

Abstract
Development of the neuromuscular junction (NMJ) requires secretion of specific isoforms of the proteoglycan agrin by motor neurons. Secreted agrin is widely expressed in the basal lamina of various tissues, whereas a transmembrane form is highly expressed in the brain. Expression in the brain is greatest during the period of synaptogenesis, but remains high in regions of the adult brain that show extensive synaptic plasticity. The well-established role of agrin in NMJ development and its presence in the brain elicited investigations of its possible role in synaptogenesis in the brain. Initial studies on the embryonic brain and neuronal cultures of agrin-null mice did not reveal any defects in synaptogenesis. However, subsequent studies in culture demonstrated inhibition of synaptogenesis by agrin antisense oligonucleotides or agrin siRNA. More recently, a substantial loss of excitatory synapses was found in the brains of transgenic adult mice that lacked agrin expression everywhere but in motor neurons. The mechanisms by which agrin influences synapse formation, maintenance and plasticity may include enhancement of excitatory synaptic signaling, activation of the “muscle-specific” receptor tyrosine kinase (MuSK) and positive regulation of dendritic filopodia. In this article I will review the evidence that agrin regulates synapse development, plasticity and signaling in the brain and discuss the evidence for the proposed mechanisms.
Keywords: agrin, brain, synapse development, synaptic activity, synaptic plasticity, filopodia
Agrin as a synapse-organizing molecule
The proteoglycan agrin was first isolated from the extracellular matrix of the Torpedo electric organ (consisting of stacks of electrocytes covered with modified neuromuscular junctions) as a high molecular weight protein factor that induced the aggregation of nicotinic acetylcholine receptors (AChRs)1 on cultured developing skeletal muscle fibers (Godfrey et al., 1984). In its original form, the “agrin hypothesis” (McMahan, 1990) postulated that motor neurons secrete a specific form of agrin, which induces de novo organization of the postsynaptic membrane of the neuromuscular junction (NMJ). With the subsequent cloning of the agrin gene in rat (Rupp et al., 1991) and chicken (Tsim et al., 1992), creation of agrin-null mice that failed to form NMJs (Gautam, et al.,1996) and discovery of the receptors and signaling pathways regulated by neuron-specific isoforms of agrin in skeletal muscle (Wu et al., 2010), this hypothesis was confirmed in its essentials and extended. The only significant modification to date of the “agrin hypothesis” is the concept that transient spontaneously formed clusters of AChRs on developing skeletal muscle fibers may serve as targets for innervating motor neurons (Sytkowski et al., 1973), and that the role of agrin secreted by motor neurons is to stabilize these clusters and to induce and maintain further postsynaptic differentiation (Kummer et al., 2006).
At the time of the first report on the cloning of rat agrin (Rupp et al., 1991), agrin -like AChR aggregating activity and immunoreactivity was reported in avian brain (Godfrey, 1991). Once agrin had been cloned, its expression in the developing rat brain was soon demonstrated (Rupp et al., 1991). The isoforms of agrin expressed in rat and mouse brain include isoforms with inserts in the “Z” site of the C-terminal moiety that are required for the induction of AChR aggregation on skeletal myotubes and formation of the NMJ (Burgess et al., 1999; Ferns et al., 1992; Hoch et al., 1993; Li et al., 1997; O'Connor et al., 1994) (see fig. 1). Expression in the brain is highest during the period of development when synaptogenesis is occurring (Li et al., 1997; Ma et al., 1994); Stone and Nikolics, 1995) and, in adult mice, expression remains high in the hippocampus and cortex, regions that show extensive synaptic plasticity (O'Connor et al., 1994). Moreover, a few groups have demonstrated localization of agrin immunoreactivity at interneuronal synapses (Gingras and Ferns, 2001; Hoover et al., 2003; Ksiazek et al., 2007; Mann and Kroger, 1996). It is noteworthy that the forms of agrin expressed in neurons of the brain have an N-terminal domain resulting from an alternative start site that makes them type II transmembrane proteoglycans rather than the secreted form found in basal laminae of skeletal muscle, blood vessels and the kidney glomerulus (Burgess et al., 2000; Neumann et al., 2001) (fig 1). The discovery of agrin protein, AChR aggregating activity and gene expression in the brain led to speculation that agrin might act as a synapse-organizing molecule in interneuronal synapses as it does at the NMJ.
Figure 1.
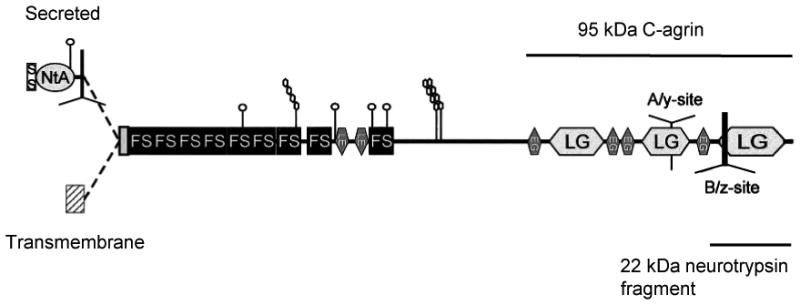
Schematic of agrin structure modified from figure 1A of Neumann et al., 2001. The schematic shows the alternative secretory (NtA) and transmembrane N-termini, the various structural motifs, the splice sites, and the extent of 95 kDa C-agrin and the 22 kDa C-terminal fragment generated by neurotrypsin, as referred to in this review. Splice sites are indicated by Y shapes. The indicated motifs are follistatin-like (FS), laminin EGF-like (LE), EGF-like and laminin G-like (LG). Vertical lines with chains of oval shapes represent GAG side chains.
