

Abstract
BACKGROUND AND PURPOSE
Hydrogen sulphide (H2S) is gaining acceptance as a gaseous signal molecule. However, mechanisms regarding signal termination are not understood. We used stigmatellin and antimycin A, inhibitors of sulphide quinone reductase (SQR), to test the hypothesis that the catabolism of H2S involves SQR.
EXPERIMENTAL APPROACH
H2S production and consumption were determined in living and intact mouse brain, liver and colonic muscularis externa using gas chromatography and HPLC. Expressions of SQR, ethylmalonic encephalopathy 1 (Ethe1) and thiosulphate transferase (TST; rhodanese) were determined by RT-PCR and immunohistochemistry.
KEY RESULTS
In the colonic muscularis externa, H235S was catabolized to [35S]-thiosulphate and [35S]-sulphate, and stigmatellin reduced both the consumption of H235S and formation of [35S]-thiosulphate. Stigmatellin also enhanced H2S release by the colonic muscularis externa. In the brain, catabolism of H235S to [35S]-thiosulphate and [35S]-sulphate, which was stigmatellin-insensitive, partially accounted for H235S consumption, while the remainder was captured as unidentified 35S that was probably bound to proteins. Levels of mRNA encoding SQR were higher in the colonic muscularis externa and the liver than in the brain.
CONCLUSIONS AND IMPLICATIONS
These data support the concept that termination of endogenous H2S signalling in the colonic muscularis externa occurs via catabolism to thiosulphate and sulphate partially via a mechanism involving SQR. In the brain, it appears that H2S signal termination occurs partially through protein sequestration and partially through catabolism not involving SQR. As H2S has beneficial effects in animal models of human disease, we suggest that selective inhibition of SQR is an attractive target for pharmaceutical development.
Keywords: hydrogen sulfide (H2S), sulphide quinone reductase, thiosulphate, gas chromatography, protein sequestration, gasotransmitter, sulphide metabolism, disulphide oxidoreductase (DiSR), sulphide oxidase, enteric nervous system
Introduction
In the past 15 years, H2S has become recognized as an important endogenous gaseous signal molecule that functions as a neuromodulator in the brain (Abe and Kimura, 1996; Ishigami et al., 2009), a smooth muscle relaxant (Hosoki et al., 1997; Zhao et al., 2001; Zhao and Wang, 2002; Cheng et al., 2004; Yang et al., 2008) and pro-angiogenic (Papapetropoulos et al., 2009; Szabo and Papapetropoulos, 2011) in the vascular system, a smooth muscle relaxant (Hosoki et al., 1997; Teague et al., 2002; Gallego et al., 2008; Gil et al., 2011) and prosecretory molecule (Schicho et al., 2006; Hennig and Diener, 2009; Krueger et al., 2010; Pouokam and Diener, 2011) in the gastrointestinal tract, and as an anti-inflammatory (Wallace et al., 2009, 2010; Ekundi-Valentim et al., 2010) and cellular protective molecule (Esechie et al., 2009; Suzuki et al., 2011; Taniguchi et al., 2011). Enzymes involved in trans-sulphuration are involved in the synthesis of H2S, and biochemical alterations (e.g. increased glucose or decreased oxygen) regulate the expression or activity of the H2S-releasing enzymes (Linden et al., 2010). Although vascular KATP channels appear to be a major target of H2S (Whiteman and Moore, 2009) putative cellular targets of H2S in other systems, which include diverse proteins such as KATP channels, NMDA channels, transient receptor potential (TRP) channels, T-type voltage-gated calcium channels and sodium channels, remain controversial (Kimura, 2010; Linden et al., 2010). The mechanism by which the H2S-mediated signalling is terminated also remains controversial. While several mechanisms have been proposed, two are substantiated with relevant physiological evidence. First, because H2S can be released from sulphane–sulphur pools via acidification (Ishigami et al., 2009) and from bound stores via reducing agents such as dithiothreitol (DTT) (Ishigami et al., 2009; Wallace et al., 2009), it has been proposed that enzymatically synthesized H2S can be removed via binding in these pools. Second, H2S signalling may be terminated by rapid enzymatic catabolism of H2S to thiosulphate and sulphate (Levitt et al., 1999). The molecular identity of the enzyme responsible for this catabolism has been controversial with both thiosulphate sulphur transferase (TST; rhodanese) (Picton et al., 2002) and an unknown sulphide oxidase (Levitt et al., 1999; Wilson et al., 2008) proposed as the rate-limiting step in the oxidation of H2S to thiosulphate. A recent study using rat liver demonstrated that H2S oxidation to thiosulphate involves the serial action of the enzymes sulphide quinone reductase (SQR), sulphur dioxygenase and TST (Hildebrandt and Grieshaber, 2008). Mice lacking ethylmalonic encephalopathy 1 (Ethe1) exhibit elevated sulphide levels, suggesting that Ethe1 is the sulphur dioxygenase involved in H2S metabolism, and that sulphide toxicity is the cause of ethylmalonic encephalopathy in patients with truncated, non-functional mutations in ETHE1 (Tiranti et al., 2009). Recombinant expression of human SQR enhances sulphide oxidation in a mammalian cell line (Lagoutte et al., 2010).
