

Abstract
Objective:
To describe the brain MRI characteristics of hereditary diffuse leukoencephalopathy with spheroids (HDLS) with known mutations in the colony-stimulating factor 1 receptor gene (CSF1R) on chromosome 5.
Methods:
We reviewed 20 brain MRI scans of 15 patients with autopsy- or biopsy-verified HDLS and CSF1R mutations. We assessed sagittal T1-, axial T1-, T2-, proton density-weighted and axial fluid-attenuated inversion recovery images for distribution of white matter lesions (WMLs), gray matter involvement, and atrophy. We calculated a severity score based on a point system (0−57) for each MRI scan.
Results:
Of the patients, 93% (14 of 15) demonstrated localized WMLs with deep and subcortical involvement, whereas one patient revealed generalized WMLs. All WMLs were bilateral but asymmetric and predominantly frontal. Fourteen patients had a rapidly progressive clinical course with an initial MRI mean total severity score of 16.7 points (range 10−33.5). Gray matter pathology and brainstem atrophy were absent, and the corticospinal tracts were involved late in the disease course. There was no enhancement, and there was minimal cerebellar pathology.
Conclusion:
Recognition of the typical MRI patterns of HDLS and the use of an MRI severity score might help during the diagnostic evaluation to characterize the natural history and to monitor potential future treatments. Indicators of rapid disease progression were symptomatic disease onset before 45 years, female sex, WMLs extending beyond the frontal regions, a MRI severity score greater than 15 points, and mutation type of deletion.
Adult-onset leukoencephalopathies encompass a diverse group of disorders affecting the brain's white matter.1,2 Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is an autosomal dominant white matter (WM) disease first described in 1984.3 It is characterized by depression, memory decline, executive dysfunction, motor impairments, and seizures. The disease is caused by mutations in the protein tyrosine kinase domain of the colony-stimulating factor 1 receptor (CSF1R), encoded by the CSF1R gene on chromosome 5q32.4
HDLS can mimic many neurologic diseases.5 MRI has significant diagnostic value in leukodystrophies.1,2 Currently, MRI patterns for different stages of HDLS are unknown. This report describes the neuroimaging characteristics of patients with pathologically proven HDLS, all carriers of the CSF1R mutation. We have identified potential diagnostic indicators of HDLS and indicators of whether the disease will be progressive or static. We created a scoring system for MRI findings. The MRI severity score might help track the natural history of HDLS and evaluate the efficacy of potential treatments.
METHODS
Protocol approvals.
The study was approved by the Mayo Clinic Institutional Review Board and the institutional review board committees of the participating institutions. Informed consent was obtained from the participants, their relatives, or both.
Clinical and pathologic studies.
We reviewed the medical records of 15 patients from 9 families, all of Caucasian descent.4 Family members were cared for in the United States (12 patients), Norway (2 patients), and Germany (1 patient). All patients had neuropathologic examinations; 9 had autopsies and 6, who are currently alive, had biopsies.
MRI studies.
There were 26 brain MRI studies performed on 15 CSF1R mutation carriers, 6 of which were not scored because of poor image quality. Longitudinal MRI studies were performed on 3 patients. The MRI examinations were performed for diagnostic purposes at multiple centers using 1.5-T MRI scanners. Sagittal T1-weighted, axial T1- and T2-weighted, and axial fluid-attenuated inversion recovery (FLAIR) images were obtained for all patients except one, who was imaged without FLAIR sequences but with an axial proton density series. All patients had contrast-enhanced studies. We used axial FLAIR and axial T2-weighted sequences for scoring. Two readers with expertise in HDLS reviewed the MRI examinations in a blinded fashion; each MRI scan was evaluated twice, 3 months apart. We resolved differences in interrater scoring by adjudication between the reviewers.
We defined white matter lesions (WMLs) as areas of prominent T2 hyperintensity and assessed signal intensity (SI) visually. We considered WM signals with intensity higher than that of gray matter as pathologic. We recorded the presence, distribution, and pattern of SI abnormalities in the cerebrum, cerebellum, and brainstem and compared them with results for healthy controls.6,7 We divided the patterns of WML distribution into localized (involving 1–3 lobes) or generalized (all lobes involved). We scored the WMLs according to the degree of SI abnormalities, where 0 indicated none, 1 indicated mild, and 2 indicated marked.
We scored atrophy as focal or generalized. We analyzed all lobes for focal atrophy, defined as a definite loss of parenchymal volume and sulcal dilation.8 Cortex atrophy was scored as 0 (none), 1 (mild), or 2 (marked). The genu, body, and splenium of the corpus callosum, cerebellum, and brainstem were analyzed and scored as 0 (absence of atrophy) or 1 (presence of atrophy). We viewed enlarged ventricles as indicative of central atrophy. One point was given for a third ventricular diameter between 5 and 10 mm, and 2 points were given for a third ventricular diameter greater than 10 mm.