The earliest studies testing the idea that agrin plays a role in interneuronal synaptogenesis did not support it. Brains of agrin-null mouse embryos, although smaller, showed normal gross architecture and synapse structure (Serpinskaya et al., 1999). Synapse formation also appeared to be normal in cultures of hippocampal and cortical neurons from agrin-null embryos (Li et al, 1999; Serpinskaya et al., 1999). However, three different groups subsequently demonstrated strongly reduced synapse formation in cultures of hippocampal neurons in which agrin expression was suppressed by agrin antisense oligonucleotides added to the culture medium (Bose et al., 2000; Ferreira, 1999) or by infection of the neurons with a lentiviral vector for expression of agrin siRNA (McCroskery et al., 2009). The discrepancy between these results and the results with agrin-null mouse embryos or neurons suggests the possibility that other molecules, such as the syndecan proteoglycans (Hsueh and Sheng, 1999), may compensate for the chronic absence of agrin in the developing brain. Subsequent studies of sympathetic ganglion neurons from agrin-null embryos in vivo and in culture revealed a reduction in cholinergic synapse formation and synaptic activity between spinal neurons and sympathetic neurons, suggesting that formation of this type of interneuronal synapse is in part dependent on agrin (Gingras et al., 2002). Most recently, synapse formation and activity was examined in the brains of adult transgenic mice that express chicken secretory agrin only in motor neurons but do not express mouse agrin (Ksiazek et al., 2007). Agrin expression in motor neurons allows sufficient NMJ formation for survival of these mice to adulthood. This study revealed a 30% reduction in the number of excitatory synapses on the dendrites of cortical pyramidal neurons, a reduction in dendritic spine density and a reduction in miniature excitatory postsynaptic potentials, with no effect on the number of inhibitory synapses or miniature inhibitory postsynaptic potentials. These results suggested agrin-dependent formation and/or maintenance in a subset of excitatory synapses in the mouse brain.
Together, the in vivo and in vitro studies support the notion that agrin plays a role in the formation and/or maintenance of interneuronal synapses, including excitatory synapses in the brain. However, relatively little is known about the mechanisms by which agrin can support interneuronal synaptogenesis. The subsequent sections of this review deal with some possible mechanisms by which agrin may support synaptogenesis as well as synaptic plasticity and signaling in the brain.
Does agrin act through activation of MuSK in the brain?
Originally, it was thought that the receptor tyrosine kinase, muscle specific kinase (MuSK), was strongly expressed and functional only in skeletal muscle, where its activation by agrin is essential to formation of the NMJ (Glass et al., 1996a,b; Valenzuela et al., 1995). Thus, despite the expression in the brain of Z+ isoforms of agrin (see above) that can activate MuSK (Glass et al., 1996a), little consideration was given to the possibility that agrin in the brain could act through MuSK. More recently, an extensive study has shown that MuSK mRNA and protein are widely expressed in neurons of adult rat brain, including those of the hippocampus, cortex and cerebellum (Garcia-Osta et al., 2006). Some of this MuSK protein appears to be concentrated at excitatory synapses in the mouse cortex (Ksiazek et al., 2007), where it is possibly colocalized with agrin. Garcia-Osta and colleagues further demonstrated that inhibition of MuSK expression by infusion of MuSK antisense oligonucleotides into the hippocampus interfered with memory consolidation associated with inhibitory avoidance (IA) training in adult rats. Inhibition of MuSK expression during training also suppressed phosphorylation of the transcription factor cyclic AMP response element binding protein (CREB) and expression of the transcription factor CCAAT enhancer binding protein β (C/EBPβ), both of which are in the signal transduction pathways underlying memory consolidation in IA training (Kandel, 2001). Additionally, electrophysiological investigations on brain slices showed that inhibition of MuSK expression interfered with the cholinergic oscillatory activity in area CA1 of the hippocampus and with long term potentiation (LTP), both activities associated with synaptic plasticity and learning (Buzsaki, 2002; Huang and Stevens, 1998).
The above study has implicated MuSK in modulation of synaptic signaling and plasticity in the rat brain, particularly in learning and memory (see fig. 2C). As yet, there is no direct evidence that MuSK is involved in interneuronal synapse formation or maintenance analogous to its role in NMJ development, but this has been suggested as a possible mechanism for the involvement of agrin in cortical synapse maintenance (Ksiazek et al., 2007) (see fig. 2A). An important unresolved question is whether the reported activities of MuSK in brain result from activation of MuSK by agrin or from some as yet unknown mode of activation. Alternate modes of MuSK activation exist, since spontaneous formation of AChR clusters on developing skeletal muscle fibers occurs in the absence of agrin and is MuSK dependent (Lin et al., 2001).
Figure 2.
