

Abstract
BACKGROUND AND PURPOSE
Glycine receptor α1 subunit R271Q and R271L (α1R271Q/L) mutations cause the neuromotor disorder, hereditary hyperekplexia. Studies suggest that the 271 residue is located within the allosteric signalling pathway linking the agonist binding site to the channel gate. The present study aimed to investigate a possible mechanism for restoring the function of the α1R271Q/L glycine receptor.
EXPERIMENTAL APPROACH
A 12-amino-acid segment incorporating the 271 residue on the glycine receptor α1271Q/L subunit was replaced by the homologous segment from the glycine receptor β subunit (α1Ch271Q/L). The function of the α1Ch271Q/L glycine receptor was examined by whole-cell patch-clamp recording and voltage-clamp fluorometry techniques.
KEY RESULTS
The function of the α1Ch271Q/L glycine receptor was restored to the level of the wild-type (WT) α1 glycine receptor. Moreover, in the α1Ch glycine receptor, in contrast to the α1 glycine receptor, the channel function was not sensitive to various substitutions of the 271 residue, and the conformational change in the vicinity of the 271 residue was uncoupled from the channel gating.
CONCLUSIONS AND IMPLICATIONS
The 271 residue is shifted out of the allosteric signalling pathway in the α1Ch glycine receptor. We propose that this mechanism provides a novel drug design strategy not only for glycine receptor α1R271Q/L-caused hereditary hyperekplexia, but also for any pathological condition that is caused by missense mutation- or covalent modification-induced disorders involving residues in allosteric signalling pathways. Such a strategy makes it possible to design an ideal drug, which only corrects the function of the mutant or modified protein without affecting the WT or naive protein.
LINKED ARTICLE
This article is commented on by Nussinov, pp. 2110–2112 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01793.x
Keywords: glycine receptor, hereditary hyperekplexia, allosteric signalling pathway, channel gating, drug design, mutation corrector
Introduction
Missense mutations and abnormal covalent modifications of certain residues in proteins are causes of a huge body of pathological conditions. Hereditary hyperekplexia (startle disease), which is a neuromotor disorder characterized by exaggerated startle reflexes and hypertonia in response to sudden unexpected auditory or tactile stimuli, is mainly caused by hereditary mutations to the inhibitory postsynaptic neurotransmitter receptor, the glycine receptor chloride channel (Harvey et al., 2008; Chung et al., 2010).
The glycine receptor exists as a pentamer. Each subunit is composed of an N-terminal extracellular domain (ECD) and four transmembrane domains (TMD) M1–4. Agonist binding to the ECDs (Brejc et al., 2001; Unwin, 2005; Hibbs and Gouaux, 2011), via an allosteric signalling pathway (channel-gating pathway), leads to the opening of the channel pore, which is lined by the M2 TMDs (Figure 1A) (Bouzat et al., 2004; Lummis et al., 2005; Unwin, 2005; Hilf and Dutzler, 2008; 2009; Bocquet et al., 2009; Lee et al., 2009; Hibbs and Gouaux, 2011).
Figure 1.
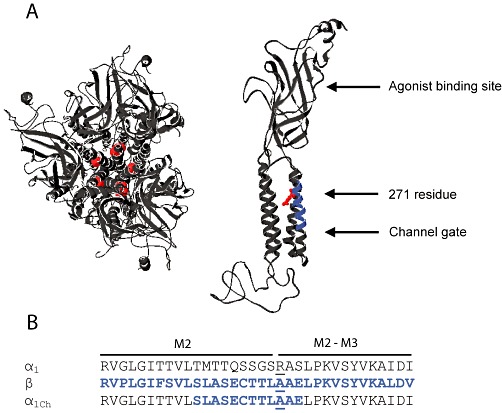
Location of the 271 residue on the glycine receptor α1 subunit. (A) In structural models of the pentameric glycine receptor (top view, left panel) and single α1 subunit (side-view, right panel) (Chung et al., 2010), the 271 residue (red) is physically located between the agonist binding site and the channel gate. The 12-amino-acid segment incorporating the 271 residue is highlighted in blue in the model of single α1 subunit. (B) Sequences of the M2 and M2–M3 domains of the glycine receptor α1, β and α1Ch subunits are shown. The 271 residues are underlined.
The most commonly occurring hyperekplexia-causing mutations are R271Q and R271L (R271Q/L) in the glycine receptor α1 subunit (Zhou et al., 2002). This residue lies at the extracellular mouth of the channel pore, physically located between the agonist-binding sites and channel gate (Unwin, 2005; Hilf and Dutzler, 2008; 2009; Hibbs and Gouaux, 2011) (Figure 1A). The R271Q/L mutations exert their pathological effects by reducing agonist glycine sensitivity (Figure 2A and B) (Lynch, 2004). Many other residue substitutions at this site, such as R271A, also reduce glycine sensitivity (Figure 2B) (Langosch et al., 1994; Rajendra et al., 1994; Lynch et al., 1997; 2001). Furthermore, taurine, which is a low-efficacy glycine receptor agonist, completely fails to activate the α1R271Q/L/A glycine receptor channel opening (Figure 3A and B) (Rajendra et al., 1995). Moreover, this residue and those in its vicinity also experience a conformational change during channel gating and more importantly this change is coupled to the channel-gating process (Pless et al., 2007). Taken together, these results suggest that the 271 residue is located within the channel-gating pathway that functionally links the agonist-binding site to the channel gate in the glycine receptor (Figure 1A).
Figure 2.