We calculated a severity score (0−57) for each MRI scan based on a point system, derived by assessing the location and distribution of WMLs, atrophy, and abnormal T2 SI in gray matter. We modified the MRI scoring systems for X-linked adrenoleukodystrophy (X-ALD),9 Krabbe disease,10 and metachromatic leukodystrophy (MLD).11 We recorded structural changes and other additional features. We designed the scoring system to evaluate all cerebral regions with respect to abnormal SI and atrophy. If a specific region had unilateral involvement, a score of 0.5 was given. We evaluated and scored the basal ganglia and thalamus for abnormal T2 hyperintensities as 0 (absent) and 1 (present). The total MRI severity score was further subdivided to analyze the different components of the score such as total WML score and total atrophy score (table 1).
Table 1.
Brain MRI scoring system for HDLS
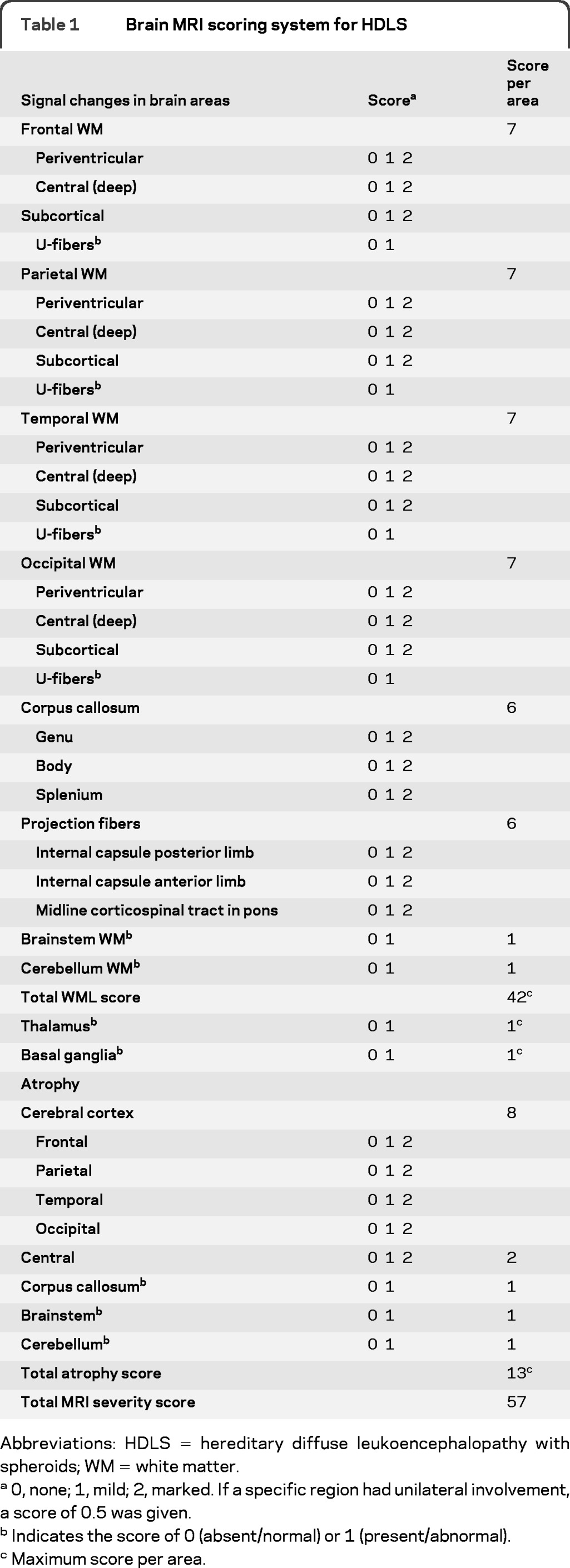
Abbreviations: HDLS = hereditary diffuse leukoencephalopathy with spheroids; WM = white matter.
0, none; 1, mild; 2, marked. If a specific region had unilateral involvement, a score of 0.5 was given.
Indicates the score of 0 (absent/normal) or 1 (present/abnormal).
Maximum score per area.
Statistical analysis.
Numerical variables were summarized with a sample mean and range. We assessed associations between total MRI severity score, total WML score, total atrophy score, age at initial MRI, and disease duration at initial MRI using the Spearman test of correlation. Spearman r was estimated along with a 95% confidence interval (CI). p ≤ 0.05 was considered statistically significant.
RESULTS
Clinical and pathologic studies.
