecular modelling studies. Bioorg Med Chem. 2005;13:4679–4693. doi: 10.1016/j.bmc.2005.04.063. [DOI] [PubMed] [Google Scholar]
- Cacciari B, Bolcato C, Spalluto G, Klotz KN, Bacilieri M, Deflorian F, Moro S. Pyrazolotriazolo-pyrimidines as adenosine receptor antagonists: a complete structure–activity profile. Purinergic Signal. 2007;3:183–193. doi: 10.1007/s11302-006-9027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarzi D, Colotta V, Varano F, Calabri FR, Lenzi O, Filacchioni G, Trincavelli L, Martini C, Tralli A, Christian M, Moro S. 2-Aryl-8-chloro-1,2,4-triazolo[1,5-a]quinoxalin-4-amines as highly potent A1 and A3 adenosine receptor antagonists. Bioorg Med Chem. 2005a;13:705–715. doi: 10.1016/j.bmc.2004.10.050. [DOI] [PubMed] [Google Scholar]
- Catarzi D, Colotta V, Varano F, Lenzi O, Filacchioni G, Trincavelli L, Martini C, Montopoli C, Moro S. 1,2,4-Triazolo[1,5-a]quinoxaline as a versatile tool for the design of selective human A3 adenosine receptor antagonists: synthesis, biological evaluation, and molecular modeling studies of 2-(hetero)aryl- and 2-carboxy-substitued derivatives. J Med Chem. 2005b;48:7932–7945. doi: 10.1021/jm0504149. [DOI] [PubMed] [Google Scholar]
- Chang LC, von Frijtag Drabbe Künzel JK, Mulder-Krieger T, Spanjersberg RF, Roerink SF, van den Hout G, Beukers MW, Brussee J, IJzerman AP. A series of ligands displaying a remarkable agonistic–antagonistic profile at the adenosine A1 receptor. J Med Chem. 2005;48:2045–2053. doi: 10.1021/jm049597+. [DOI] [PubMed] [Google Scholar]
- Colotta V, Catarzi D, Varano F, Cecchi L, Filacchioni G, Martini C, Trincavelli L, Lucacchini A. Synthesis and structure–activity relationships of a new set of 2-arylpyrazolo [3,4-c]quinoline derivatives as adenosine receptor antagonists. J Med Chem. 2000;43:3118–3124. doi: 10.1021/jm000936i. [DOI] [PubMed] [Google Scholar]
- Colotta V, Catarzi D, Varano F, Calabri FR, Lenzi O, Filacchioni G, Martini C, Trincavelli L, Deflorian F, Moro S. 1,2,4-Triazolo[4,3-a]quinoxalin-1-one moiety as an attractive scaffold to develop new potent and selective human A3 adenosine receptor antagonists: synthesis, pharmacological, and ligand–receptor modeling studies. J Med Chem. 2004;47:3580–3590. doi: 10.1021/jm031136l. [DOI] [PubMed] [Google Scholar]
- Colotta V, Catarzi D, Varano F, Capelli F, Lenzi O, Filacchioni G, Martini C, Trincavelli L, Ciampi O, Pugliese AM, Pedata F, Schiesaro A, Morizzo E, Moro S. New 2-arylpyrazolo[3,4-c]quinoline derivatives as potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligand–receptor modeling studies. J Med Chem. 2007;50:4061–4074. doi: 10.1021/jm070123v. [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Alexander SP, Kellam B, Hill SJ. Agonist-occupied A3 adenosine receptors exist within heterogeneous complexes in membrane microdomains of individual living cells. FASEB J. 2008;22:850–860. doi: 10.1096/fj.07-8180com. [DOI] [PubMed] [Google Scholar]
- Costanzi S, Tikhonova IG, Harden TK, Jacobson KA. Ligand and structure-based methodologies for the prediction of the activity of G protein-coupled receptor ligands. J Comput Aided Mol Des. 2008 doi: 10.1007/s10822-008-9218-3. doi:10.1007/s10822-008-9218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosyn L, Gao ZG, Van Rompaey P, Lu C, Jacobson KA, Van Calenbergh S. Synthesis of hypermodified adenosine derivatives as selective adenosine A3 receptor ligands. Bioorg Med Chem. 2006a;14:1403–1412. doi: 10.1016/j.bmc.2005.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA, Van Calenbergh S. 2-Triazole-substituted adenosines: a new class of selective A3 adenosine receptor agonists, partial agonists, and antagonists. J Med Chem. 2006b;49:7373–7383. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Settimo F, Primofiore G, Taliani S, Marini AM, La Motta C, Simorini F, Salerno S, Sergianni V, Tuccinardi T, Martinelli A, Cosimelli B, Greco G, Novellino E, Ciampi O, Trincavalle ML, Martini C. 5-Amino-2-phenyl[1,2,3]triazolo[1,2-a][1,2,4]benzotriazin-1-one: a versatile scaffold to obtain potent and selective A3 adenosine receptor antagonists. J Med Chem. 2007;50:5676–5684. doi: 10.1021/jm0708376. [DOI] [PubMed] [Google Scholar]
- DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ. 3′ -Aminoadenosine-5′ - uronamides: discovery of the first highly selective agonist at the human adenosine A3 receptor. J Med Chem. 2003;46:353–355. doi: 10.1021/jm0255724. [DOI] [PubMed] [Google Scholar]
- DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ. The synthesis of highly potent, selective, and water-soluble agonists at the human adenosine A3 receptor. Bioorg Med Chem Lett. 2006;16:2525–2527. doi: 10.1016/j.bmcl.2006.01.088. [DOI] [PubMed] [Google Scholar]
- Drabczyńska A, Schumacher B, Müller CE, Karolak-Wojciechowska J, Michalak B, Pȩkala E, Kieć-Kononowicz K. Impact of the aryl substituent kind and distance from pyrimido[2,1-f ]purindiones on the adenosine receptor selectivity and antagonistic properties. Eur J Med Chem. 2003;38:397–402. doi: 10.1016/s0223-5234(03)00051-5. [DOI] [PubMed] [Google Scholar]
- Elzein E, Palle V, Wu Y, Maa T, Zeng D, Zablocki J. 2-Pyrazolyl-N6-substituted adenosine derivatives as high affinity and selective adenosine A3 receptor agonists. J Med Chem. 2004;47:4766–4773. doi: 10.1021/jm049682h. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Gallo-Rodriguez C, Ji X-D, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q-L, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure–activity relationships of N6-benzyladenosine-5′ -uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Blaustein J, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Mamedova L, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Kim SK, IJzerman AP, Jacobson KA. Allosteric modulation of the adenosine family of receptor. Mini Rev Med Chem. 2005;5:545–553. doi: 10.2174/1389557054023242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Joshi BV, Klutz A, Kim SK, Lee HW, Kim HO, Jeong LS, Jacobson KA. Conversion of A3 adenosine receptor agonists into selective antagonists by modification of the 5′ -ribofuran-uronamide moiety. Bioorg Med Chem Lett. 2006a;16:596–601. doi: 10.1016/j.bmcl.2005.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Duong HT, Sonina T, Lim SK, Van Rompaey P, Van Calenbergh S, Mamedova L, Kim HO, Kim MJ, Kim AY, Liang BT, Jeong LS, Jacobson KA. Orthogonal activation of the reengineered A3 adenosine receptor (neoceptor) using tailored nucleoside agonists. J Med Chem. 2006b;49:2689–2702. doi: 10.1021/jm050968b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta F, Del Giudice MR, Borioni A, Borea PA, Dionisotti S, Ongini E. Synthesis of imidazo[1,2-c]pyrazolo[4,3-e]pyrimidines, pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidines and 1,2,4-triazolo[5,1-i]purines: new potent adenosine A2 receptor antagonists. Eur J Med Chem. 1993;28:569–576. [Google Scholar]
- Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Goblyös A, Gao ZG, Brussee J, Connestari R, Santiago S Neves, Ye K, IJzerman AP, Jacobson KA. Structure–activity relationships of 1H-imidazo[4,5-c]quinolin-4-amine derivatives new as allosteric enhancers of the A3 adenosine receptor. J Med Chem. 2006;49:3354–3361. doi: 10.1021/jm060086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JW. A role for central A3-adenosine receptors: mediation of behavioral depressant effects. FEBS Lett. 1993;336:57–60. doi: 10.1016/0014-5793(93)81608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Siddiqi SM, Olah ME, Ji XD, Melman N, Bellamkonda K, Meshulmam Y, Stiles GL, Kim HO. Structure–activity relationships of 9-alkyladenine and ribose modified adenosine derivatives at rat A3 adenosine receptors. J Med Chem. 1995;38:1720–1735. doi: 10.1021/jm00010a017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Park KS, Jiang J-L, Kim YC, Olah ME, Stiles GL, Ji X-D. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Ji X-d, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, Van Rompaey P, Van Calenbergh S, Liang BT. Neoceptor concept based on molecular complementarity in GPCRs: a mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J Med Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chem Biol. 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, Gao ZG, Lu C, Duong HT, Gunaga P, Lee SK, Jin DZ, Chun MW, Moon HR. Structure–activity relationships of 2-chloro-N6-substituted-4′ -thioadenosine-5′ -uronamides as highly potent and selective agonists at the human A3 adenosine receptor. J Med Chem. 2006a;49:273–281. doi: 10.1021/jm050595e. [DOI] [PubMed] [Google Scholar]