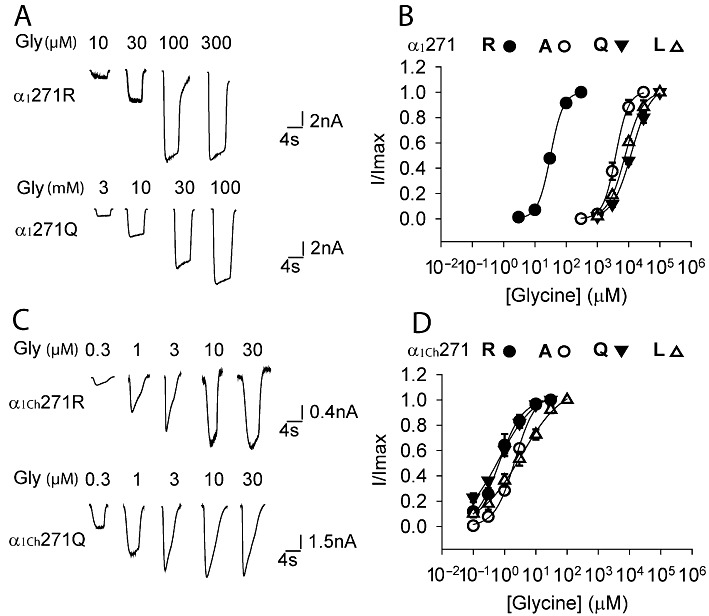
Effects of various substitutions of the 271 residue on the glycine (Gly) sensitivity of the α1 and α1Ch glycine receptors. Example traces of currents induced by increasing glycine concentrations in the indicated constructs of the α1 and α1Ch glycine receptors are shown in (A) and (C), respectively. Averaged normalized glycine dose–response curves for various substitutions of the 271 residue of the α1 and α1Ch glycine receptors are shown in (B) and (D), respectively (n= 3 or 4).
Figure 3.
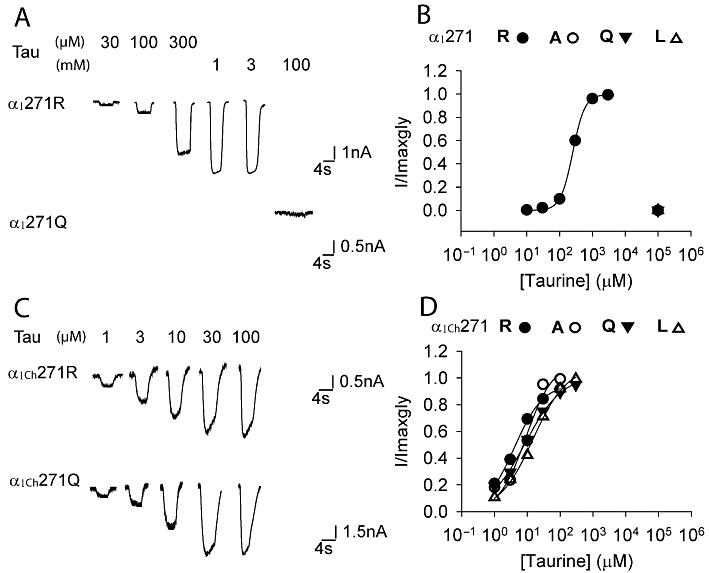
Effects of various substitutions of the 271 residue on the taurine sensitivity of the α1 and α1Ch glycine receptors. Example traces of currents induced by increasing taurine concentrations in the indicated constructs of the α1 and α1Ch glycine receptors are shown in (A) and (C), respectively. Averaged normalized taurine dose–response curves for various substitutions of the 271 residue of the α1 and α1Ch glycine receptors are shown in (B) and (D), respectively (n= 3 or 4).
Hereditary hyperekplexia, including those resulting from the R271Q/L glycine receptor mutations, are currently treated by using benzodiazepines, such as clonazepam, which act presumably by potentiating another inhibitory postsynaptic receptor, the type A GABA (GABAA) receptor (Zhou et al., 2002; Bakker et al., 2009; Thomas et al., 2010). However, the treatment is non-specific and symptomatic. Although there are barely any case reports, due to the limited literature, on the side effects of using clonazepam to treat hyperekplexia, drowsiness, ataxia and behaviour problems have often been listed as side effects when using clonazepam to treat other more common neurological disorders, such as epilepsy (Browne, 1976). Moreover, in contrast to the majority of hyperekplexia-causing mutations, which are recessive and do not require life-long treatment, the R271L/Q glycine receptor mutations are dominant, present life-long symptoms and require long-term treatment (Rees et al., 2006; Harvey et al., 2008; Chung et al., 2010). This posits a high chance of potential serious side effects if the benzodiazepine clonazepam is used. To minimize the occurrence of side effects, the ideal treatment would be one that specifically corrects the structural or functional defect imposed by the disease mutation.
Here we report that the replacement of a 12-amino-acid (12-AA) segment incorporating the 271 residue on the glycine receptor α1 subunit with the homologous segment from the glycine receptor β subunit restores the function of the α1R271Q/L glycine receptor. Further experiments suggest that such a restoration is achieved by altering the local microenvironment in the vicinity of the 271 residue and in consequence shifting this residue out of the dominant channel-gating pathway.
Like residue replacement, the binding of a small molecule could also alter local conformation (Todd and Freire, 1999; Kumar et al., 2000; del Sol et al., 2009; Kar et al., 2010), and therefore, our proposal could form the basis for a universal mutant or modified residue-specific drug design strategy: an allosteric drug (Kar et al., 2010) can be designed to alter the microenvironment in the vicinity of the affected residue and thereby eliminate the residue from the dominant allosteric signalling pathway. Such a strategy may make it possible to design an ‘ideal’ drug that simply corrects the function of the mutant or modified protein without affecting the wild-type (WT) or naive protein.
Methods
Mutagenesis and chimera construction of the glycine receptor cDNAs
Nomenclature used in this article conforms to the Guide to Receptors and Channels published in the British Journal of Pharmacology (Alexander et al., 2011).
The human glycine receptor α1 cDNAs were subcloned into the pcDNA3.1zeo+ (Invitrogen, Carlsbad, CA, USA) or pGEMHE (Liman et al., 1992) plasmid vectors for expression in HEK293 cells or Xenopus oocytes, respectively. Site-directed mutagenesis and chimera construction were performed using the QuickChange (Stratagene, La Jolla, CA, USA) mutagenesis and multiple-template-based sequential PCR protocols, respectively.
The multiple-template-based sequential PCR protocol for chimera construction was developed in our laboratory and has recently been described in detail elsewhere (Shan and Lynch, 2010). This procedure does not require the existence of restriction sites or the purification of intermediate PCR products, and needs only two or three simple PCRs followed by general subcloning steps. Most importantly, the chimera joining sites are seamless and the success rate for construction is nearly 100% (Shan and Lynch, 2010).