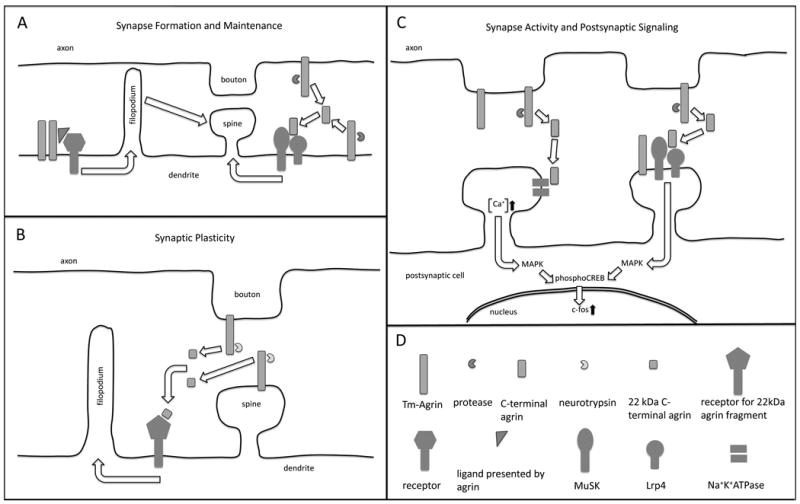
A simplified and somewhat speculative schematic of the ways in which agrin may influence synapse formation, maintenance, plasticity, activity and postsynaptic signaling, as suggested by the published findings reviewed in this article. (A left) Tm-agrin promotes the formation of dendritic filopodia by acting as a transmembrane receptor itself or by facilitation of ligand binding to an undetermined transmembrane receptor. Filopodia in turn promote the formation of spine synapses. (A right) Tm-agrin or a C-terminal proteolytic fragment activates MuSK through Lrp4 binding to promote synapse formation and/or maintenance. (B) Under conditions that promote LTP, secreted neurotrypsin generates a 22 kDa agrin C-terminal fragment. This fragment induces filopodia formation through an undetermined receptor, thus promoting new spine synapse formation. (C-left) Agrin or a C-terminal proteolytic fragment binds to the α3NKA, inhibiting the activity of Na+K+ ATPase and thus increasing excitatory synaptic activity, intracellular [Ca+] and downstream signal transduction through the MAP kinase pathway. (C-right) Tm-agrin or a C-terminal fragment activates MuSK through Lrp4 binding to elicit downstream signal transduction involved in synaptic plasticity. (D) Key to representations of the molecules mentioned in this legend. Note that Tm-agrin is actually large enough to span the synaptic cleft and act across it.
Agrin modulation of excitatory signaling through the α3 subunit of Na+K+ ATPase (α3NKA)
A convergence between studies characterizing the effects of agrin on excitatory signaling by brain neurons and a search for binding sites of agrin fragments that could elicit these effects led to the identification of the only known functional neuronal receptor for agrin, the α3 subunit of Na+K+ ATPase (α3NKA). Among the first functional effects of soluble agrin fragments to be discovered on cultured brain neurons were an increase in phosphorylation of CREB in cultured hippocampal neurons (Ji et al., 1998) and expression of the immediate early gene c-fos in cultured cortical and other neuron types (Hilgenberg et al., 1999). Increased phosphorylation of CREB and expression of c-fos have been associated with the effects of excitatory synaptic activity as seen, for example, in LTP, and are thought to be involved in synaptic plasticity and memory formation (Kaczmarek, 1993; Kandel, 2001). The elevation of CREB and c-fos expression was obtained with the soluble C-terminal 95 kDa moiety of agrin (fig. 1) (Ji et al., 1998; Hilgenberg et al., 1999). However, whereas the induction of CREB expression was dependent on the Z-site inserts (Z+ agrin) required for MuSK activation, the induction of c-fos expression was not, suggesting that different receptors are involved. Subsequently, it was shown that cultured cortical neurons from agrin-null mouse embryos had reduced c-fos induction by activation of nicotinic and glutaminergic receptors. Moreover, kainic acid-induced c-fos expression and excitotoxic neuronal cell death were reduced in brain slices of adult agrin knockout heterozygous mice, indicating that endogenous agrin enhances excitatory signaling in vivo (Hilgenberg et al., 2002). Further studies indicated that a 20 kDa fragment of the C-terminus of agrin was sufficient to induce the in vitro effects described above, and that this fragment bound specifically to the α3NKA concentrated at neuronal synapses, inhibiting the activity of the Na+K+ ATPase and thus decreasing the resting membrane potential of the affected neurons and increasing action potential frequency (Hilgenberg et al., 2006). Moreover, a 15 kDa fragment that acts as a competitive antagonist of the 20 kDa fragment depressed action potential frequency. These findings suggest that endogenous neuronal agrin enhances excitatory synaptic signaling through its binding to and inhibition of Na+K+ ATPase (see fig. 2C).