Patient characteristics are summarized in table e-1 on the Neurology® Web site at www.neurology.org. The mean age at onset was 44.3 years; the mean disease duration, from symptom onset to death, was 5.8 years; and the mean age at death was 53.2 years. Mean disease duration in the 4 deceased patients (all women) with onset before 45 years of age was 3.8 years. In the 5 deceased patients with onset after 45 years (all male), the mean disease duration was 7.4 years. All patients were initially misdiagnosed. Neuropathologically, all had myelin loss and axonal spheroids in cerebral WM, confirming the diagnosis of HDLS.3,5
MRI characteristics.
We categorized the patients into 3 groups: mild (total MRI severity score, 1.0−6.0 points), moderate (total MRI severity score, 7.0−15.0 points), and severe disease burden (total MRI severity score, 16.0−57.0 points). One patient had mild disease burden (4 points) on the initial MRI scan, 4 patients had moderate disease burden (mean points 12.7, range 10.0−14.5), and 10 patients had severe disease burden (mean points 20.5, range 16.5−33.5). The moderate group had a mean disease duration of 6.7 years (range 5.0−8.0 years) compared with the severe group, who had a mean disease duration of 5.3 years (range 3.0−11.0 years). Only the patient with mild disease burden had a stable clinical disease course during the study period; the other 14 patients had a progressive clinical course. For patient 6 (highest total MRI severity score), the clinical course was also one of the shortest. Four patients with the shortest disease duration (4 years or less) all had a total MRI severity score greater than 16 points (mean score 22.1, range 16.0−33.5).
The distribution of MRI lesions is summarized in table 2. MRI images from representative patients are shown in figures 1 and 2 and figure e-1. All patients demonstrated bilateral, asymmetric WMLs, and all had frontal predominance of WMLs. Of the patients, 93% (14 of 15) had localized WMLs on the initial MRI examination. One patient had generalized WMLs (patient 6). All had deep and subcortical WM involvement, and 14 of 15 had periventricular involvement. The one patient without periventricular involvement had the lowest total MRI severity score of 4 points (patient 1).
Table 2.
MRI characteristics of the different patientsa

Abbreviations: A = apraxia; BBG = broad-based gait; BR = bradykinesia; CP = cognitive problems; CSP = cavum septum pellucidum; CV = cavum vergae; D = depression; DA = dysarthria; DLF = dystonia left foot; DP = dysphasia; DPG = dysphagia; ED = executive dysfunction; FAC = frontal arachnoid cyst; FEC = frontal ependymal cyst; H = hypophonia; HF = hypomimic face; MHPL = mild hemiparesis left side; MP = memory problems; P = pyramidal signs; PC = personality changes; PDLA = pronator drift left arm; PG = parkinsonian gait; PSB = pyramidal signs bilateral; R = rigidity; RP = retropulsion; T = tremor; WM = white matter.
−, indicates normal or absent; +, mild; ++, marked.
Figure 1. MRI scans of patients 4, 6, 12, and 14.

(A) Patient 4 (axial fluid-attenuated inversion recovery [FLAIR]-weighted). Localized confluent white matter lesion (WML) with interspersed multifocal bifrontal WML (arrow). WML signal intensity is marked (MRI performed 2.5 years after the onset of the symptoms). (B) Patient 4 (axial T2-weighted). Involvement of the corticospinal tracts bilaterally (arrows) at the level of mesencephalon (MRI performed 2.5 years after the onset of the symptoms). (C) Patient 6 (axial FLAIR-weighted). Bilateral confluent WML frontoparietally (arrows), most pronounced on the right where it also affects the U-fibers (lower arrow). WML extending into the corpus callosum (MRI performed 1.9 years after the onset of the symptoms). (D) Patient 6 (axial FLAIR-weighted). Generalized confluent WML, most pronounced on the right (arrow) (MRI performed 1.9 years after the onset of the symptoms). (E) Patient 12 (axial FLAIR-weighted). Localized confluent bifrontoparietal WMLs with frontal predominance involving the periventricular, deep and subcortical regions but sparing of the U-fibers (arrow) (MRI performed 3.9 years after the onset of the symptoms). (F) Patient 14 (axial FLAIR-weighted). Localized confluent bifrontal WML (thin upper arrow) with involvement of the anterior corpus callosum (thick arrow) and localized focal WML in the biparietal periventricular and deep regions (lower thin arrow) (MRI performed 3.5 years after the onset of the symptoms). For additional images of patients 1, 2, 5, 7−11, and 15, see figure e-1.
Figure 2. Longitudinal MRI scans of patients 2 and 13.