In the voltage-clamp fluorometry (VCF) experiments, to eliminate non-essential background cysteines, the C41A mutation was introduced into the glycine receptor α1 cDNAs in the pGEMHE vector (Shan et al., 2003), and a further C267S mutation was introduced into the 12-AA region of the glycine receptor α1Ch cDNA. This manipulation did not alter channel function.
HEK293 cell culture, expression and electrophysiological recording
The effects of various substitutions of the 271 residue on the glycine and taurine sensitivity of the α1 and α1Ch glycine receptors were determined by experiments on HEK293 cells. Details of the HEK293 cell culture, glycine receptor expression and electrophysiological recording of the HEK293 cells are described elsewhere (Shan et al., 2001b). Briefly, HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were transfected using a calcium phosphate precipitation protocol. In addition, the pEGFP-N1 (Clontech, Mountain View, CA, USA) was co-transfected to facilitate identifying the transfected cells.
Glycine and taurine-induced currents were measured using the whole cell patch-clamp configuration. Cells were treated with external Ringer's solution and internal CsCl solution (Shan et al., 2001b). Cells were voltage-clamped at −40 mV.
Xenopus oocyte preparation, expression and VCF recording
VCF experiments were performed on glycine receptors expressed in Xenopus oocytes. Details of oocyte preparation, glycine receptor expression and VCF recording are described elsewhere (Pless et al., 2007). Briefly, the mMessage mMachine kit (Ambion, Austin, TX, USA) was used to generate capped mRNA. The mRNA was injected into oocytes of the female Xenopus laevis frog with 10 ng per oocyte. After the injection, the oocytes were incubated in ND96 solution (Pless et al., 2007) for 3–4 days at 18°C before recording.
The sulphhydryl-reactive reagents, sulphorhodamine methanethiosulphonate (MTSR, Toronto Research Chemicals, North York, Ontario, Canada) and tetramethylrhodamine methyl ester (TMRM; Invitrogen), were used to label the 271C residues. On the day of recording, the oocytes were labelled with 10 µM MTSR for 25 s or 10 µM TMRM for 60 min, either in the absence or presence of glycine. The oocytes were then transferred to the recording chamber and perfused with ND96 solution. The current was recorded by the two-electrode voltage-clamp configuration and the recording electrode was filled with 3 M KCl. Cells were voltage-clamped at −40 mV. The fluorescence was recorded using the PhotoMax 200 photodiode detection system (Dagan Corp., Minneapolis, MN, USA).
Data analysis
Results are expressed as mean ± SEM of three or more independent experiments. The empirical Hill equation, fitted by a non-linear least squares algorithm (SigmaPlot 9.0; Systat Software, Point Richmond, CA, USA), was used to calculate the EC50 values for glycine- or taurine-induced current and fluorescence changes. Statistical significance was determined using Student's t-test.
Results
Replacement of the 12-AA segment incorporating the 271 residue restores the function of α1R271Q/L glycine receptor
The glycine receptor and the GABAA receptor, two major chloride-permeable postsynaptic neurotransmitter receptors, share common structural and functional characteristics and possibly even the same evolutionary origin (Lynch, 2004; Miller and Smart, 2010; Thompson et al., 2010). It has long been recognized that, with few exceptions, an Arg at sites corresponding to the 271 position of the glycine receptor α1 subunit is a signature of both the glycine receptor and GABAA receptor subunit members (including the glycine receptor α1 subunit) (Supporting Information Figure S1). One of the exceptions is the glycine receptor β subunit, where an Ala exists at this position (Figure 1B). The heteromeric glycine receptor that incorporates three Ala-carrying β subunits together with two α1 subunits exhibits a glycine sensitivity similar to that of the homomeric α1 glycine receptor (Shan et al., 2001b; Grudzinska et al., 2005). On the other hand, replacing the Arg in the α1 glycine receptor with Ala compromises channel function and mimics the phenotype of the α1R271Q/L glycine receptor (Figure 2A and B) (Lynch et al., 1997).
Supposing that this paradox might be due to a local effect, we replaced the 12-AA segment (262–273 residues) incorporating the 271 residue in the glycine receptor α1 subunit with the homologous segment from the glycine receptor β subunit (Figure 1B). The modified subunit was named the glycine receptor α1Ch subunit (Ch is short for chimera) (Figure 1B). Surprisingly, the α1Ch glycine receptor, which has an Ala at the 271 position, showed a glycine sensitivity 2600 times higher than the α1R271A glycine receptor and even an order of magnitude higher than the α1WT glycine receptor (Figure 2B and D, Table 1). Because the α1R271A glycine receptor mimics the phenotype of α1R271Q/L glycine receptors, we wondered whether this 12-AA segment replacement also restored the function of α1R271Q/L glycine receptors. We next introduced either Gln or Leu to the 271 position of the α1Ch glycine receptor. Both constructs demonstrated glycine sensitivities 20000 (Gln) and 2500 (Leu) times higher than their corresponding substitutions in the α1 glycine receptor (Figure 2B–D, Table 1). We concluded that the 12-AA segment replacement restored the function of the α1R271Q/L glycine receptors.
Table 1.
Properties of glycine- and taurine- induced currents of glycine receptors
| Glycine | Taurine | ||||
|---|---|---|---|---|---|
| Glycine receptor | EC50 (µM) | n | EC50 (µM) | Imax,tau/Imax,gly (%) | n |
| α1 | |||||
| 271R(WT) | 33 ± 2 | 4 | 261 ± 32 | 99 ± 1 | 3 |
| 271A | 5700 ± 1600 | 4 | N.D. | 0 | 3 |
| 271Q | 13200 ± 2000 | 4 | N.D. | 0 | 3 |
| 271 L | 8000 ± 490 | 3 | N.D. | 0 | 3 |
| α1Ch | |||||
| 271R | 0.87 ± 0.19 | 4 | 3.7 ± 0.3 | 92 ± 5 | 4 |
| 271A(WT) | 2.2 ± 0.2 | 4 | 8.7 ± 0.9 | 99 ± 1 | 4 |
| 271Q | 0.65 ± 0.06 | 4 | 7.3 ± 0.7 | 95 ± 2 | 4 |
| 271 L | 3.4 ± 1.1 | 4 | 16 ± 3 | 100 ± 0 | 4 |
N.D., not determined because taurine exhibited no agonist efficacy.