The present study was designed to test the hypothesis that SQR plays a role in the catabolism of H2S in the mouse colonic muscularis externa, liver and brain. In these experiments, living and intact tissues were used in order to determine the physiological relevance of the findings. These tissues were not amenable to gene knockdown approaches, so we used a pharmacological approach by utilizing two known inhibitors of SQR in H2S production and consumption assays. Additionally, the expressions of SQR, Ethe1 and TST were investigated using RT-PCR and immunohistochemistry.
Methods
Animals
All methods used in this study were approved by the Mayo Clinic and VA Medical Center Animal Care and Use Committees. Adult C57Bl/6 (Jackson Laboratories, Bar Harbor, ME, USA), 129Sv (in house breeding at Mayo Clinic) and C57BL/6 (Harlan, Indianapolis, IN, USA) mice of either sex, aged 6–10 weeks, weighing 17 to 30 g, were housed five per cage in plastic cages with soft bedding. The animals had access to food and water ad libitum and were maintained at 23–24°C on a 12:12 h light–dark cycle. At the time of tissue collection, animals were killed by CO2 asphyxiation.
Tissue preparation
A 5 cm segment of the muscularis externa of the mouse colon containing circular muscle, longitudinal muscle and the myenteric plexus was isolated from the mucosal and submucosal layers in a sterile manner such that the muscle layers were never exposed to the luminal contents of the colon, as previously described (Linden et al., 2008). Briefly, the anus, intact with surrounding skin, was sutured and hung outside of a 100 × 15 mm plastic Petri dish. The colon and caecum were placed into a dish with iced normal Krebs solution (in mM: Na+, 137.4; K+, 5.9; Ca2+, 2.5; Mg2+, 1.2; Cl-, 134; HCO3-, 15.5; H2PO4-, 1.2; and glucose, 11.5). A small incision made around the circumference of the colon allowed the muscle to be dissected away from the mucosa, keeping the muscularis mucosa and the barrier to the lumen and luminal bacteria intact. These large segments of muscularis externa were cut into either six (for H2S release assays) or eight (for tissue H2S concentration assays and H235S consumption assays) evenly sized pieces (∼0.5–0.75 cm in length).
After the mice had been killed their brain and liver tissue were immediately removed and homogenized in ice-cold RPMI at a 1:10 dilution. Homogenization was performed with a Duall grinder (Kontes, Vineland, NJ, USA) with a Teflon pestle, using 8–10 strokes.
Following their death, mice that were used to rapidly dissect tissue for RNA analysis or immunohistochemistry were transcardially perfused with 60 mL ice-cold PBS (0.1 M, pH 7.4). While the entire brain was used to extract RNA, liver samples were obtained only from distal lobes. The muscularis externa of the colon including the myenteric plexus were dissected free from the mucosal and submucosal layers.
H2S release assays
Intact living colonic muscularis externa tissue was assayed for H2S release using a previously described method (Linden et al., 2008). Briefly, colonic muscularis externa tissue was incubated in 1 mL Krebs solution containing 10 mM l-cysteine. A gas mixture containing 97% O2 and 3% CO2 was bubbled through this solution, which maintained a pH of 7.4. The gas coming off this solution was subsequently bubbled through a 1% solution of zinc acetate. Modifications were made to the previously described experimental setup to decrease variability and improve the detection of H2S. The newly designed apparatus was constructed of polysulphone with sensitive rotometers to more precisely control gas flow (4 mL·min−1). It contained a thin nozzle in the tissue chamber to ensure that gas bubbles formed a column in the centre of the chamber and tissue hooks to ensure the suspended tissue was within the column of gas bubbles. The connecting tubing was made of Teflon instead of silastic to reduce loss of H2S via diffusion through the tubing during transit to a sealed solution of zinc acetate. The H2S stored as stable zinc sulphide was released via acidification with 50% HCl into a sealed gas space that was analysed using a gas chromatograph (model 5890; Hewlett-Packard, Palo Alto, CA, USA) equipped with a Teflon column [8 ft × 0.125 in, packed with Chromosil 330 (Supelco), maintained at 80°C with a flow rate of 20 mL·m−1] and a sulphur chemiluminescence detector (model 355; Sievers Instruments, Boulder, CO, USA), which is specific for sulphur-containing gases. Two small pieces of colon muscularis externa from each of two mice (four pieces total) were incubated in a single chamber to form a single data point. The data from six tissue chambers, three with vehicle (0.1% EtOH) and three with vehicle plus stigmatellin (3 µM), from each pair of mice were averaged to single paired observations for each condition.