(A) Patient 2 (axial fluid-attenuated inversion recovery [FLAIR]-weighted). Minimal localized multifocal periventricular white matter lesion (WML) frontoparietally (arrows) (MRI performed 1.8 years after the onset of the symptoms). (B) Patient 2 (axial FLAIR-weighted). Increase in WML frontoparietally (arrows) with involvement of deep regions (MRI performed 2.8 years after the onset of the symptoms). (C) Patient 2 (axial FLAIR-weighted). Progression of WML frontoparietally, becoming more confluent (arrows) with involvement of the corpus callosum (thick arrowhead) (MRI performed 3.5 years after the onset of the symptoms). (D−G) Patient 2 (coronal FLAIR-weighted). Increase in periventricular, deep, and subcortical involvement of WML (arrows) with progressive involvement of the corpus callosum (arrowheads). (MRI performed 1.8 [D], 2.8 [E], 3.3 [F] ,and 3.5 [G] years after the onset of the symptoms). (H, I) Patient 13 (axial FLAIR-weighted). Progression of generalized cortical atrophy with slight progression of bifrontal WML (arrows) (MRI performed 4.0 [H] and 5.0 [I] years after the onset of the symptoms).
Thirteen patients had bilateral frontoparietal involvement, whereas 2 had only frontal involvement: mild in one (patient 1) and marked in the other (patient 4). All patients had preservation of the subcortical U-fibers, except patient 6. Temporal lobe WMLs were seen in 3 patients (20%); one of these also had occipital WM involvement (patient 6). The average total MRI severity score for these patients was 25.2 points (range 19−33.5).
There was no evidence of an association between total MRI severity score and age at the initial MRI (r = 0.04, 95% CI −0.49 to 0.54, p = 0.89) or disease duration at the initial MRI (r = −0.33, 95% CI −0.72 to 0.22, p = 0.23), as shown in figure e-2, A and B, respectively. However, there was evidence that the total WML score decreases as disease duration at the time of MRI increases (r = −0.54, 95% CI −0.82 to −0.04, p = 0.040), as displayed in figure e-2C.
Eleven patients had increased T2 SI in the corpus callosum. By using the categorization of mild, moderate, and severe disease burden with regard to the corpus callosum, there was a trend of increasing involvement in the severe disease burden group; however, 2 patients in the moderate group and 1 patient in the severe group did not have involvement of the corpus callosum.
Three patients (20%) had involvement of the projection fibers and a total MRI severity score greater than 18 points. The posterior limb of the internal capsule was affected in 1 patient and the pontine corticospinal tract in 2 patients. None of the 15 patients had an affected anterior limb of the internal capsule. The brainstem was affected in 3 patients (20%). The WM signal was due to corticospinal tract involvement in 2 patients (patients 4 and 6), with more diffuse WM signal involvement of anterolateral pons in the third patient (patient 13). Abnormal SI was seen in the cerebellar peduncle only in patient 7, whose severity score was 16 points.
In all 14 patients with cortical atrophy, the atrophy was in the same region as the WML, except for patients 1 and 4, in whom there was atrophy of the parietal cortex without WML involvement. In addition, central atrophy was present in all patients, except for patients 1 and 14, who had a mean total MRI severity score of 8.5 points. There was no evidence of an association between the total WML score and total atrophy score (r = 0.29, 95% CI −0.26 to 0.70, p = 0.29) (figure e-2D).
Eight patients (53%) had atrophy of the corpus callosum. Three of these patients did not have WM SI changes in the corpus callosum. Although none of the patients had brainstem atrophy, only 3 patients had slight cerebellar atrophy (20%). One of these patients had concomitant minimal cerebellar WML. None of the patients had gray matter lesions, abnormal contrast enhancement, or enlarged perivascular spaces.
Two of the 3 patients studied longitudinally (patients 2 and 13) had progression of the WML and atrophy (figure 2). The average total MRI severity score for these patients was 20 points, the average follow-up score was 25 points, and the average follow-up interval was 18 months. The interval between the initial MRI and follow-up ranged from 13 to 24 months. Patient 1 had a stable disease burden with a 24-month interval between the initial MRI and the last follow-up.
Radiologic-genotype comparison.
In each of the 9 families studied, a different heterozygous mutation in CSF1R was identified (table e-1). All mutations affect the protein tyrosine kinase domain of CSF1R either by changing a single amino acid (6 families) or by small in-frame deletions resulting from splice-site mutations, leading to single exon deletions (3 families) (figure e-3). Overall, the patients from the same family, carriers of the same mutation, have a similar MRI pattern, particularly in the same disease stage (table 2).
When dividing the cases into 2 groups according to the CSF1R mutation type, exon deletions, or single amino acids changes, we observed a lower mean disease onset age at 36.8 years (compared with 47.1 years), a lower mean disease duration of 4.0 years (compared with 6.7 years), and a lower mean age of death of 45.3 years (compared with 57.2 years) in the deletion group. This finding suggests that CSF1R mutation affecting multiple amino acids may be associated with a more aggressive disease course. Interestingly, the total WML score was also higher in the deletion group (16.1 compared with 11.5). In contrast, the total atrophy score was slightly higher in the patients with a single amino acid change (5.1 vs 3.4). None of the other brain MRI features were differently affected between the groups.