As noted above, the α1R271Q/L glycine receptors are completely insensitive to activation by the low-efficacy agonist, taurine (Figure 3A and B) (Rajendra et al., 1995). We thus investigated whether the 12-AA replacement also restored the taurine sensitivity of α1R271Q/L glycine receptors to WT levels. As shown in Figure 3C and D and Table 1, taurine behaved as a full-agonist in activating the α1Ch271Q/L/A glycine receptors with a sensitivity even higher than in the WT α1 glycine receptor. We therefore concluded that the 12-AA replacement also restored taurine sensitivity of the α1R271Q/L glycine receptors.
Replacement of the 12-AA segment incorporating the 271 residue diminishes the residue's contribution to channel gating
To further characterize the 271 residue in the α1Ch glycine receptor, we replaced the 271 Ala with Arg, which is the residue at the 271 position of the α1WT glycine receptor. Surprisingly, the α1Ch271R glycine receptor indicated a glycine sensitivity similar to that of the α1Ch271Q/L/A glycine receptors (Figure 2C and D, Table 1), in sharp contrast to the case of the α1 glycine receptor, where the α1271R(WT) glycine receptor showed a glycine sensitivity 180–400 times higher than the α1R271Q/L/A glycine receptors (Figure 2A and B, Table 1). Consistently, the α1Ch271R glycine receptor also demonstrated taurine sensitivity and maximal response similar to those of the α1Ch271Q/L/A glycine receptors (Figure 3C and D, Table 1). Such insensitivity of the α1Ch glycine receptor to various residue substitutions at the 271 position implies that this residue might have a diminished contribution to channel gating in the chimeric receptor.
However, it is also possibly because the energy barrier of the channel-gating pathway, which is reflected by the glycine EC50s in this case (Colquhoun, 1998), has reached its lowest limit (‘ceiling effect’) in the α1Ch271Q/L/A glycine receptors, as these constructs had very low glycine EC50s, around 1 µM (Table 1). In this scenario, the energy barrier (glycine EC50) would not reduce further when a more gating-favourable Arg is in place. If that is the case, we argue that the channel function would not be enhanced by a potentiator. To test this possibility, we applied the glycine receptor potentiator, propofol (PPF), to the α1Ch271Q glycine receptor, which exhibited the lowest glycine EC50 among the Q/L/A substitutions (Table 1). As shown in Figure 4A, PPF enhanced the sub-saturating glycine induced current by 93 ± 10 % (n= 4). Moreover, PPF left-shifted the glycine dose–response curve of the α1Ch271Q glycine receptor (EC50 < 0.3 µM, n= 4 in the presence of PPF vs. EC50= 0.65 ± 0.06 µM, n= 4 in the absence of PPF, Figure 4B and C). Note that it was not possible to quantify glycine concentrations less than 0.3 µM due to a variable contribution from the glycine that inevitably contaminates salt solutions (0.01–0.1 µM). These data imply that the energy barrier of the channel-gating pathway of the α1Ch271Q glycine receptor has not reached the lowest limit, confirming that the insensitivity of the α1Ch glycine receptor to various residue substitutions at the 271 position is due to this residue's diminished contribution to channel gating.
Figure 4.
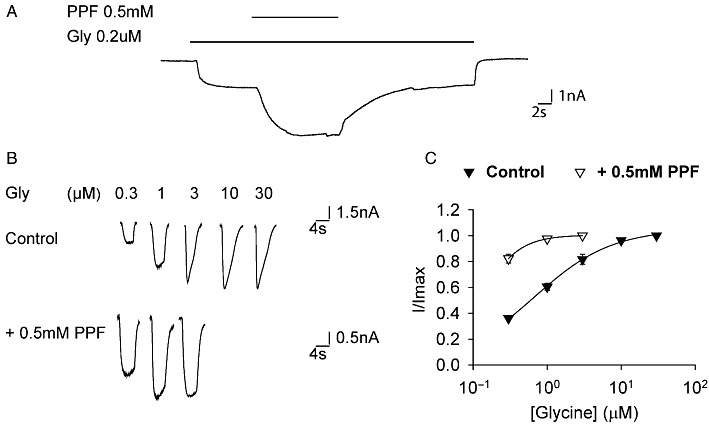
Propofol (PPF) potentiation of α1Ch271Q glycine receptor function. (A) Example of propofol potentiating sub-saturating glycine-induced α1Ch271Q glycine receptor currents. (B) Example traces of α1Ch271Q glycine receptor currents induced by increasing glycine concentrations in the absence and presence of propofol. (C) Averaged normalized glycine dose–response curves of the α1Ch271Q glycine receptor in the absence and presence of propofol (n= 4).
Replacement of the 12-AA segment incorporating the 271 residue alters its local microenvironment
We next sought to determine the underlying mechanism for the different contributions of the 271 residue to channel gating in the α1 and α1Ch glycine receptors. To achieve this, we turned to the VCF technique. VCF detects local conformational changes in the vicinity of a residue when the residue is labelled with a fluorescent dye (Gandhi and Isacoff, 2005; Pless and Lynch, 2008). Rhodamine fluorescent dyes are usually used, because rhodamine fluorescence exhibits an increase in quantum efficiency as the hydrophobicity of its environment is increased. Thus, rhodamine fluorescence intensity reports the change of hydrophobicity of its immediate microenvironment, which is often caused by local conformational changes. The VCF experiments were carried out in Xenopus oocytes, as fluorescence detection is not routinely possible in glycine receptors expressed in HEK293 cells (Pless and Lynch, 2008).
To label the 271 position with a rhodamine fluorescent dye, a cysteine was introduced to this position so that the dye can be attached through a disulphide bond (Gandhi and Isacoff, 2005; Pless and Lynch, 2008). Interestingly, the α1271C and α1Ch271C glycine receptors exhibited glycine EC50 values of 4300 ± 200 µM (n= 4) and 2.1 ± 0.4 µM (n= 5), respectively. It is thus evident that the 271C residue behaves in the same manner as the Q/L substitutions, in both the α1Ch and α1 glycine receptors. The result of the VCF investigation is therefore expected to reflect the behaviour of the 271Q/L substitutions.