Modulation of synapse formation and plasticity by agrin through regulation of dendritic filopodia
Dendritic filopodia are thought to promote synapse formation and act as precursors of dendritic spines, the sites of excitatory synapses on dendrites (Takahashi et al., 2003; Yuste and Bonhoeffer, 2004; Ziv and Smith, 1996). Two research groups have provided evidence that the transmembrane form of agrin (Tm-agrin), the predominant form expressed by neurons in the brain (Burgess et al., 2000; Neumann et al., 2001) positively regulates filopodia in cultured neurons. In one study, it was shown that clustering of endogenous Tm-agrin induced the formation of filopodia along the axonal processes of cultured chick peripheral and central neurons as well as on the axons and dendrites of mouse hippocampal neurons (Annies et al., 2002). In another, the overexpression of TM-agrin in rat hippocampal neurons cultured for 1-6 days was shown to increase the number and stability of filopodia on neurites (both the presumptive axons and presumptive dendrites in early stage cultures). Moreover, transfection of these neurons with an shRNA plasmid to suppress agrin expression reduced filopodia density and stability on neurites, indicating a positive role for endogenous Tm-agrin in regulating the filopodia on neurites (McCroskery et al., 2006). In low-density hippocampal neuron cultures such as used in this study, filopodia appear along developing axons and dendrites from the earliest days of culture, but most synapse formation occurs in the second and third week in culture. A more recent study showed that inhibition of agrin expression beginning at 7 days in culture by infection at > 95% efficiency with a shRNA lentiviral vector reduced filopodia density on dendrites by 37% compared to controls at 21 days in culture (McCroskery et al, 2009). Moreover, the density of PSD95-synaptotagmin positive synapses on dendrites was reduced by 55% at 21 days. The effect on synapse number is in general agreement with the in vivo observations of Ksiazek et al. in the mouse cortex, described above. McCroskery et al. (2009) also examined the density of synapses on dendrites in cultures infected with lower efficiency, in cases where an axon-dendrite segment of contact consisted of an infected dendrite and uninfected axon or vice versa. They observed a greater reduction of synaptic density when the dendrite was infected than when the axon was. This result is consistent with a role for dendritic Tm-agrin in supporting synaptogenesis through positive regulation of dendritic filopodia, but suggests that axonal Tm-agrin may also play a role in synaptogenesis (see fig. 2A).
Recently, it was discovered that the neuronal protease, neurotrypsin, specifically cleaves agrin at two sites to form a 90 and a 22 kDa fragment (fig. 1), the smaller fragment consisting of the C-terminal laminin G domain (Reif et al., 2007). Neurotrypsin is located in and released from axonal boutons in an activity dependent manner (Frischknecht et al., 2008). A study in isolated hippocampi and hippocampal slices of wild-type and neurotrypsin deleted mice showed that coincidence of presynaptic firing and postsynaptic activation of glutamate receptors is required for release of neurotrypsin and for proteolysis of agrin to produce the 22 kDa fragment, respectively (Matsumoto-Miyai et al., 2009). Moreover, this study showed that production of this fragment by neurotrypsin is required for the LTP dependent induction of dendritic filopodia. Since, as mentioned earlier, dendritic filopodia are involved in the formation of new spine synapses, these findings strongly implicate neurotrypsin and this 22 kDa proteolytic fragment of Tm-agrin that it produces, in de novo synapse formation associated with synaptic plasticity (see fig. 2B).
How does agrin regulate dendritic filopodia? – early clues
The mechanisms by which agrin regulates dendritic filopodia are not well understood, but it seems clear that the receptors and upstream signaling pathways differ from those utilized in the developing NMJ. Agrin exerts its NMJ organizing activity by binding to the LDL receptor-related protein 4 (Lrp4), resulting in activation of MuSK (Kim et al., 2008; Zhang et al., 2008). However, whereas activation of MuSK to induce AChR aggregation and NMJ formation is dependent on the presence of an 8, 11 or 19 amino acid insert in the C-terminal “Z” site of agrin (Z+ agrin) (Burgess et al., 1999; Ferns et al., 1992; Glass et al., 1996a,b), the filopodia inducing activity of the 22 kDa neurotrypsin-dependent C-terminal agrin fragment does not require Z+ agrin (Matsumoto-Miyai et al., 2009). Likewise, induction of filopodia in neurons or filopodia-like processes in non-neuronal cells by overexpression of Tm-agrin is also independent of Z-site inserts (McCroskery et al., 2006). In fact, this latter activity is dependent on the N-terminal moiety of Tm-agrin, importantly including the transmembrane domain (McCroskery et al., 2006) and 3 of the 8 follistatin-like domains (fig.1) in the N-terminal extracellular moiety (Porten et al., 2010). There is also strong evidence that the glycosaminoglycan (GAG) chains (fig. 1) attached to the N-terminal moiety (Winzen et al., 2003) play an important role in the induction of filopodia-like processes on non-neuronal cells and filopodia in neurons. Few or none of these processes are induced in non-neuronal cells or neurons, respectively, overexpressing Tm-agrin with the GAG chains deleted (Lin et al., 2010).