DISCUSSION
Our report describes the MRI pattern of HDLS in patients with CSF1R gene mutations. MRI findings in patients with HDLS have been described previously in 20 reports,5,12–30 however, without knowledge of the CSF1R gene mutation carrier status.4 We developed an HDLS MRI severity scoring system, modified from those devised for X-ALD, Krabbe disease, and MLD.9–11 Our scoring system provides quantitative markers to track HDLS progression and characterize the natural history of HDLS MRI lesions.
WMLs were localized bifrontally and asymmetrically in 93% of the patients. Patient 6 (highest total MRI severity score) showed generalized confluent WMLs, possibly reflecting disease severity. Current evidence suggests that HDLS initially manifests focally in the WM and becomes confluent.5,16,26 Our study demonstrates that in some patients, lesions develop deep in the WM, spreading to periventricular and subcortical regions, and finally becoming confluent and generalized. Focal lesions may occur in the early stages of HDLS. In the advanced stages, WM abnormalities are all confluent. This is also true for other leukoencephalopathies including leukodystrophies and the advanced stages of multiple sclerosis (MS), which may mimic HDLS radiologically. Some vasculopathies might also reveal confluent WMLs in advanced stages but usually have additional signal changes and microhemorrhages in the basal ganglia, thalamus, and brainstem.31 The punctate WMLs in patient 1 could be mistaken for ischemia and the multifocal lesions in patient 8 for small-vessel disease. Both patients may have been imaged at an early stage of the disease burden, because each lived 2 years or more after the initial MRI examination, and their mean total MRI severity score was 8.5 points.
Fourteen patients had corpus callosum involvement (table 2). This finding may suggest MS or Susac syndrome. However, in MS, the ependymal corpus callosal undersurface is involved and characteristic right angle lesions are seen in the sagittal sections, reflecting the perivenular inflammation.32 In Susac syndrome, the corpus callosal lesions are centrally located without atrophy.33 They are of variable shapes and sizes, and they will eventually evolve into pathognomic central callosal holes. There is a characteristic “string of pearls” in the internal capsule and leptomeningeal enhancement in Susac syndrome, which is not expected in MS or HDLS.33 Some vasculopathies can involve the corpus callosum but usually include neighboring structures.31
Brain atrophy on MRI has shown to correlate with clinical parameters in MS. Cortical atrophy present in patients with MS is partially independent of WML load, and the pattern of atrophy differs from normal aging.34 The cortical atrophy in MS may have regional variation, more prominent within the regions of most corticocortical connections. Atrophy is limited in primary motor, sensory, and visual cortices, even though these areas often have prominent WMLs.34 This does not completely fit with findings in HDLS; although cortical atrophy was present in 93% of our patients, the WMLs in the specific distributions were more prominent than the atrophy. Twelve of the patients had localized atrophy in the same region as the WMLs, and of those, patient 6 trended toward more generalized atrophy. Interestingly, when the whole group was compared for cortical atrophy, the parietal lobe had the greatest atrophy (mean atrophy score 22 points) compared with the frontal lobe (17 points). We hypothesize that the cortical atrophy on MRI may reflect irreversible tissue loss demonstrated neuropathologically in all deceased patients, reflecting the extent of axonal damage and loss. The loss of axonal density has been proposed as the cause of transition into progressive MS and of the continuous and irreversible neurologic decline seen in MS.34,35 It is not known whether this phenomenon is true for HDLS.
Atrophy of the anterior and posterior aspects of the corpus callosum has recently shown significant correlation to the increasing load of age-related WM changes in elderly neurologically normal subjects with leukoaraiosis.36 In our study, 8 patients had atrophy of the corpus callosum, which is also reported in MS and dementia, a finding only partially explained by Wallerian degeneration.37,38
There was U-fiber preservation in 93% of the patients with HDLS. The diagnostic utility of this finding is limited, because it is found in other WM disorders.1,2 Importantly, none of our patients had gray matter pathology on MRI. Many of the disorders that can demonstrate WMLs also have abnormalities in the gray matter, including MS, Alzheimer disease, certain leukodystrophies, Parkinson disease, atypical Parkinson disease and vasculopathies,1,31,39 and other non-neurodegenerative disorders, distinguishing them from HDLS.
Age at onset and disease course varied among patients with different mutations. This may be related to specific mutations; however, even onset ages varied significantly within the same family, potentially reflecting the influence of currently unknown genetic modifiers or environmental factors. Individuals from the same families can show unique MRI findings, including corpus callosal WMLs, atrophy, or both (table 2). These features may be useful markers to follow the disease course in different family members and to evaluate response to future clinical research trials.