As previously reported (Pless et al., 2007), we confirmed that the rhodamine fluorescent dye MTSR, when attached to the 271C residue in the α1 glycine receptor, exhibited an increase in fluorescence intensity (reflected by the upwards step of the fluorescence trace) upon glycine application (Figure 5A). This implies that MTSR detected an increase of hydrophobicity in the vicinal microenvironment due to a local conformational change during channel gating. Moreover, as the fluorescence and current glycine dose–response relationships overlapped, we concluded that the local conformational change is coupled with a channel-gating process. This conclusion is consistent with the suggestion that the 271 residue in the α1 glycine receptor lies within the dominant channel-gating pathway, as previously proposed (Langosch et al., 1994; Rajendra et al., 1994; 1995; Lynch et al., 1997; 2001).
Figure 5.
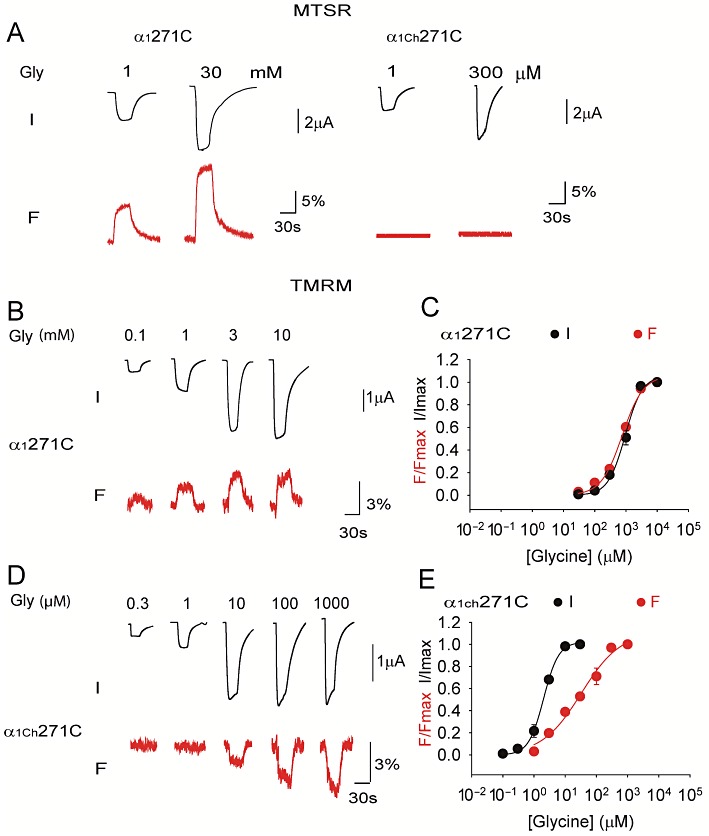
VCF of the α1 and α1Ch glycine receptors. Example current (I) and fluorescence (F) traces of the α1271C and α1Ch271C glycine receptors labelled with MTSR or TMRM are shown in (A), (B) and (D). Averaged normalized glycine dose-response curves of current (I) and fluorescence (F) of the α1271C and α1Ch271C glycine receptors labelled with TMRM are shown in (C) and (E), respectively (n= 4 or 5).
Following the same protocol, the α1Ch271C glycine receptor was labelled with MTSR and subjected to VCF investigation. Surprisingly, no fluorescence change was detected upon glycine application (Figure 5A). The 271C residue was possibly not labelled by the MTSR due to structural inaccessibility. Alternatively, this residue was labelled, but during channel gating, either no conformational change occurred in the vicinity of the 271 residue, or the microenvironment hydrophobicity detected by the MTSR fluorophore was not altered even though a local conformational change took place. Nevertheless, such different behaviours of the 271 residue between the α1 and α1Ch glycine receptors suggest that either the static microenvironment or the dynamic microenvironment change during channel gating, or both, in the vicinity of the 271 residue in the α1Ch glycine receptor are altered by the 12-AA segment replacement from those in the α1 glycine receptor.
Considering that rhodamine fluorophores are structurally different and may thus respond differently to a given conformational change when attached to the α1271C glycine receptor (Pless et al., 2007), we next investigated the response of another rhodamine fluorescent dye TMRM in the α1271C and α1Ch271C glycine receptors. In the TMRM-labelled α1271C glycine receptor, the fluorescence intensity was increased upon glycine application (reflected by the upwards step of the fluorescence trace, Figure 5B). In contrast, in the TMRM-labelled α1Ch271C glycine receptor, the fluorescence intensity was decreased upon glycine application (reflected by the downwards step of the fluorescence trace, Figure 5D). Such different direction of fluorescence intensity change provides a more direct indication that either the static microenvironment or the dynamic microenvironment change, or both, during channel gating, in the vicinity of the 271 residue in the α1Chglycine receptor, are distinct from those in the α1 glycine receptor.
More interestingly, the dose–response curve of fluorescence was right-shifted from that of the current in the α1Ch271C glycine receptor when TMRM was used (fluorescence EC50= 36 ± 8 µM, n= 4 vs. current EC50= 2.0 ± 0.2 µM, n= 5, P < 0.01, Figure 5E). This is in contrast with the α1271C glycine receptor, where the dose–response curves of fluorescence and current overlapped (fluorescence EC50= 770 ± 150 µM, n= 5 vs. current EC50= 960 ± 120 µM, n= 5, P > 0.05, Figure 5C), consistent with what was observed when MTSR was used (Pless et al., 2007). These data suggest that the conformational change in the vicinity of the 271 residue in the α1Ch271C glycine receptor, unlike in the α1271C glycine receptor, is uncoupled from the channel-gating process. We hence propose that, in the α1Ch glycine receptor, the 271 residue is not essential for channel gating and might not reside within the dominant channel-gating pathway. Such a proposal is also supported by the fact that the α1Ch glycine receptor channel function is not sensitive to various residue substitutions at the 271 position, as described earlier.