The difference in the agrin domains required for neurotrypsin-dependent induction of dendritic filopodia and those required for the formation of filopodia in neurons and filopodia-like processes in non-neuronal cells induced by Tm-agrin overexpression suggests that different receptors and downstream signal transduction mechanisms may be involved. Some progress has been made in understanding the signal transduction mechanisms involved in filopodia induction by overexpression or clustering of Tm-agrin, but it must be kept in mind that the mechanisms uncovered in this way are not necessarily the ones involved in the positive regulation of neuronal filopodia by endogenous Tm-agrin. The formation of filopodia-like processes in non-neuronal cells and neuronal cell lines transfected with Tm-agrin is dependent on the activation of Rac1 and Cdc42 (Lin et al., 2010; McCroskery et al., 2006), Ras family GTPases well known to be involved in protrusive activity of the actin cytoskeleton (Hall, 1998; Hall, 2005; Ridley, 1999). However, the upstream receptors and signal transduction pathways for this process are unknown. Filopodia formation induced by clustering of endogenous Tm-agrin has recently been shown to involve sequential clustering of Tm-agrin in lipid rafts, activation of the tyrosine kinase Fyn, and activation of mitogen activated protein kinase (MAPkinase) (Ramseger et al., 2009). In view of the similarity to other cellular responses that can be mediated either by clustering or ligand-induced dimerization of transmembrane receptors, Ramseger et al. proposed that Tm-agrin itself acts as a receptor in this process. If this is the case, the endogenous ligand for Tm-agrin with respect to regulation of filopodia remains to be identified. Alternatively, Tm-agrin may facilitate a receptor-ligand interaction that promotes filopodia formation and stabilization, perhaps through the ability of the agrin GAG chains to bind and present various ligands to their cell surface receptors (see, for example, Kim et al., 2003) (see fig. 2A).
Does neuronal agrin regulate synapse formation, synaptic plasticity and signaling through diverse receptors and common signal transduction mechanisms in the brain?
As described above, there is strong evidence that agrin regulates formation or stabilization of a subclass of excitatory synapses in the murine brain and that it also regulates excitatory synaptic activity and synaptic plasticity in areas including the hippocampus and cortex. Two broad questions remain unanswered and should be the subject of future research: 1) Are distinct receptors acted upon by different isoforms or fragments of agrin to regulate these processes? 2) Are there common signal transduction pathways for these diverse roles of agrin?
Known and putative receptors for agrin in the brain
The predominant form of agrin expressed by neurons in the brain is a type II transmembrane proteoglycan (Burgess et al., 2000; Neumann et al., 2001). However, as described above, a 20 kDa fragment of agrin, consisting of the C-terminal-most laminin G domain, binds to the α3NKA, inhibiting the activity of Na+K+ ATPase and thus reducing the membrane potential of neurons and increasing excitability (Hilgenberg et al., 2006). A C-terminal 22kDa fragment containing the same laminin G domain, produced by neurotrypsin under conditions favoring synaptic plasticity, induces dendritic filopodia formation in brain slices (Matsumoto-Miyai et al., 2009). It would be tempting to suggest that the filopodia-inducing activity was somehow related to increased excitability. However, the induction of c-fos in cortical neurons, for which the 20 kDa fragment is sufficient, was initially demonstrated with the 95 kDa soluble C-agrin fragment (Hilgenberg et al., 1999), whereas neurotrypsin proteolysis to produce the 22kDa fragment was required for filopodia induction (Matsumoto-Miyai et al., 2009).
Thus it seems likely that a receptor other than the α3NKA is involved in excitatory synaptic activity-related filopodia induction. On the other hand, one might speculate that increased excitability due to binding of the 22 kDa fragment to the α3NKA would enhance the synaptic activity required for neurotrypsin-dependent production of the 22 kDa fragment, thus creating a positive feedback loop. With regard to the role of agrin in supporting synaptogenesis through positive regulation of dendritic filopodia during development, it remains to be determined whether the active form of agrin involved in this process is the intact transmembrane proteoglycan or a fragment produced by proteolytic activity. As mentioned above, the induction of filopodia on neurons by Tm-agrin overexpression appears to require the transmembrane domain and the N-terminal moiety of Tm-agrin, including 3 of the 8 follistatin-like domains (McCroskery et al., 2006; Porten et al., 2010), suggesting that a different receptor is involved in this process than in the neurotrypsin-dependent induction of dendritic filopodia in adult brain slices. However, it remains to be determined if induction by overexpression utilizes the same receptor as the regulation of neuronal filopodia by endogenous Tm-agrin. Finally, it would be of interest to determine whether or not the 22 kDa fragment of agrin produced by neurotrypsin digestion would induce filopodia on the dendrites of hippocampal or cortical neurons in culture and to identify the relevant receptor.
As described above, there is good evidence that MuSK is involved in synaptic activity and downstream signaling in the rat brain (Garcia-Osta et al., 2006), but it has not been determined if this activity requires activation by agrin. The phosphorylation of CREB induced by soluble C-terminal agrin in cultured hippocampal neurons appears to require activation of MuSK, since this effect was observed with a Z+ isoform of C-terminal agrin but not with the Z- form (Ji et al., 1993). It will be important to determine if MuSK activation by agrin is required for the phosphorylation of CREB in vivo, where the phosphorylation of CREB is thought to be involved in activity-dependent regulation of synaptic strength (Kandel, 2001; Weller and Waltereit, 2003). In skeletal muscle cells, agrin activates MuSK by binding to Lrp4 (Kim et al., 2008; Zhang et al., 2008). If agrin indeed activates MuSK in brain, the relevant binding site remains to be identified. Interestingly, Lrp4 has been found in synaptic fractions of brain and appears to interact with the postsynaptic density proteins PSD95 and SAP97 (Tian et al., 2006). Thus it is positioned also to interact with synaptic agrin and MuSK.
Does agrin act through the mitogen-activated protein kinase (MAP kinase) pathway in the brain?