In our study, indicators of rapid progressive disease were disease onset before 45 years, female sex, WMLs extending beyond the frontal regions, and a total MRI severity score greater than 15 points. There was significant evidence that the total WML score decreases as disease duration at the time of MRI increases. The individual with the highest total MRI severity score also had one of the shortest durations, the highest total WML score, and a mutation of deletion type. Overall, a total MRI severity score less than 15 was suggestive of longer disease duration.
Our study demonstrates that the MRI abnormalities in patients with HDLS have a characteristic pattern, most recognizable in the middle stages. Minor WMLs in early stages lack diagnostic specificity. End-stage HDLS is characterized by confluent involvement of the cerebral WM, which may also be nonspecific. The MRI abnormalities are generally similar among patients with the same mutation and slightly different in patients with different mutations. There are limitations in interpreting these results, because this study did not use the same imaging technique protocols for all patients, and the MRI studies were performed at different time points in disease progression. The number of patients included in this study is small and MRI examinations were limited. Prospective longitudinal studies with a more uniform imaging protocol are warranted.
Typical MRI findings for patients with HDLS with CSF1R gene mutations include T2 hyperintense foci in the periventricular, deep, and subcortical bifrontal or bifrontoparietal cerebral WM, as well as involvement of the corpus callosum and corticospinal tracts; central atrophy; and no significant gray matter pathology, no brainstem atrophy, and no enhancement. Cerebellar pathology is minimal. Knowledge of the MRI appearance may assist in correctly diagnosing HDLS with CSF1R gene mutations.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the patients and their families for their participation. This research would not have been possible without their consistent support. The authors also thank Victoria L. Jackson, MLIS (Academic and Research Support, Mayo Clinic, Jacksonville, FL) for providing editorial assistance in proofreading, manuscript formatting, and submitting our manuscript to the editorial office.
GLOSSARY
- CI
confidence interval
- CSF1R
colony-stimulating factor 1 receptor
- FLAIR
fluid-attenuated inversion recovery
- HDLS
hereditary diffuse leukoencephalopathy with spheroids
- MLD
metachromatic leukodystrophy
- MS
multiple sclerosis
- SI
signal intensity
- WM
white matter
- WNL
white matter lesion
- X-ALD
X-linked adrenoleukodystrophy
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
C. Sundal: radiologic analysis; drafting and revising the manuscript; design and conceptualization of the study; analysis and interpretation of the data; final approval of manuscript. J.A. Van Gerpen: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. A.M. Nicholson: data interpretation; manuscript revision for intellectual content; final approval of manuscript. C. Wider: data interpretation; manuscript revision for intellectual content; final approval of manuscript. E.A. Shuster: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. J. Aasly: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. S. Spina: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. B. Ghetti: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. S. Roeber: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. J. Garbern: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. A. Borjesson-Hanson: data interpretation; manuscript revision for intellectual content; final approval of manuscript. A. Tselis: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. R.H. Swerdlow: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. B.B. Miller: data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. S. Fujioka: data interpretation; manuscript revision for intellectual content; final approval of manuscript. M.G. Heckman: statistical analysis; data interpretation; manuscript revision for intellectual content; final approval of manuscript. R.J. Uitti: Data interpretation; manuscript revision for intellectual content; final approval of manuscript; contributed patients. K.A. Josephs: data interpretation; manuscript revision for intellectual content; final approval of manuscript. M. Baker: Data interpretation; manuscript revision for intellectual content; final approval of manuscript. O. Andersen: data interpretation; manuscript revision for intellectual content; final approval of manuscript. R. Rademakers: genetic data analysis; data interpretation; manuscript revision for intellectual content; final approval of manuscript. D.W. Dickson: neuropathologic analysis; data interpretation; manuscript revision for intellectual content; final approval of manuscript. D. Broderick: radiologic analysis; drafting of the manuscript; data interpretation; manuscript revision for intellectual content; final approval of manuscript. Z.K. Wszolek: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript; final approval of manuscript; head of HDLS consortium.
Study Funding
C.S. was partially supported by Sven and Dagmar Saléns Stiftelse, Signe och Olof Wallenius Stiftelse, Stiftelsen for Gamla Tjänarinnor, and Anna-Lisa och Bror Björnsson Stiftelse (2008−2010), Sweden, The Swedish Society of Medicine Gothenburg (GLS), the Swedish Society of Medicine Sweden (2010), The Swedish and Gothenburg Societies for the Neurologically Disabled, the Gothenburg Foundation for Neurological Research, Stiftelson Edit Jacobsons Donationsfond Sweden, and the American Scandinavian Foundation: Haakon Styri Fund. A.M.N. was partially supported by an Association for Frontotemporal Degeneration Postdoctoral Fellowship. R.H.S. was partially supported by the University of Kansas Alzheimer's Disease Core Center Funding Agency (P30-AG035982). B.G. was partially supported by the National Institutes of Health, Public Health Service (P30-AG10133). J.A.V.G., R.R., D.W.D., and Z.K.W. were partially supported by Mayo Clinic Florida (MCF) Research Committee CR program (MCF #90052030). R.R. was partially supported by the National Institutes of Health (R01-NS065782, R01-AG26251, and P50-AG16574). D.W.D. and Z.K.W. were partially supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke (P50-NS072187–01S2). D.W.D. was partially supported by an anonymous Mayo benefactor. Z.K.W. was also partially supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke (1RC2-NS070276 and R01-NS057567) and the Dystonia Medical Research Foundation.