Discussion
The function of α1R271Q/L glycine receptors is restored by shifting the affected residue out of the dominant channel-gating pathway
Here we report that replacement of a 12-AA segment incorporating the 271 residue of the glycine receptor α1 subunit with the homologous segment of the glycine receptor β subunit restores channel function of the hereditary hyperekplexia-causing α1R271Q/L glycine receptors. More interestingly, through residue substitution and VCF investigation, we concluded that this rescue effect is achieved by adjusting the local microenvironment and in consequence, diminishing the 271 residue's contribution to channel gating. It has been proposed that multiple allosteric signalling pathways exist in proteins, and which pathways dominate is determined by protein topologies, specific binding events, covalent modifications and cellular conditions (del Sol et al., 2009). Residue replacement, which potentially changes the protein topology (Sinha and Nussinov, 2001), can shift the dominant signalling pathway from one pathway to another. In our experiment, the 271 residue lies within the dominant channel-gating pathway in the α1 glycine receptor. However, the 12-AA segment replacement induces a local conformational change and, in consequence, shifts the dominant channel-gating pathway to an alternative one, where the 271 residue does not reside (Figure 6A). The hypothesis that the 271 residue does not reside within the dominant channel-gating pathway is reminiscent of ivermectin-induced glycine receptor channel activation. Ivermectin is a glycine receptor agonist that binds to the glycine receptor and gates the channel opening in a manner distinct from the physiological agonist glycine (Shan et al., 2001a; Pless et al., 2007; Hibbs and Gouaux, 2011). For example, the α1 glycine receptor function activated by ivermectin is almost conserved when the R271Q mutation is introduced (Shan et al., 2001a). Moreover, the MTSR-labelled α1271C glycine receptor does not show any fluorescence change upon ivermectin application (Pless et al., 2007). Both observations imply that the 271 residue does not reside within the ivermectin-mediated channel-gating pathway.
Figure 6.
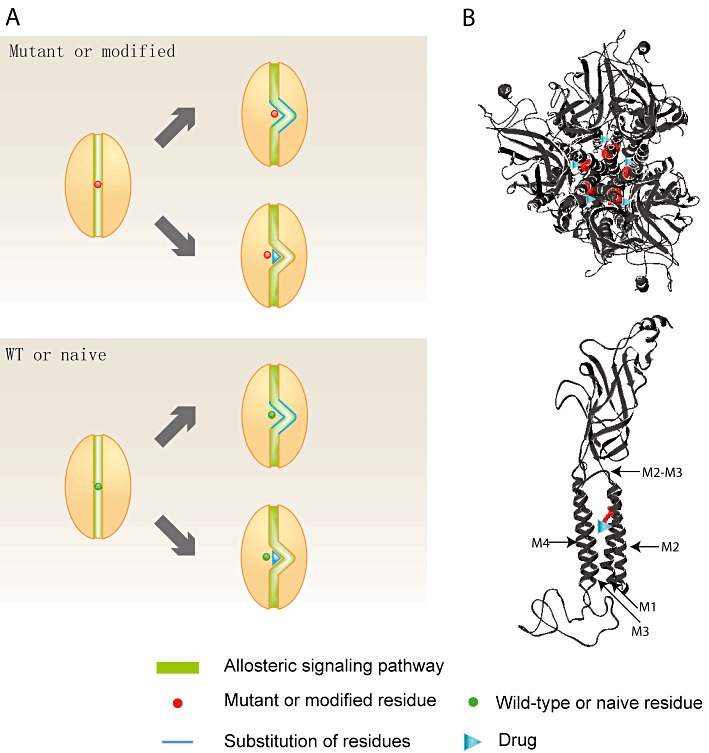
Model of the residue-specific drug design strategy. (A) In a protein with a certain residue, either mutant or modified (red circle), the protein function is compromised because the affected residue blocks the dominant allosteric signalling pathway (green strip). The protein function can be restored by activating an alternative allosteric signalling pathway that does not include the affected residue. This restoration can be achieved through adjusting the local microenvironment, either internally, by substituting the amino acids in the vicinity of the affected residue (blue line), or externally, by applying a drug (blue triangle) that has an equivalent effect as the vicinal amino acid substitution. If the newly activated alternative allosteric signalling pathway has equivalent strength as the original one in the WT or naïve protein, neither the vicinal amino acid substitution nor external drug application apparently affects the WT or naïve protein function. (B) When designing a drug (blue triangle) that specifically corrects the glycine receptor 271Q/L mutations (red residue), a possible docking site for this drug is the cavity formed by the extracellular halves of the M1, M2 and M3 segments and the M2–M3 domain, which is the binding site of many clinically related drugs and substances. The location of the 271 residue and the potential drug molecule are indicated in the structural models of the pentameric glycine receptor (top view, top panel) and single α1 subunit (side view, bottom panel) (Chung et al., 2010).
Implications for a residue-specific drug design strategy
Many pathophysiological conditions are caused by residues being either missense mutated or abnormally covalently modified (for example, by phosphorylation). The relevant treatment strategy is usually symptomatic. For example, to treat glycine receptor mutation-caused hereditary hyperekplexia, benzodiazepines, such as clonazepam, are used (Zhou et al., 2002; Thomas et al., 2010). The benzodiazepines, which are GABAA receptor potentiators, can counter the overexcitation symptoms due to the compromised glycine receptor function. However, such an ‘off-target’ treatment strategy is the source of a wide range of side effects.
A more specific treatment strategy is to directly target the affected protein. A drug is usually designed either to enhance (in loss-of-function) or to inhibit (in gain-of-function) the function of the affected protein. However, these effects are usually global rather than mutation- or modification-specific, as the drug affects the WT or naïve protein as well as the mutant or modified protein (Wang et al., 2003; Joerger and Fersht, 2007). This will lead to a lack of specificity as proteins usually have multiple subtypes (e.g. α1, α2 and α3 glycine receptors) of different genomic origins, which share a high degree of homology and, in consequence, similar structure and function relationships. Any drug acting on one subtype (e.g. the mutant protein, glycine receptor α1R271Q/L) has a very high chance of affecting other subtypes (e.g. other WT subtypes of the mutant protein such as glycine receptor α2 and α3) as well. As protein subtypes are usually distributed in various tissues and thus have different physiological or pathological roles from each other, a drug that is supposed to only act on the specific target subtype in the ideal state but affects multiple other subtypes in reality, will cause undesirable side effects. Another consideration is that abnormal residue covalent modification of a given protein under a certain pathological condition usually only occurs in a localized region of the human body. A drug that affects the naïve as well as the modified proteins may correct the modifications in the localized region, but would also interfere with processes in other regions where the target protein expresses but without any modification. This is another source of undesirable side effects.