Tissue H2S concentration assay
The concentration of H2S in colonic muscularis externa was determined using gas chromatography, with minor modifications to a previously described method (Furne et al., 2008). Briefly, the colonic muscularis externa was rapidly homogenized in ice-cold glycine–NaOH buffer (pH 9.3). An aliquot of the homogenate was then transferred to a 3 mL polypropylene syringe, sealed with a plunger and a three-way stopcock attached to an empty, sealed 3 mL syringe and acidified to pH 5.8 to aid the conversion of HS- to dissolved H2S. The homogenate in solution was vigorously mixed between the two syringes for 1 min to speed the equilibriums between HS- and dissolved H2S and between dissolved H2S and H2S released into the gas space. The gas space was then assayed immediately using gas chromatography as described above. Control syringes containing the buffers without tissue were used to calculate background H2S concentrations, which were subtracted from concentrations obtained in the presence of tissue. Concentrations of H2S in the gas space (minus controls) were used to calculate total molar content of H2S in the sample using known physical constants for H2S including the water dissolved-H2S to H2S gas solubility ratio (2.2 at 4°C) and dissociation to HS- at pH 5.8, normalized to the weight of the tissue to yield values in units of fmol·mg−1.
H2S consumption assays
H2S consumption assays were done similar to previously described methods (Levitt et al., 1999). Tissue was placed into 20 mL polypropylene syringes containing 36 µL Krebs solution and 4 µL of either 1% ethanol (in H2O) or 30 µM stigmatellin (in 1% ethanol/H2O) to achieve final concentrations of 0.1% ethanol and 3 µM stigmatellin, respectively. The syringes were then filled with 20 mL of gas containing 5 ppm H235S, 97% O2 and 3% CO2, sealed with the plunger and a three-way stopcock, and incubated at 37°C for 30 min. When using liver, unlabelled H2S was delivered to the gas space instead of H235S because further characterization of H2S metabolites was not carried out. The gas space was analysed for H2S (or H235S) using gas chromatography as described above. The reduction in H2S concentration, minus that observed in no tissue controls, and the volume of gas space were used to calculate the quantity of H2S removed by the homogenate. Values were normalized for weight of the tissue sample and incubation time to yield H2S consumption in units of pmol·mg−1·min−1.
For the brain and colonic muscularis externa tissues, further analyses of H235S metabolites were performed. After removing the remaining H235S by flushing the gas space with air several times, the samples were weakly acidified with HCl (to pH ∼5.8) to convert HS- to volatile H2S. The gas space was again flushed with air several times before the addition of 20 mM sodium hydroxide, 2 mM sodium sulphate and 2 mM sodium thiosulphate. Samples were then sonicated (Misonix Sonicator 3000, Farmingdale, NY, USA) and analysed for protein, [35S]-thiosulphate and [35S]-sulphate content. Following sonication, the solution was passed over a filter that limits the passage of proteins greater than 30 kDa (Millipore, Billerica, MA, USA). The resulting solution was analysed by HPLC (model C-R3A Chromatopac; Shimadzu Corp., Kyoto, Japan), run at 2 mL·min−1 and 13.8 MPa using an anion–ion exchange column (IonPac AS16; Dionex Corp., Salt Lake City, UT, USA) and a conductivity monitor (Amersham Pharmacia Biotech, Piscataway, NJ, USA) for mass measurements. The column eluate was collected in 2 mL fractions in individual scintillation vials, and the radioactivity of these fractions was determined by scintillation counting. The retention times of the unknowns were compared with retention times of authentic thiosulphate and sulphate standards.