The MAP kinase signal transduction pathway mediates multiple effects of tyrosine kinase activation in mammalian cells, in particular the activation that occurs when membrane receptors bind their ligands (McKay and Morrison, 2007). In skeletal muscle, this pathway mediates the effects of MuSK activation by agrin, including AChR clustering and NMJ-specific gene expression (Rimer, 2011). A few lines of evidence described above suggest that the MAP kinase pathway may mediate the effects of agrin and MuSK in the brain and in cultured cortical and hippocampal neurons. MAP kinase phosphorylation has been implicated as a downstream step in the phosphorylation of CREB induced by Z+ agrin in hippocampal neurons in culture (Ji et al, 1998; Karasewski and Ferreira, 2003), and in the expression of c-fos induced by either Z+ or Z- agrin in cultured cortical neurons (Hilgenberg et al., 1999). The latter effect of agrin was triggered by an elevation of intracellular [Ca2+], but also required Ca2+entry through voltage-sensitive Ca2+ channels. As described above, the neuronal receptor responsive to Z- as well as Z+ agrin is the α3NKA, to which the binding of agrin results in decreased membrane potential and increased excitability (Hilgenberg et al., 2006). It is possible that increased excitability of cortical neurons exposed to agrin might increase the Ca2+ flux through voltage gated Ca2+ channels, thus stimulating the downstream pathways. As cited above, the induction of filopodia-like processes by the clustering of endogenous Tm-agrin also requires activation of MAP kinase subsequent to Fyn activation (Ramseger et al., 2009). It is of interest here that suppression of MuSK expression in the rat hippocampus causes a reduction in CREB phosphorylation (Garcia-Osta et al., 2006). However, in the case of agrin function in the brain, it remains to be determined if the MAP kinase pathway is involved in CREB phosphorylation and the consequences to synaptic plasticity and memory consolidation (see fig. 2C).
Conclusions
In vivo and culture studies, particularly in the last 10-15 years, have provided strong evidence for a role of agrin in synaptic development, plasticity and signaling in the brain. Much work remains to be done to elucidate the specific actions of agrin in different brain areas, stages of development and types of synapses. It will be important to identify additional receptors for agrin and the relevant downstream signal transduction pathways.
Highlights.
Agrin is expressed in neurons of developing and adult brain
Reduced agrin expression inhibits synapse formation in vivo and in vitro
Receptors and co-receptors of agrin regulate synaptic activity and plasticity
Agrin may promote synapse formation and plasticity by inducing dendritic filopodia
Future goal is to better understand relevant agrin receptors and downstream signals
Acknowledgments
I am grateful to Dr. Earl Godfrey for critical reading of the manuscript and to Dr. Markus Ruegg for permission to use the agrin structure schematic.
Footnotes
Non-standard abbreviations used in this article: AChR, nicotinic acetylcholine receptor; NMJ, neuromuscular junction; Tm-agrin, transmembrane agrin; MuSK, muscle-specific kinase; IA training, inhibitory avoidance training; C/EBPβ, CCAAT enhancer binding protein β; α3NKA, α3 subunit of Na+K+ ATPase; Lrp4, LDL receptor-related protein 4; MAP kinase, mitogen-activated protein kinase.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annies M, Bittcher G, Ramseger R, Loschinger J, Woll S, Porten E, Abraham C, Ruegg MA, Kroger S. Clustering transmembrane-agrin induces filopodia-like processes on axons and dendrites. Mol Cell Neurosci. 2006;31:515–524. doi: 10.1016/j.mcn.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Bose CM, Qiu D, Bergamaschi A, Gravante B, Bossi M, Villa A, Rupp F, Malgaroli A. Agrin controls synaptic differentiation in hippocampal neurons. J Neurosci. 2000;20:9086–9095. doi: 10.1523/JNEUROSCI.20-24-09086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR. Alternatively spliced isoforms of nerve- and muscle-derived agrin: Their roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/s0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Skarnes WC, Sanes JR. Agrin isoforms with distinct amino termini: differential expression, localization, and function. J Cell Biol. 2000;151:41–52. doi: 10.1083/jcb.151.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Ferns M, Hoch W, Campanelli JT, Rupp F, Hall ZW, Scheller RH. RNA splicing regulates agrin-mediated acetylcholine receptor clustering activity on cultured myotubes. Neuron. 1992;8:1079–1086. doi: 10.1016/0896-6273(92)90129-2. [DOI] [PubMed] [Google Scholar]
- Ferreira A. Abnormal synapse formation in agrin-depleted hippocampal neurons. J Cell Sci. 1999;112:4729–4738. doi: 10.1242/jcs.112.24.4729. [DOI] [PubMed] [Google Scholar]