DISCLOSURE
C. Sundal reports no disclosures. J. Van Gerpen reports receiving funding from the Mayo Clinic Florida Research Committee CR program for research on hereditary leukoencephalopathy with axonal spheroids (MCF #90052030). A. Nicholson, C. Wider, E. Shuster, J. Aasly, S. Spina, B. Ghetti, and S. Roeber report no disclosures. J. Garbern is now deceased. We cannot confirm his disclosures, but as far as we know he had no disclosures relevant to this manuscript. A. Borjesson-Hanson, A. Tselis, R. Swerdlow, B. Miller, S. Fujioka, and M. Heckman report no disclosures. R. Uitti serves as an associate editor for Neurology®. K. Josephs, M. Baker, and O. Andersen report no disclosures. R. Rademakers reports receiving funding from the Mayo Clinic Florida Research Committee CR program for research on hereditary leukoencephalopathy with axonal spheroids (MCF #90052030). D. Dickson reports receiving funding from the Mayo Clinic Florida Research Committee CR program for research on hereditary leukoencephalopathy with axonal spheroids (MCF #90052030). D. Broderick reports no disclosures. Z. Wszolek reports receiving funding from the Mayo Clinic Florida Research Committee CR program for research on hereditary leukoencephalopathy with axonal spheroids (MCF #90052030). Go to Neurology.org for full disclosures.
REFERENCES
- 1. van der Knaap M, Valk J, Barkhof F, eds. Magnetic Resonance of Myelination and Myelin Disorders, 3rd ed New York: Birkhäuser; 2005 [Google Scholar]
- 2. Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Axelsson R, Roytta M, Sourander P, Akesson HO, Andersen O. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl 1984;314:1–65 [PubMed] [Google Scholar]
- 4. Rademakers R, Baker M, Nicholson AN, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) cause hereditary diffuse leukoencephalopathy with spheroids Nature Genetics 2011;44:200–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sundal C, Lash J, Aasly J, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci 2012;314:130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salonen O, Autti T, Raininko R, Ylikoski A, Erkinjuntti T. MRI of the brain in neurologically healthy middle-aged and elderly individuals. Neuroradiology 1997;39:537–545 [DOI] [PubMed] [Google Scholar]
- 7. Raininko R, Autti T, Vanhanen SL, Ylikoski A, Erkinjuntti T, Santavuori P. The normal brain stem from infancy to old age: a morphometric MRI study. Neuroradiology 1994;36:364–368 [DOI] [PubMed] [Google Scholar]
- 8. Pasquier F, Leys D, Weerts JG, Mounier-Vehier F, Barkhof F, Scheltens P. Inter- and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol 1996;36:268–272 [DOI] [PubMed] [Google Scholar]
- 9. Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol 1994;15:1761–1766 [PMC free article] [PubMed] [Google Scholar]
- 10. Loes DJ, Peters C, Krivit W. Globoid cell leukodystrophy: distinguishing early-onset from late-onset disease using a brain MR imaging scoring method. AJNR Am J Neuroradiol 1999;20:316–323 [PMC free article] [PubMed] [Google Scholar]
- 11. Eichler F, Grodd W, Grant E, et al. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. AJNR Am J Neuroradiol 2009;30:1893–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol 2006;111:300–311 [DOI] [PubMed] [Google Scholar]
- 13. Yamashita M, Yamamoto T. Neuroaxonal leukoencephalopathy with axonal spheroids. Eur Neurol 2002;48:20–25 [DOI] [PubMed] [Google Scholar]
- 14. Itoh K, Shiga K, Shimizu K, Muranishi M, Nakagawa M, Fushiki S. Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: clinical and neuropathological characteristics. Acta Neuropathol 2006;111:39–45 [DOI] [PubMed] [Google Scholar]
- 15. Mendes A, Pinto M, Vieira S, Castro L, Carpenter S. Adult-onset leukodystrophy with axonal spheroids. J Neurol Sci 2010;297:40–45 [DOI] [PubMed] [Google Scholar]
- 16. Van Gerpen JA, Wider C, Broderick DF, Dickson DW, Brown LA, Wszolek ZK. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology 2008;71:925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hancock N, Poon M, Taylor B, McLean C. Hereditary diffuse leucoencephalopathy with spheroids. J Neurol Neurosurg Psychiatry 2003;74:1345–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Browne L, Sweeney BJ, Farrell MA. Late-onset neuroaxonal leucoencephalopathy with spheroids and vascular amyloid. Eur Neurol 2003;50:85–90 [DOI] [PubMed] [Google Scholar]