Although the experiments were not initially designed as recovery assays, differences in the results obtained using colonic muscularis externa and brain prompted us to calculate the recovery of H235S metabolites in our studies. Percentage recovery of the 35S that was added to the syringe was determined at various steps: (i) in the gas space after incubation, (ii) in the homogenate just before filtration, (iii) in the homogenate after filtration (30 kDa cut-off) and (iv) in [35S]-sulphate and [35S]-thiosulphate after HPLC analysis. The difference between the pre- and post-filter counts was used to calculate the percentage of [35S] caught within the filter (molecules >30 kDa). The difference between the post-filter counts and sum of the [35S]-sulphate and [35S]-thiosulphate counts weas used to calculate the percentage of 35S that was injected into the HPLC column but not recovered as an identified molecule (i.e. not sulphate or thiosulphate).
RT-qPCR
RNA was extracted and converted to cDNA as described previously (Linden et al., 2008) using RNA-Bee (Tel-Test INC., Friendswood, TX, USA), RNAeasy Plus (Qiagen, Valencia, CA, USA) and GeneAmp Gold RNA PCR (Applied Biosystems, Foster City, CA, USA) kit systems according to the manufacturers' instructions. Real-time PCR reactions were completed using an Applied Biosystems Prism 7000 Sequence Detector. Forward and reverse primer sets for mouse sulphide quinone reductase domain-like (Sqrdl), Ethe1 and TST (rhodanese), as well as the housekeeping genes (HKG) β-actin and GAPDH were purchased from SuperArray Bioscience (Frederick, MD, USA). A 1:5.55 dilution of the RT reaction and 0.4 nM of the appropriate primer set in 1× CyberGreen master mix (SuperArray) were first subjected to 10 min at 95°C to activate the Taq polymerase followed by 40 cycles of 15 s at 95°C and 2 min at 60°C. Melting curves generated with a dissociation protocol were used to ensure the specificity of primers for a single product for each reaction (Ririe et al., 1997). Plasmid DNA (TOPO2.1 vector, Invitrogen, Carlsbad, CA, USA) containing the inserted PCR products were constructed to verify the sequence of the products and used to generate standard curves for each gene product. The cycle number at which the fluorescence intensity crosses a standard threshold value (Ct) for each sample was converted to transcript copy number using the standard curves of log10 concentration of plasmid DNA versus Ct. These standard curves were linear between 30 and 3 million copies with slopes (r2) of these lines for β-actin, GAPDH, SQR, TST and Ethe1-specific amplification of −3.32 ± 0.18 (r2: 0.9826), −3.59 ± 0.37 (r2: 0.9947), −3.45 ± 0.06 (r2: 0.9937), −3.58 ± 0.16 (r2: 0.9979) and −3.13 ± 0.18 (r2: 0.9826), respectively, indicating amplification efficiency within the acceptable range for absolute quantification. The molar quantity of SQR, TST and Ethe1, calculated for each sample, was normalized to the average molar quantity of both β-actin and GAPDH and presented as pmol gene of interest (GOI) over nmol HKG.
Immunohistochemistry
Colon, liver and brain were obtained from mice transcardially perfused with 20 mL PBS and 6 mL 4% paraformaldehyde and immersion fixed in 4% paraformaldehyde for 2 h. Cryostat sections of tissue (15 µm) were blocked with PBS containing 4% normal donkey serum and 0.5% triton X-100 and stained with 1:100 dilutions of rabbit anti SQR antisera (Sigma, St. Louis, MO, USA; Proteintech, Chicago, IL, USA) or 1:100 dilutions of rabbit anti Ethe1 (Sigma). Following three 15 min washes in PBS, tissue sections were incubated with FITC-conjugated donkey anti-rabbit IgG (1:200; Millipore) or Cy3-conjugated donkey anti Rabbit IgG (1:800; Jackson Immunoresearch, West Grove, PA, USA). Immunostained tissues were viewed on an Olympus BXS1W1 epifluorescence microscope, and images were taken using MagnaFire software (Optronics, Goleta, CA, USA).