- Jeong LS, Lee HW, Kim HO, Jung JY, Gao ZG, Duong HT, Rao S, Jacobson KA, Shin DH, Lee JA, Gunaga P, Lee SK, Jin DZ, Chun MW. Design, synthesis, and biological activity of N6-substituted-4′ -thioadenosines at the human A3 adenosine receptor. Bioorg Med Chem. 2006b;14:4718–4730. doi: 10.1016/j.bmc.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Jeong LS, Choe SA, Gunaga P, Kim HO, Lee HW, Lee SK, Tosh DK, Patel A, Palaniappan KK, Gao ZG, Jacobson KA, Moon HR. Discovery of a new nucleoside template for human A3 adenosine receptor ligands: D-4′ -thioadenosine derivatives without 4′ -hydroxymethyl group as highly potent and selective antagonists. J Med Chem. 2007;50:3159–3162. doi: 10.1021/jm070259t. [DOI] [PubMed] [Google Scholar]
- Jeong LS, Lee HW, Kim HO, Tosh D, Pal S, Choi WJ, Gao ZG, Patel AR, Williams W, Jacobson KA, Kim HD. Structure–activity relationships of 2-chloro-N6-substituted-4′ -thioadenosine-5′ -N, N-dialkyluronamides as human A3 adenosine receptor antagonists. Bioorg Med Chem. 2008;18:1612–1616. doi: 10.1016/j.bmcl.2008.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Xd, Melman N, Jacobson KA. Interactions of flavonoids and other phytochemicals with adenosine receptors. J Med Chem. 1996;39:781–788. doi: 10.1021/jm950661k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K-Y, Kim S-K, Gao Z-G, Gross AS, Melman N, Jacobson KA, Kim YC. Structure– activity relationships of thiazole and thiadiazole derivatives as potent and selective human adenosine A3 receptor antagonists. Bioorg Med Chem. 2004;12:613–623. doi: 10.1016/j.bmc.2003.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji X-D, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5′ -uronamides enhances selectivity for A3-adenosine receptors. J Med Chem. 1994a;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji X-D, Melman N, Olah ME, Stiles GL, Jacobson KA. Selective ligands for rat A3-adenosine receptors: structure–activity relationships of 1,3-dialkylxanthine-7-riboside derivatives. J Med Chem. 1994b;37:4020–4030. doi: 10.1021/jm00049a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Ji X-D, Jacobson KA. Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem. 1996;39:4142–4148. doi: 10.1021/jm960482i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Gao Z-G, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, Jacobson KA. Modeling the adenosine receptors: comparison of binding domains of A2A agonist and antagonist. J Med Chem. 2003;46:4847–4859. doi: 10.1021/jm0300431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Gao ZG, Jeong LS, Jacobson KA. Docking studies of agonists and antagonists suggest an activation pathway of the A3 adenosine receptor. J Mol Graph Model. 2006;25:562–577. doi: 10.1016/j.jmgm.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ravi RG, Ji X-D, Marquez VE, Jacobson KA. Ring-constrained (N)methanocarba-nucleosides as adenosine receptor agonists: independent 5′ -uronamide and 2′ -deoxy modifications. Bioorg Med Chem Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi O, Colotta V, Catarzi D, Varano F, Filacchioni G, Martini C, Trincavelli L, Ciampi O, Varani K, Marighetti F, Morizzo E, Moro S. 4-Amido-2-aryl-1,2,4-triazolo[4,3-a]quinoxalin-1-ones as new potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligand–receptor modeling studies. J Med Chem. 2006;49:3916–3925. doi: 10.1021/jm060373w. [DOI] [PubMed] [Google Scholar]