One way to circumvent this ‘global effect’ is to design a mutant or modified residue-specific drug. This ideal drug should affect the mutant or modified protein but not the WT or naïve protein. Despite many attempts, this goal has been successfully achieved in only a few cases. One successful case is the mutant p53-targeting drug, PRIMA-1. PRIMA-1 affects the function of mutant p53 but not the WT p53 (Bykov et al., 2002a,b), through a mechanism involving modification of thiol groups within the protein (Lambert et al., 2009). However, such a mechanism apparently cannot become a universal strategy for mutant or modified protein-specific drug design.
We proposed in this article that the affected residue could be shifted out of the dominant allosteric signalling pathway by the local conformational change induced by residue substitutions. Since binding of a small molecule, like residue substitutions, can also induce conformational change and redistribute the dominant signalling pathway (Todd and Freire, 1999; Kumar et al., 2000; del Sol et al., 2009; Kar et al., 2010), our proposal could form the basis for a universal mutant or modified residue-specific drug design strategy: an allosteric drug (Kar et al., 2010) can be designed to alter the microenvironment in the vicinity of the affected residue and to activate an alternative allosteric signalling pathway that excludes the affected residue (Figure 6A). This drug action can be realized to have a neutral effect on the WT or naïve protein through activating the alternative allosteric signalling pathway with a strength equivalent to the original one (Figure 6A). However, the drug should restore the function of the mutant or modified proteins to the WT level, because the affected residue is no longer within the dominant allosteric signalling pathway and hence does not affect the protein function (Figure 6A). This missense mutation- or covalent modification-specific drug design strategy would help tackle one of the most serious problems existing among the drugs clinically used today: lack of specificity.
Possible drug design strategy for the glycine receptor R271Q/L hereditary hyperekplexia
The 12-AA segment that restores the function of the α1R271Q/L glycine receptor is located along the extracellular half of the M2 segment and the M2-M3 domain (Figures 1A and 6B). Both domains, together with the extracellular halves of the M1 and M3 segments, form a cavity, which contains the binding site of many clinically related drugs or substances including alcohol (Mihic et al., 1997), neurosteroids (Hosie et al., 2006), general anaesthetics (Nury et al., 2011) and ivermectin (Collins and Millar, 2010; Lynagh and Lynch, 2010; Hibbs and Gouaux, 2011), and therefore can be used as the potential docking site for drugs that specifically correct the glycine receptor R271Q/L mutations. Interestingly, the general anaesthetic, PPF, which binds into this cavity and potentiates the glycine receptor function, restores the WT phenotype of the hyperekplexic glycine receptor R271Q transgenic mice (O'Shea et al., 2004), although a wide range of side effects would be expected, since PPF also potentiates the GABAA receptor and inhibits the nAChR (Franks, 2008). Nevertheless, PPF could possibly serve as the seeding backbone for designing a drug specifically correcting the glycine receptor R271Q/L mutations. The final ideal glycine receptor R271Q/L mutation corrector, by exploiting the novel drug design strategy proposed in the article, could be achieved to affect the function of the α1R271Q/L glycine receptor but not any other protein including the α1WT, α2 and α3 glycine receptors and closely related GABAA receptor and nAChR.
It should be noted however that this mutation corrector is only effective in treating hereditary hyperekplexia caused by α1R271Q/L mutations, but not by any mutation arising from other sites of the glycine receptor α1 subunit, from the glycine receptor β subunit or from the SLC6A5 glycine transporter. Considering that the absolute number of patients diagnosed with hyperekplexia caused by α1R271Q/L mutations is low, it might not be commercially feasible to develop a specific α1R271Q/L mutation corrector. Instead, the target-specific drug design strategy we propose here provides a general principle for developing drugs that correct mutations or abnormal residue-modifications in proteins.
Acknowledgments
This study was supported by a research grant from the National Health and Medical Research Council of Australia. We thank J. Mullins for kindly sharing with us the model of the glycine receptor (Swansea University, UK).
Glossary
- ECD
extracellular domain
- MTSR
sulphorhodamine methanethiosulphonate
- PPF
propofol
- TMD
transmembrane domain
- TMRM
tetramethylrhodamine methyl ester
- VCF
voltage-clamp fluorometry
- WT
wild-type
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Alignment of the protein sequences covering the M2 segments and M2–M3 domains of human glycine and GABAA receptor subunits. The residues corresponding to the 271 position of the glycine receptor α1 subunit are high-lighted in bold.