Chemicals
All drugs and molecular targets conform to BJP's Guide to Receptors and Channels (Alexander et al., 2011). Stigmatellin, the myxobacteria-derived antibiotic (Kunze et al., 1984), antimycin A, the Streptomyces-derived antibiotic (Dunshee et al., 1949), both quinone substrate inhibitors, and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), the protonophore that uncouples oxidative phosphorylation (Heytler and Prichard, 1962) were purchased from Sigma, and dissolved in 100% EtOH to form a stock concentration of 3 mM for stigmatellin and antimycin A and 10 mM for FCCP. Vehicle controls were 0.1% EtOH for stigmatellin and antimycin A and 0.01% EtOH for FCCP. Neither antibiotic-containing nor vehicle stock solutions changed the pH of buffer solutions in which they were used. H235S was synthesized by heating to 37°C 10 µL of a solution containing 1 mg [35S]-cysteine (1075 Ci·mol−1; Moravek Biological, Brea, CA) in glass tubing and capturing the liberated H235S gas in a 2% zinc acetate solution. Before the experiments, an aliquot of the zinc acetate was acidified, releasing H235S into a gas space that was collected in a Teflon gas storage bag equipped with a stopcock. The concentration of H235S was determined by gas chromatography (as described above) and diluted to the final concentration using 97% O2 and 3% CO2. In addition, the radioactivity of the H235S was determined by scintillation counting. All other chemicals were purchased from Sigma.
Statistical analyses
All statistical analyses were completed using GraphPad Prism software, using tests described in the results section. For all tests, P-values less than 0.05 were considered significant.
Results
Stigmatellin enhances the release of H2S by the colonic muscularis externa
The muscularis externa of the mouse colon generated and released detectable levels of H2S (Figure 1), in agreement with our previous study (Linden et al., 2008). In the present experiments, the rate of H2S production and release was greater than previously reported due to the improvements in the methodology. H2S release increased in the presence of tissue by nearly sixfold over that of the control Krebs solution containing 10 mM cysteine. In the presence of cysteine and 3 µM stigmatellin, the rate of H2S release increased twofold over tissue incubated with vehicle alone such that the release rate was 11-fold increased over the vehicle without tissue (control Krebs solutions with cysteine). When colonic muscularis externa was assayed to measure the concentration of free H2S using previously described techniques (Furne et al., 2008), the tissue concentration was determined to be 76 ± 25 fmol·mg−1 tissue (n= 4). This concentration is higher than concentrations reported for brain and liver (Furne et al., 2008) but less than the concentration reported for the aorta (Levitt et al., 2011).
Figure 1.
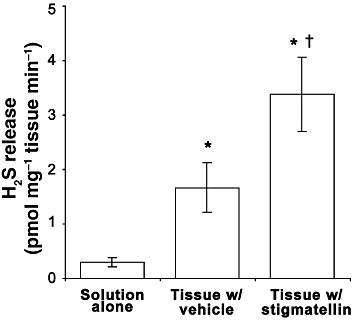
The rate of H2S generation in normal Krebs solution containing 10 mM cysteine (solution alone), and the rate of H2S release in the same solution containing colonic muscularis externa with vehicle (0.1% EtOH) alone or with 3 µM stigmatellin. H2S release was greater in the presence of tissue, which was further increased by the presence of stigmatellin. Data are the mean ± SEM values for three independent experiments run in triplicate. *P < 0.05 compared with no tissue control; †P < 0.05 compared with vehicle treated control; repeated-measures anova, Neuman–Keuls post test.
Stigmatellin reduces H2S consumption and thiosulphate production but does not alter sulphate production in the colonic muscularis externa
Analysis of the gas space over muscularis externa of the mouse colon showed that the muscularis externa consumed H235S, and that this consumption was reduced by 33% during incubation with stigmatellin (Figure 2A). Homogenates (tissue plus incubating solution) were analysed for the conversion of H235S to [35S]-sulphate and [35S]-thiosulphate (Figure 2B and C). Stigmatellin reduced the conversion of H235S to [35S]-thiosulphate by 47% but did not affect the conversion of H235S to [35S]-sulphate. Data from these experiments were analysed to determine the proportion of H235S that was converted to [35S]-sulphate and [35S]-thiosulphate or remained as H235S at the end of the experiment (Figure 3). The proportion of H235S converted to [35S]-sulphate or [35S]-thiosulphate was significantly higher in samples that contained tissue compared with samples that did not, and the proportion of H235S that remained as H235S was significantly lower in samples that contained tissue compared with samples that did not (P < 0.05, Kruskal–Wallis test followed by Dunn's test). The total proportion of H235S recovered as H235S, [35S]-sulphate or [35S]-thiosulphate was not affected by the presence of tissue (P > 0.05, Kruskal–Wallis test). Stigmatellin significantly reduced the proportion of H235S converted by tissue to [35S]-thiosulphate compared with vehicle-treated tissue (P < 0.05, Kruskal–Wallis test followed by Dunn's test). When tested with Kruskal–Wallis test (including the samples that did not contain tissue), the proportion of H235S that remained as H235S was not significantly affected by stigmatellin. However, when only the two tissue-containing experiments were compared, stigmatellin caused a significant increase in the proportion of H235S that remained H235S (P < 0.05, Mann–Whitney U-test). Both statistical tests showed that the proportion of H235S recovered as [35S]-sulphate was unaffected by stigmatellin. There were no differences between groups in either the proportion of H235S recovered on the filter (>30 kDa) (vehicle no tissue: −0.8 ± 0.4%; vehicle plus tissue: −1.6 ± 1.2%; stigmatellin plus tissue: −1.6 ± 0.9%) or the proportion of H235S injected onto but not recovered from the HPLC column (vehicle no tissue: 0.7 ± 0.5%; vehicle plus tissue: 2.3 ± 0.7%; stigmatellin plus tissue: 2.3 ± 0.5%) (P > 0.05, Kruskal–Wallis test followed by Dunn's test).