- Li AH, Moro S, Melman N, Ji XD, Jacobson KA. Structure–activity relationships and molecular modeling of 3,5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem. 1998;41:3186–3201. doi: 10.1021/jm980093j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Rebola N, Pinheiro PC, Richardson PJ, Oliveira CR, Cunha RA. Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport. 2003;14:1645–1648. doi: 10.1097/00001756-200308260-00021. [DOI] [PubMed] [Google Scholar]
- Maconi A, Pastorin G, Da Ros T, Spalluto G, Gao ZG, Jacobson KA, Baraldi PG, Cacciari B, Varani K, Moro S, Borea PA. Synthesis, biological properties, and molecular modeling investigation of the first potent, selective, and water-soluble human A3 adenosine receptor antagonist. J Med Chem. 2002;45:3579–82. doi: 10.1021/jm020974x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides. J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- Matot I, Weininger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 Adenosine receptors and mitogen activated protein kinases in lung injury following in-vivo reperfusion. Crit Care. 2006;10:R65. doi: 10.1186/cc4893. doi:10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Design of (N)-methanocarba adenosine 5′ -uronamides as species-independent A3 receptorselective agonists. Bioorg Med Chem Lett. 2008a;18:2813–2819. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Selective A3 adenosine receptor antagonists derived from nucleosides containing a bicyclo[3.1.0]hexane ring system. Bioorg Med Chem. 2008b;16:8546–8556. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhof W, Müller-Brechlin R, Richter D. Molecular cloning of a novel putative G-protein coupled receptor expressed during rat spermiogenesis. FEBS Lett. 1991;284:155–160. doi: 10.1016/0014-5793(91)80674-r. [DOI] [PubMed] [Google Scholar]
- Moro S, Braiuca P, Deflorian F, Ferrari C, Pastorin G, Cacciari B, Baraldi PG, Varani K, Borea PA, Spalluto G. Combined target-based and ligand-based drug design approach as a tool to define a novel 3D-pharmacophore model of human A3 adenosine receptor antagonists: pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as a key study. J Med Chem. 2005;48:152–162. doi: 10.1021/jm049662f. [DOI] [PubMed] [Google Scholar]
- Moro S, Spalluto G, Gao ZG, Jacobson KA. Progress in pursuit of therapeutic adenosine receptor antagonists. Med Res Rev. 2006;26:131–159. doi: 10.1002/med.20048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CE, Thorand M, Qurishi R, Diekmann M, Jacobson KA, Padgett WL, Daly JW. Imidazo[2,1-i]purin-5-ones and related tricyclic water-soluble purine derivatives: potent A2A- and A3-adenosine receptor antagonists. J Med Chem. 2002a;45:3440–3450. doi: 10.1021/jm011093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CE, Diekmann M, Thorand M, Ozola V. 3H 8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H [ ] a onist ] PSB-11), novel high affinity antag-radioligand for human A3 adenosine receptors. Bioorg Med Chem Lett. 2002b;12:501–503. doi: 10.1016/s0960-894x(01)00785-5. [DOI] [PubMed] [Google Scholar]
- Müller CE. Medicinal chemistry of adenosine A3 receptor ligands. Curr Top Med Chem. 2003;3:445–462. doi: 10.2174/1568026033392174. [DOI] [PubMed] [Google Scholar]
- Novellino E, Barbara Cosimelli, Marina Ehlardo, Giovanni Greco, Manuela Iadanza, Antonio Lavecchia, Rimoli MG, Sala A, Da Settimo A, Primofiore G, Da Settimo F, Taliani S, La Motta C, Klotz KN, Tuscano D, Trincavelli ML, Martini C. 2-(Benzimidazol-2-yl)quinoxalines: a novel class of selective antagonists at human A1 and A3 adenosine receptors designed by 3D database searching. J Med Chem. 2005;48:8253–8260. doi: 10.1021/jm050792d. [DOI] [PubMed] [Google Scholar]