- Frischknecht R, Fejtova A, Viesti M, Stephan A, Sonderegger P. Activity-induced synaptic capture and exocytosis of the neuronal serine protease neurotrypsin. J Neurosci. 2008;28:1568–1579. doi: 10.1523/JNEUROSCI.3398-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Osta A, Tsokas P, Pollonini G, Landau ML, Blitzer R, Alberini CM. MuSK expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation. J Neurosci. 2006;26:7919–7932. doi: 10.1523/JNEUROSCI.1674-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gingras J, Ferns M. Expression and localization of agrin during sympathetic synapse formation in vitro. J Neurobiol. 2001;48:228–242. doi: 10.1002/neu.1053. [DOI] [PubMed] [Google Scholar]
- Gingras J, Rassadi S, Cooper E, Ferns M. Agrin plays an organizing role in the formation of sympathetic synapses. J Cell Biol. 2002;158:1109–1118. doi: 10.1083/jcb.200203012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996a;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Glass DJ, DeChiara TM, Stitt TN, DiStefano PS, Valenzuela DM, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation and is a functional receptor for agrin. Cold Spring Harb Symp Quant Biol. 1996b;61:435–444. [PubMed] [Google Scholar]
- Godfrey EW. Comparison of agrin-like proteins from the extracellular matrix of chicken kidney and muscle with neural agrin, a synapse organizing protein. Exp Cell Res. 1991;195:99–109. doi: 10.1016/0014-4827(91)90504-n. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Nitkin RM, Wallace BG, Rubin LL, McMahan UJ. Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J Cell Biol. 1984;99:615–627. doi: 10.1083/jcb.99.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Hilgenberg LG, Hoover CL, Smith MA. Evidence of an agrin receptor in cortical neurons. J Neurosci. 1999;19:7384–7393. doi: 10.1523/JNEUROSCI.19-17-07384.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenberg LG, Ho KD, Lee D, O'Dowd DK, Smith MA. Agrin regulates neuronal responses to excitatory neurotransmitters in vitro and in vivo. Mol Cell Neurosci. 2002;19:97–110. doi: 10.1006/mcne.2001.1056. [DOI] [PubMed] [Google Scholar]
- Hilgenberg LGW, Su H, Gu H, O'Dowd DK, Smith MA. Alpha3Na+/K+- ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Hoch W, Ferns M, Campanelli JT, Hall ZW, Scheller RH. Developmental regulation of highly active alternatively spliced forms of agrin. Neuron. 1993;11:479–490. doi: 10.1016/0896-6273(93)90152-h. [DOI] [PubMed] [Google Scholar]
- Hoover CL, Hilgenberg LG, Smith MA. The COOH-terminal domain of agrin signals via a synaptic receptor in central nervous system neurons. J Cell Biol. 2003;161:923–932. doi: 10.1083/jcb.200301013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh YP, Sheng M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J Neurosci. 1999;19:7415–7425. doi: 10.1523/JNEUROSCI.19-17-07415.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EP, Stevens CF. The matter of mind: molecular control of memory. Essays Biochem. 1998;33:165–178. doi: 10.1042/bse0330165. [DOI] [PubMed] [Google Scholar]
- Ji RR, Bose CM, Lesuisse C, Qiu D, Huang JC, Zhang Q, Rupp F. Specific agrin isoforms induce cAMP response element binding protein phosphorylation in hippocampal neurons. J Neurosci. 1998;18:9695–9702. doi: 10.1523/JNEUROSCI.18-23-09695.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: A dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Karasewski L, Ferreira A. MAPK signal transduction pathway mediates agrin effects on neurite elongation in cultured hippocampal neurons. J Neurobiol. 2003;55:14–24. doi: 10.1002/neu.10197. [DOI] [PubMed] [Google Scholar]
- Kaczmarek L. Molecular biology of vertebrate learning: is c-fos a new beginning? J Neurosci Res. 1993;34:377–381. doi: 10.1002/jnr.490340402. [DOI] [PubMed] [Google Scholar]
- Kim MK, Cotman SL, Halfter W, Cole GJ. The heparan sulfate proteoglycan agrin modulates neurite outgrowth mediated by FGF-2. J Neurobiol. 2003;55:261–277. doi: 10.1002/neu.10213. [DOI] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, D ML, Burden SJ. Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek I, Burkhardt C, Lin S, Seddik R, Maj M, Bezakova G, Jucker M, Arber S, Caroni P, Sanes JR, Bettler B, Ruegg MA. Synapse loss in cortex of agrin-deficient mice after genetic rescue of perinatal death. J Neurosci. 2007;27:7183–7195. doi: 10.1523/JNEUROSCI.1609-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Current Opinion in Neurobiology. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Li Z, Massengill JL, O'Dowd DK, Smith MA. Agrin gene expression in mouse somatosensory cortical neurons during development in vivo and in cell culture. Neuroscience. 1997;79:191–201. doi: 10.1016/s0306-4522(96)00654-9. [DOI] [PubMed] [Google Scholar]
- Li Z, Hilgenberg LG, O'Dowd DK, Smith MA. Formation of functional synaptic connections between cultured cortical neurons from agrin-deficient mice. J Neurobiol. 1999;39:547–557. doi: 10.1002/(sici)1097-4695(19990615)39:4<547::aid-neu8>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- Lin L, McCroskery S, Ross JM, Chak Y, Neuhuber B, Daniels MP. Induction of filopodia-like protrusions by transmembrane agrin: role of agrin glycosaminoglycan chains and Rho-family GTPases. Exp Cell Res. 2010;316:2260–2277. doi: 10.1016/j.yexcr.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, Morgan R, Godfrey EW. Distribution of agrin mRNAs in the chick embryo nervous system. J Neurosci. 1994;14:2943–2952. doi: 10.1523/JNEUROSCI.14-05-02943.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S, Kroger S. Agrin is synthesized by retinal cells and colocalizes with gephyrin [corrected] Mol Cell Neurosci. 1996;8:1–13. doi: 10.1006/mcne.1996.0039. [DOI] [PubMed] [Google Scholar]