Figure 2.
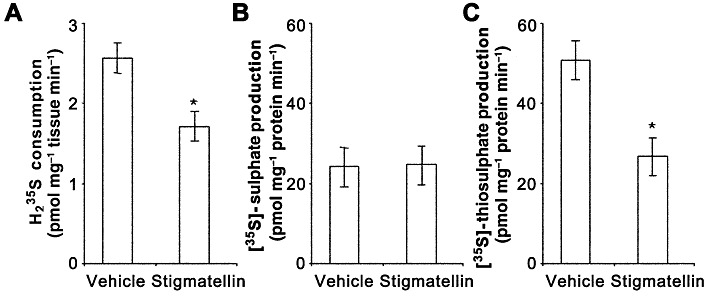
The rates of consumption of H235S (A) and conversion of H235S to [35S]-sulphate (B) and [35S]-thiosulphate (C) by colonic muscularis externa incubated with vehicle (0.1% EtOH) alone or with 3 µM stigmatellin. Stigmatellin reduced H235S consumption and the conversion of H235S to [35S]-thiosulphate production but did not affect the conversion of H235S to [35S]-sulphate. Data are the mean ± SEM values for six independent experiments run in duplicate. *P < 0.05 compared with vehicle-treated control; paired t-test.
Figure 3.
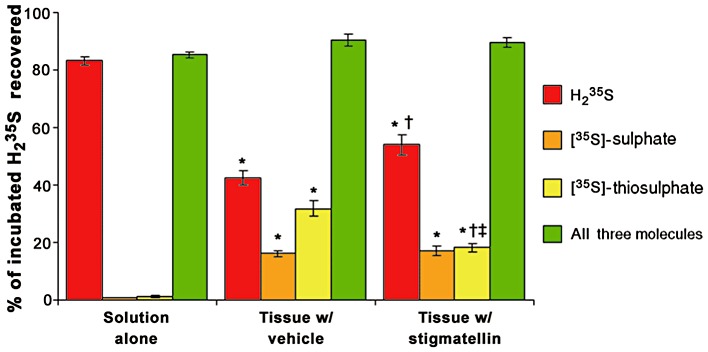
The proportion of H235S delivered to colonic muscularis externa incubated with vehicle (0.1% EtOH) or 3 µM stigmatellin that was recovered as either [35S]-sulphate, [35S]-thiosulphate or H235S or all three of these molecules. Nearly all of the supplied H235S was recovered as H235S when added to syringes containing the incubation solution without tissue (solution alone). The presence of tissue reduced the proportion of supplied H235S recovered as H235S and increased the proportion of H235S recovered as [35S]-sulphate and [35S]-thiosulphate (*P < 0.05 compared with solution alone, Kruskal–Wallis test followed by Dunn's test). Stigmatellin (3 µM) increased the proportion of H235S recovered as H235S (†P < 0.05 compared with tissue with vehicle, Kruskal–Wallis test followed by Dunn's test) and decreased the proportion of H235S recovered as [35S]-thiosulphate (‡P < 0.05 compared with tissue with vehicle, Mann–Whitney U-test) but did not affect the proportion of H235S recovered as [35S]-sulphate (P > 0.05 compared with tissue with vehicle, Kruskal–Wallis test and Mann–Whitney U-test). Data are the mean ± SEM proportions for 8–12 observations.