Please note: Wiley–Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker MJ, Peeters EAJ, Tijssen MAJ. Clonazepam is an effective treatment for hyperekplexia due to a SLC6A5 (GlyT2) mutation. Mov Disord. 2009;24:1852–1854. doi: 10.1002/mds.22493. [DOI] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, et al. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, et al. Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature. 2004;430:896–900. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Browne TR. Clonazepam: a review of a new anticonvulsant drug. Arch Neurol. 1976;33:326–332. doi: 10.1001/archneur.1976.00500050012003. [DOI] [PubMed] [Google Scholar]
- Bykov VJN, Issaeva N, Selivanova G, Wiman KG. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis. 2002a;23:2011–2018. doi: 10.1093/carcin/23.12.2011. [DOI] [PubMed] [Google Scholar]
- Bykov VJN, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002b;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- Chung SK, Vanbellinghen JF, Mullins JG, Robinson A, Hantke J, Hammond CL, et al. Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J Neurosci. 2010;30:9612–9620. doi: 10.1523/JNEUROSCI.1763-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T, Millar NS. Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol Pharmacol. 2010;78:198–204. doi: 10.1124/mol.110.064295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Gandhi CS, Isacoff EY. Shedding light on membrane proteins. Trends Neurosci. 2005;28:472–479. doi: 10.1016/j.tins.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H, et al. The beta subunit determines the ligand binding properties of synaptic glycine receptors. Neuron. 2005;45:727–739. doi: 10.1016/j.neuron.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Harvey RJ, Topf M, Harvey K, Rees MI. The genetics of hyperekplexia: more than startle! Trends Genet. 2008;24:439–447. doi: 10.1016/j.tig.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- Hilf RJC, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HMA, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- Kar G, Keskin O, Gursoy A, Nussinov R. Allostery and population shift in drug discovery. Curr Opin Pharmacol. 2010;10:715–722. doi: 10.1016/j.coph.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Ma B, Tsai C-J, Sinha N, Nussinov R. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JMR, Gorzov P, Veprintsev DB, Soerqvist M, Segerbak D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Langosch D, Laube B, Rundstrom N, Schmieden V, Bormann J, Betz H. Decreased agonist affinity and chloride conductance of mutant glycine receptors associated with human hereditary hyperekplexia. EMBO J. 1994;13:4223–4228. doi: 10.1002/j.1460-2075.1994.tb06742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Free CR, Sine SM. Binding to gating transduction in nicotinic receptors: Cys-loop energetically couples to Pre-M1 and M2-M3 regions. J Neurosci. 2009;29:3189–3199. doi: 10.1523/JNEUROSCI.6185-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 2005;438:248–252. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- Lynagh T, Lynch JW. A glycine residue essential for high ivermectin sensitivity in Cys-loop ion channel receptors. Int J Parasitol. 2010;40:1477–1481. doi: 10.1016/j.ijpara.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Lynch JW. Molecular structure and function of the glycine receptor chloride channel. Physiol Rev. 2004;84:1051–1095. doi: 10.1152/physrev.00042.2003. [DOI] [PubMed] [Google Scholar]
- Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR. Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 1997;16:110–120. doi: 10.1093/emboj/16.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW, Han N-LR, Haddrill J, Pierce KD, Schofield PR. The surface accessibility of the glycine receptor M2-M3 loop is increased in the channel open state. J Neurosci. 2001;21:2589–2599. doi: 10.1523/JNEUROSCI.21-08-02589.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, et al. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci. 2010;31:161–174. doi: 10.1016/j.tips.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- O'Shea SM, Becker L, Weiher H, Betz H, Laube B. Propofol restores the function of “hyperekplexic” mutant glycine receptors in Xenopus oocytes and mice. J Neurosci. 2004;24:2322–2327. doi: 10.1523/JNEUROSCI.4675-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless SA, Lynch JW. Illuminating the structure and function of Cys-loop receptors. Clin Exp Pharmacol Physiol. 2008;35:1137–1142. doi: 10.1111/j.1440-1681.2008.04954.x. [DOI] [PubMed] [Google Scholar]
- Pless SA, Dibas MI, Lester HA, Lynch JW. Conformational variability of the glycine receptor M2 domain in response to activation by different agonists. J Biol Chem. 2007;282:36057–36067. doi: 10.1074/jbc.M706468200. [DOI] [PubMed] [Google Scholar]
- Rajendra S, Lynch JW, Pierce KD, French CR, Barry PH, Schofield PR. Startle disease mutations reduce the agonist sensitivity of the human inhibitory glycine receptor. J Biol Chem. 1994;269:18739–18742. [PubMed] [Google Scholar]
- Rajendra S, Lynch JW, Pierce KD, French CR, Barry PH, Schofield PR. Mutation of an arginine residue in the human glycine receptor transforms beta-alanine and taurine from agonists into competitive antagonists. Neuron. 1995;14:169–175. doi: 10.1016/0896-6273(95)90251-1. [DOI] [PubMed] [Google Scholar]
- Rees MI, Harvey K, Pearce BR, Chung S-K, Duguid IC, Thomas P, et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet. 2006;38:801–806. doi: 10.1038/ng1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Lynch JW. Chimera construction using multiple-template-based sequential PCRs. J Neurosci Methods. 2010;193:86–89. doi: 10.1016/j.jneumeth.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL, Lynch JW. Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J Biol Chem. 2001a;276:12556–12564. doi: 10.1074/jbc.M011264200. [DOI] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL, Lynch JW. A single beta subunit M2 domain residue controls the picrotoxin sensitivity of alphabeta heteromeric glycine receptor chloride channels. J Neurochem. 2001b;76:1109–1120. doi: 10.1046/j.1471-4159.2001.00124.x. [DOI] [PubMed] [Google Scholar]
- Shan Q, Nevin ST, Haddrill JL, Lynch JW. Asymmetric contribution of alpha and beta subunits to the activation of alphabeta heteromeric glycine receptors. J Neurochem. 2003;86:498–507. doi: 10.1046/j.1471-4159.2003.01872.x. [DOI] [PubMed] [Google Scholar]
- Sinha N, Nussinov R. Point mutations and sequence variability in proteins: redistributions of preexisting populations. Proc Natl Acad Sci U S A. 2001;98:3139–3144. doi: 10.1073/pnas.051399098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Sol A, Tsai CJ, Ma B, Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RH, Chung SK, Hammond CL, Robinson A, Rees MI. Spectrum of clinical disease in hyperekplexia: the genotypes and phenotypes of 48 cases. J Neurol Neurosurg Psychiatry. 2010;81:e18–e19. [Google Scholar]
- Thompson AJ, Lester HA, Lummis SC. The structural basis of function in Cys-loop receptors. Q Rev Biophys. 2010;43:449–499. doi: 10.1017/S0033583510000168. [DOI] [PubMed] [Google Scholar]
- Todd MJ, Freire E. The effect of inhibitor binding on the structural stability and cooperativity of the HIV-1 protease. Proteins. 1999;36:147–156. doi: 10.1002/(sici)1097-0134(19990801)36:2<147::aid-prot2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Wang W, Rastinejad F, El-Deiry WS. Restoring p53-dependent tumor suppression. Cancer Biol Ther. 2003;2:S55–S63. [PubMed] [Google Scholar]
- Zhou L, Chillag KL, Nigro MA. Hyperekplexia: a treatable neurogenetic disease. Brain Dev. 2002;24:669–674. doi: 10.1016/s0387-7604(02)00095-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.