- Ohana G, Bar-Yehuda S, Barer F, Fishman P. Differential effect of adenosine on tumor and normal cell growth: focus on the A3 adenosine receptor. J Cell Physiol. 2001;186:19–23. doi: 10.1002/1097-4652(200101)186:1<19::AID-JCP1011>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ohno M, Gao ZG, Van Rompaey P, Tchilibon S, Kim SK, Harris BA, Blaustein J, Gross AS, Duong HT, Van Calenbergh S, Jacobson KA. Modulation of adenosine receptor affinity and intrinsic efficacy in nucleosides substituted at the 2-position. Bioorg Med Chem. 2004;12:2995–3007. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Kurogi Y, Nishikawa H, Hashimoto K, Fujiwara H, Nagao Y. 1,2,4-triazolo[5,1-i]purine derivatives as highly potent and selective human adenosine A3 receptor ligands. J Med Chem. 2002;45:3703–3708. doi: 10.1021/jm010570p. [DOI] [PubMed] [Google Scholar]
- Okamura T, Kurogi Y, Hashimoto K, Nagao Y. Facile synthesis of fused 1,2,4-triazolo[1,5-c]-pyrimidine derivatives as human adenosine A3 receptor ligands. Bioorg Med Chem Lett. 2004a;14:2443–2446. doi: 10.1016/j.bmcl.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Okamura T, Kurogi Y, Hashimoto K, Sato S, Nishikawa H, Kiryu K, Nagao Y. Structure-activity relationships of adenosine A3 receptor ligands: new potential therapy for the treatment of glaucoma. Bioorg Med Chem Lett. 2004b;14:3775–3779. doi: 10.1016/j.bmcl.2004.04.099. [DOI] [PubMed] [Google Scholar]
- Ozola V, Thorand M, Diekmann M, Qurishi R, Schumacher B, Jacobson KA, Müller CE. 2-Phenylimidazo[2,1-i]purin-5-ones: structure–activity relationships and characterization of potent and selective inverse agonists at human A3 adenosine receptors. Bioorg Med Chem. 2003;11:347–356. doi: 10.1016/s0968-0896(02)00456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorin G, Da Ros T, Bolcato C, Montopoli C, Moro S, Cacciari B, Baraldi PG, Varani K, Borea PA, Spalluto G. Synthesis and biological studies of a new series of 5 heteroarylcarbamoylaminopyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidines as human A3 adenosine receptor antagonists. Influence of the heteroaryl substituent on binding affinity and molecular modeling investigations. J Med Chem. 2006;49:1720–1729. doi: 10.1021/jm051147+. [DOI] [PubMed] [Google Scholar]
- Perreira M, Jiang J, Klutz AM, Gao ZG, Shainberg A, Lu C, Thomas CJ, Jacobson KA. Reversine and its 2-substituted adenine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem. 2005;48:4910–4918. doi: 10.1021/jm050221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press NJ, Keller TH, Tranter P, Beer D, Jones K, Faessler A, Heng R, Lewis C, Howe T, Gedeck P, Mazzoni L, Fozard JR. New highly potent and selective adenosine A3 receptor antagonists. Curr Top Med Chem. 2004;4:863–870. doi: 10.2174/1568026043451023. [DOI] [PubMed] [Google Scholar]
- Priego EM, von Frijtag Drabbe Kuenzel J, IJzerman AP, Camarasa MJ, Pérez-Pérez MJ. Pyrido[2,1-f ]purine-2,4-dione derivatives as a novel class of highly potent human A3 adenosine receptor antagonists. J Med Chem. 2002;45:3337–3344. doi: 10.1021/jm0208469. [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Coppi E, Volpini R, Cristalli G, Corradetti R, Jeong LS, Jacobson KA, Pedata F. Role of adenosine A3 receptors on CA1 hippocampal neurotransmission during oxygenglucose deprivation episodes of different duration. Biochem Pharmacol. 2007;74:768–779. doi: 10.1016/j.bcp.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saki M, Tsumuki H, Nonaka H, Shimada J, Ichimura M. KF26777 (2-(4-bromophenyl)-7,8-dihydro-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one dihydrochloride), a new potent and selective adenosine A3 receptor antagonist. Eur J Pharmacol. 2002;444:133–144. doi: 10.1016/s0014-2999(02)01662-x. [DOI] [PubMed] [Google Scholar]
- Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Shneyvais V, Mamedova L, Zinman T, Jacobson KA, Shainberg A. Activation of A3 adenosine receptor protects against doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol. 2001;33:1249–1261. doi: 10.1006/jmcc.2001.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi SM, Jacobson KA, Esker JL, Olah ME, Ji XD, Melman N, Tiwari KN, Secrist JA, III, Schneller SW, Cristalli G, Stiles GL, Johnson Cr, IJzerman AP. Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors. J Med Chem. 1995;38:1174–1188. doi: 10.1021/jm00007a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickler J, Jacobson KA, Liang BT. Direct preconditioning of cultured chick ventricular myocytes: novel functions of cardiac adenosine A2A and A3 receptors. J Clin Invest. 1996;98:1773–1779. doi: 10.1172/JCI118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafi A, Bernardini C, Botta M, Corelli F, Andreini M, Martinelli A, Ortore G, Baraldi PG, Fruttarolo F, Borea PA, Tuccinardi T. Pharmacophore based receptor modeling: the case of adenosine A3 receptor antagonists. An approach to the optimization of protein models. J Med Chem. 2006;49:4085–4097. doi: 10.1021/jm051112+. [DOI] [PubMed] [Google Scholar]
- Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein J, Gross AS, Melman N, Jacobson KA. Exploring distal regions of the A3 adenosine receptor binding site: sterically-constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg Med Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. (N)-Methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, Knight DR. Novel N6-substituted adenosine 5′ -N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury. Am J Physiol Heart Circ Physiol. 2003;285:H2780–H2787. doi: 10.1152/ajpheart.00411.2003. [DOI] [PubMed] [Google Scholar]
- van Galen PJM, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. A binding site model and structure–activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- van Muijlwijk-Koezen JE, Timmerman H, Link R, von der Goot H, Menge WMPB, von Frijtag von Drabbe Künzel JK, de Groote M, IJzerman AP. Isoquinoline and quinazoline urea analogues as antagonists for the human adenosine A3 receptor. J Med Chem. 2000;43:2227–2238. doi: 10.1021/jm000002u. [DOI] [PubMed] [Google Scholar]
- van Muijlwijk-Koezen JE, Timmerman H, Vollinga RC, von Drabbe Künzel JF, de Groote M, Visser S, IJzerman AP. Thiazole and thiazole analogues as novel class of adenosine receptor antagonists. J Med Chem. 2001;44:749–762. doi: 10.1021/jm0003945. [DOI] [PubMed] [Google Scholar]
- Van Rompaey P, Jacobson KA, Gross AS, Gao ZG, Van Calenbergh S. Exploring human adenosine A3 receptor complementarity and activity for adenosine analogues modified in the ribose and purine moiety. Bioorg Med Chem. 2005;13:973–983. doi: 10.1016/j.bmc.2004.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilburg EW, von Frijtag Drabbe Kunzel J, de Groote M, IJzerman AP. 2, 5′ -Disubstituted adenosine derivatives: evaluation of selectivity and efficacy for the adenosine A1, A2A, and A3 receptor. J Med Chem. 2002;45:420–429. doi: 10.1021/jm010952v. [DOI] [PubMed] [Google Scholar]
- Varani K, Merighi S, Gessi S, Klotz KN, Leung E, Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Borea PA. [3H]MRE 3008F20: a novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol Pharmacol. 2000;57:968–975. [PubMed] [Google Scholar]
- Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-Alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J Med Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- Volpini R, Dal Ben D, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli GJ. N6-Methoxy-2-alkynyladenosine derivatives as highly potent and selective ligands at the human A3 adenosine receptor. J Med Chem. 2007;50:1222–1230. doi: 10.1021/jm060963u. [DOI] [PubMed] [Google Scholar]
- Yaar R, Lamperti ED, Toselli PA, Ravid K. Activity of the A3 adenosine receptor gene promoter in transgenic mice: characterization of previously unidentified sites of expression. FEBS Lett. 2002;532:267–272. doi: 10.1016/s0014-5793(02)03612-8. [DOI] [PubMed] [Google Scholar]
- Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A3 adenosine-receptor antagonist MRS1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr Eye Res. 2005;30:747–754. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci USA. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Frazier CR, Linden J, Macdonald TL. N6-Ethyl-2-alkynyl NECAs, selective human A3 adenosine receptor agonists. Bioorg Med Chem Lett. 2006;16:2416–2418. doi: 10.1016/j.bmcl.2006.01.110. [DOI] [PubMed] [Google Scholar]