The consumption of H2S by the intact colonic muscularis externa was also sensitive to antimycin A, another inhibitor of SQR (Griesbeck et al., 2000). The rate of H2S consumption in the presence of 3 µM antimycin A (2.6 ± 0.2 pmol·min−1·mg−1) was significantly less than in tissue incubated with the vehicle control solution (4.3 ± 0.1 pmol·min−1·mg−1; P < 0.05, paired t-test). The consumption of H2S by the intact colonic muscularis externa was insensitive to FCCP, a mitochondrial uncoupler. The rate of H2S consumption in the presence of 1 µM FCCP (6.7 ± 1.1 pmol·min−1·mg−1) was not different from tissue incubated with the vehicle control solution (5.9 ± 1.0 pmol·min−1·mg−1; P > 0.05, paired t-test).
Stigmatellin reduced H2S consumption in the liver but not the brain
The effect of stigmatellin on the consumption of H235S by colonic muscularis externa prompted us to test the effect of stigmatellin on liver and brain. Stigmatellin (3 µM) reduced the consumption of H2S by mouse liver by approximately 50% (Figure 4A) but had little effect on H235S consumption in mouse brain (Figure 4B). Likewise, antimycin A, another inhibitor of SQR, did not significantly alter H2S consumption by mouse brain homogenate at either 3 µM (13.8 ± 0.8 pmol·min−1·mg−1) or 70 µM (19.3 ± 1.6 pmol·min−1·mg−1) when compared with tissue incubated with the vehicle control solutions (16.0 ± 1.4 pmol·min−1·mg−1, 16.5 ± 0.8 pmol·min−1·mg−1, respectively; P > 0.05, paired t-test). Like the colonic muscularis externa, FCCP did not significantly alter H2S consumption by either mouse liver homogenate (vehicle: 123 ± 3 pmol·min−1·mg−1; FCCP: 136 ± 11 pmol·min−1·mg−1; P > 0.05, paired t-test) or mouse brain homogenate (vehicle: 21 ± 1 pmol·min−1·mg−1; FCCP: 20 ± 2 pmol·min−1·mg−1; P > 0.05, paired t-test).
Figure 4.
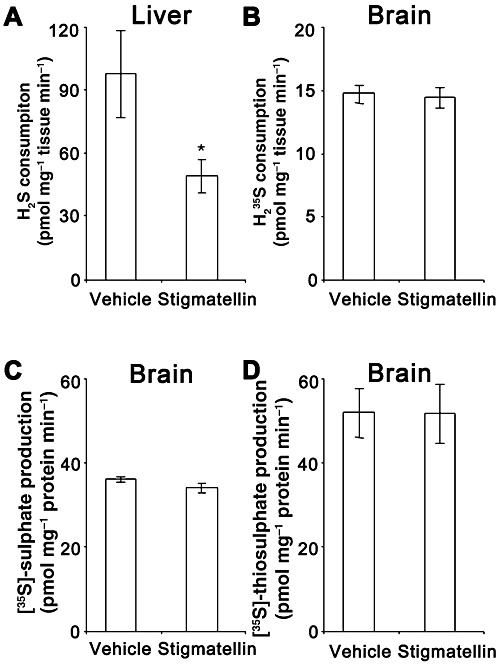
The rate of H2S consumption in liver tissue (A) and H235S consumption in brain tissue (B) incubated with vehicle (0.1% EtOH) alone or with 3 µM stigmatellin. Stigmatellin reduced H2S consumption by the liver but did not alter H235S consumption by brain tissue. Brain tissue was further analysed for the conversion of H235S to [35S]-sulphate (C) and [35S]-thiosulphate (D). Stigmatellin had no significant effect on the conversion of H235S to [35S]-sulphate or [35S]-thiosulphate. Data are the mean ± SEM values for three (liver) or five (brain) independent experiments run in duplicate. *P < 0.05 compared with vehicle-treated control; paired t-test.