- 19. Moro-de-Casillas ML, Cohen ML, Riley DE. Leucoencephalopathy with neuroaxonal spheroids (LENAS) presenting as the cerebellar subtype of multiple system atrophy. J Neurol Neurosurg Psychiatry 2004;75:1070–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Terada S, Ishizu H, Yokota O, et al. An autopsy case of hereditary diffuse leukoencephalopathy with spheroids, clinically suspected of Alzheimer's disease. Acta Neuropathol 2004;108:538–545 [DOI] [PubMed] [Google Scholar]
- 21. Mayer B, Oelschlaeger C, Keyvani K, Niederstadt T. Two cases of LENAS: diagnosis by MRI and biopsy. J Neurol 2007;254:1453–1454 [DOI] [PubMed] [Google Scholar]
- 22. Keegan BM, Giannini C, Parisi JE, Lucchinetti CF, Boeve BF, Josephs KA. Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking cerebral MS. Neurology 2008;70:1128–1133 [DOI] [PubMed] [Google Scholar]
- 23. Mateen FJ, Keegan BM, Krecke K, Parisi JE, Trenerry MR, Pittock SJ. Sporadic leucodystrophy with neuroaxonal spheroids: persistence of DWI changes and neurocognitive profiles: a case study. J Neurol Neurosurg Psychiatry 2010;81:619–622 [DOI] [PubMed] [Google Scholar]
- 24. Boisse L, Islam O, Woulfe J, Ludwin SK, Brunet DG. Neurological picture: hereditary diffuse leukoencephalopathy with neuroaxonal spheroids: novel imaging findings. J Neurol Neurosurg Psychiatry 2010;81:313–314 [DOI] [PubMed] [Google Scholar]
- 25. van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, Kamphorst W, Weinstein HC. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology 2000;54:463–468 [DOI] [PubMed] [Google Scholar]
- 26. Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 2009;19:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology 2009;72:1953–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maillart E, Rousseau A, Galanaud D, et al. Rapid onset frontal leukodystrophy with decreased diffusion coefficient and neuroaxonal spheroids. J Neurol 2009;256:1649–1654 [DOI] [PubMed] [Google Scholar]
- 29. Sundal C, Ekholm S, Nordborg C, et al. Update of the original HDLS kindred: divergent clinical courses. Acta Neurol Scand 2012;126:67−75 [DOI] [PubMed] [Google Scholar]
- 30. Sundal C, Ekholm S, Andersen O. White matter disorders with autosomal dominant heredity: a review with personal clinical case studies and their MRI findings. Acta Neurol Scand 2010;121:328–337 [DOI] [PubMed] [Google Scholar]
- 31. Patel B, Markus HS. Magnetic resonance imaging in cerebral small vessel disease and its use as a surrogate disease marker. Int J Stroke 2011;6:47–59 [DOI] [PubMed] [Google Scholar]
- 32. Palmer S, Bradley WG, Chen DY, Patel S. Subcallosal striations: early findings of multiple sclerosis on sagittal, thin-section, fast FLAIR MR images. Radiology 1999;210:149–153 [DOI] [PubMed] [Google Scholar]
- 33. Saenz R, Quan AW, Magalhaes A, Kish K. MRI of Susac's syndrome. AJR Am J Roentgenol 2005;184:1688–1690 [DOI] [PubMed] [Google Scholar]
- 34. Charil A, Dagher A, Lerch JP, Zijdenbos AP, Worsley KJ, Evans AC. Focal cortical atrophy in multiple sclerosis: relation to lesion load and disability. Neuroimage 2007;34:509–517 [DOI] [PubMed] [Google Scholar]
- 35. Schirmer L, Antel JP, Bruck W, Stadelmann C. Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol 2011;21:428–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ryberg C, Rostrup E, Sjostrand K, et al. White matter changes contribute to corpus callosum atrophy in the elderly: the LADIS study. AJNR Am J Neuroradiol 2008;29:1498–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain 2000;123:1845–1849 [DOI] [PubMed] [Google Scholar]
- 38. Di Paola M, Luders E, Di Iulio F, et al. Callosal atrophy in mild cognitive impairment and Alzheimer's disease: different effects in different stages. Neuroimage 2010;49:141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol 2008;7:841–851 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.