- Matsumoto-Miyai K, Sokolowska E, Zurlinden A, Gee CE, Luscher D, Hettwer S, Wolfel J, Ladner AP, Ster J, Gerber U, Rulicke T, Kunz B, Sonderegger P. Coincident pre- and postsynaptic activation induces dendritic filopodia via neurotrypsin-dependent agrin cleavage. Cell. 2009;136:1161–1171. doi: 10.1016/j.cell.2009.02.034. [DOI] [PubMed] [Google Scholar]
- McCroskery S, Chaudhry A, Lin L, Daniels MP. Transmembrane agrin regulates filopodia in rat hippocampal neurons in culture. Mol Cell Neurosci. 2006;33:15–28. doi: 10.1016/j.mcn.2006.06.004. [DOI] [PubMed] [Google Scholar]
- McCroskery S, Bailey A, Lin L, Daniels MP. Transmembrane agrin regulates dendritic filopodia and synapse formation in mature hippocampal neuron cultures. Neuroscience. 2009;163:168–179. doi: 10.1016/j.neuroscience.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–3121. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- McMahan UJ. The agrin hypothesis. Cold Spring Harb Symp Quant Biol. 1990;55:407–418. doi: 10.1101/sqb.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- Neumann FR, Bittcher G, Annies M, Schumacher B, Kroger S, Ruegg MA. An alternative amino-terminus expressed in the central nervous system converts agrin to a type II transmembrane protein. Mol Cell Neurosci. 2001;17:208–225. doi: 10.1006/mcne.2000.0932. [DOI] [PubMed] [Google Scholar]
- O'Connor LT, Lauterborn JC, Gall CM, Smith MA. Localization and alternative splicing of agrin mRNA in adult rat brain: transcripts encoding isoforms that aggregate acetylcholine receptors are not restricted to cholinergic regions. J Neurosci. 1994;14:1141–1152. doi: 10.1523/JNEUROSCI.14-03-01141.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porten E, Seliger B, Schneider VA, Woll S, Stangel D, Ramseger R, Kroger S. The process-inducing activity of transmembrane agrin requires follistatin-like domains. J Biol Chem. 2010;285:3114–3125. doi: 10.1074/jbc.M109.039420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramseger R, White R, Kroger S. Transmembrane form agrin-induced process formation requires lipid rafts and the activation of Fyn and MAPK. J Biol Chem. 2009;284:7697–7705. doi: 10.1074/jbc.M806719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif R, Sales S, Hettwer S, Dreier B, Gisler C, Wölfel J, Lüscher D, Zurlinden A, Stephan A, Ahmed S, Baici A, Ledermann B, Kunz B, Sonderegger P. Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J. 2007;21:3468–3478. doi: 10.1096/fj.07-8800com. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho family proteins and regulation of the actin cytoskeleton. Progress in Molecular and Subcellular Biology. 1999;22:1–22. doi: 10.1007/978-3-642-58591-3_1. [DOI] [PubMed] [Google Scholar]
- Rimer M. Emerging roles for MAP kinases in agrin signaling. Communicative &. Integrative Biol. 2011;4:143–146. doi: 10.4161/cib.4.2.14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp F, Payan DG, Magill-Solc C, Cowan DM, Scheller RH. Structure and expression of a rat agrin. Neuron. 1991;6:811–823. doi: 10.1016/0896-6273(91)90177-2. [DOI] [PubMed] [Google Scholar]
- Serpinskaya AS, Feng G, Sanes JR, Craig AM. Synapse formation by hippocampal neurons from agrin-deficient mice. Dev Biol. 1999;205:65–78. doi: 10.1006/dbio.1998.9112. [DOI] [PubMed] [Google Scholar]
- Stone DM, Nikolics K. Tissue- and age-specific expression patterns of alternatively spliced agrin mRNA transcripts in embryonic rat suggest novel developmental roles. J Neurosci. 1995;15:6767–6778. doi: 10.1523/JNEUROSCI.15-10-06767.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sytkowski AJ, Vogel Z, Nirenberg MW. Development of acetylcholine receptor clusters on cultured muscle cells. Proc Natl Acad Sci USA. 1973;70:270–274. doi: 10.1073/pnas.70.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian QB, Suzuki T, Yamauchi T, Sakagami H, Yoshimura Y, Miyazawa S, Nakayama K, Saitoh F, Zhang JP, Lu Y, Kondo H, Endo S. Interaction of LDL receptor-related protein 4 (LRP4) with postsynaptic scaffold proteins via its C-terminal PDZ domain-binding motif, and its regulation by Ca2+⁄calmodulin-dependent protein kinase. II European J Neurosci. 2006;23:2864–2876. doi: 10.1111/j.1460-9568.2006.04846.x. [DOI] [PubMed] [Google Scholar]
- Tsim KW, Ruegg MA, Escher G, Kroger S, McMahan UJ. cDNA that encodes active agrin. Neuron. 1992;8:677–689. doi: 10.1016/0896-6273(92)90089-v. [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Weller M, Waltereit R. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol. 2003;27:99–106. doi: 10.1385/MN:27:1:99. [DOI] [PubMed] [Google Scholar]
- Winzen U, Cole GJ, Halfter W. Agrin is a chimeric proteoglycan with the attachment sites for heparan sulfate/chondroitin sulfate located in two multiple serine-glycine clusters. J Biol Chem. 2003;278:30106–30114. doi: 10.1074/jbc.M212676200. [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]