Brain homogenates (tissue plus incubating solution) were analysed for the conversion of H235S to [35S]-sulphate and [35S]-thiosulphate (Figure 4C and D). Consistent with a lack of effect of H235S consumption, stigmatellin had no effect on the conversion of H235S to either [35S]-thiosulphate or [35S]-sulphate. Data from these experiments were combined to determine the proportion of the supplied H235S that was converted to [35S]-sulphate and [35S]-thiosulphate or remained as H235S at the end of the experiment (Figure 5). The proportions of H235S converted to [35S]-sulphate or [35S]-thiosulphate were significantly higher in samples that contained tissue compared to samples that did not (P < 0.05, Kruskal–Wallis test followed by Dunn's test). Likewise, the proportion of H235S that remained as H235S was significantly lower in samples that contained tissue compared with samples that did not (P < 0.05, Kruskal–Wallis test followed by Dunn's test). Of particular interest, the total proportion of H235S recovered as H235S, [35S]-sulphate or [35S]-thiosulphate was also lower in samples that contained tissue (P < 0.05, Kruskal–Wallis test followed by Dunn's test). The proportion of 35S recovered on the filter (>30 kDa) (vehicle no tissue: −1.6 ± 0.4%; vehicle plus tissue: 7.4 ± 1.0%; stigmatellin plus tissue: 8.0 ± 1.3%) and the proportion of H235S injected onto but not recovered from the HPLC column (vehicle no tissue: 2.2 ± 0.5%; vehicle plus tissue: 9.0 ± 1.5%; stigmatellin plus tissue: 9.2 ± 1.4%) were greater in solutions containing tissue compared with samples that did not (P < 0.05, Kruskal–Wallis test followed by Dunn's test). The molar quantities of 35S captured on the filter or unidentified on the HPLC column were combined and used to calculate the proportion of H235S (Figure 5). When these measurements were included in the calculation of total recovery (Figure 5), the proportion of H235S recovered was not affected by the presence of tissue (P > 0.05, Kruskal–Wallis test). A 50 µL sample of a 1:5 dilution of pre-filtered solutions was also subjected to chloroform extraction to assess elemental sulphur (and other lipid soluble metabolites) of H2S. The proportion of H235S recovered as lipid soluble metabolites (vehicle no tissue: 1.6 ± 0.1%; vehicle plus tissue: 2.7 ± 0.4%; stigmatellin plus tissue: 2.9 ± 0.3%) was not different between groups (P > 0.05, Kruskal–Wallis test) and was not included in the calculations for Figure 5. In brain tissue, the proportion of H235S recovered as H235S, [35S]-sulphate, [35S]-thiosulphate, [35S]-sulphur or unidentified [35S]-containing molecules in the filter or HPLC column was unaffected by stigmatellin.
Figure 5.
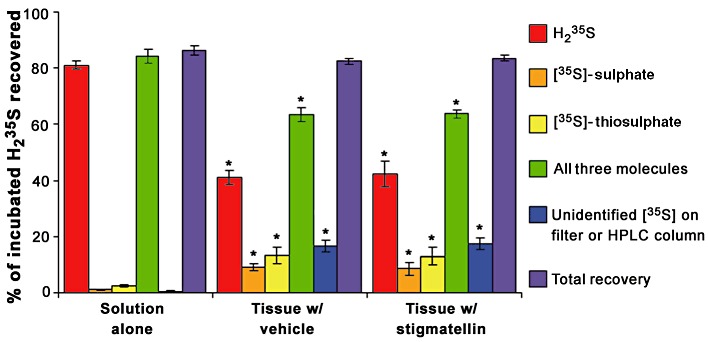
The proportion of H235S delivered to brain tissue with vehicle (0.1% EtOH) or 3 µM stigmatellin that was recovered as either [35S]-sulphate, [35S]-thiosulphate or H235S, all three of these molecules, unidentified 35S recovered from the filter (>30 kDa) or HPLC column or a combination of the three identified molecules, and the 35S that was not identified. Nearly all of the supplied H235S was recovered as H235S when added to syringes containing the incubation solution without tissue (solution alone). Brain tissue reduced the proportion of H235S recovered as H235S and increased the proportions of H235S recovered as [35S]-sulphate, [35S]-thiosulphate, and unidentified [35S] (*P < 0.05 compared with solution alone, Kruskal–Wallis test followed by Dunn's test). Stigmatellin (3 µM) had no effect on any of these measurements. Data are the mean ± SEM proportions for 4–10 observations.
Mouse tissues express various levels of the mRNA encoding SQR
RNA encoding SQR was expressed at relatively high levels, approximately 40% and 20% of the expression levels of the HKG Gapdh and Actb, in the liver and colonic muscularis externa, respectively (Figure 6), and at especially high levels in the colonic mucosa at 220% the levels of HKG. The brain contained relatively low levels of SQR (approximately 1% of HKG). RNA encoding Ethe1, the sulphur dioxygenase and TST, implicated in H2S catabolism was also expressed by these tissues. In the colonic muscularis externa and brain, the expression levels of Ethe1 were at relatively low levels (0.009% and 6% of HKG, respectively) as were levels of Tst (0.2% and 3% of HKG, respectively). In the liver, Ethe1 and Tst expression were relatively high (approximately 130% and 50% of HKG, respectively) as were expression of these genes in the colonic mucosa (approximately 11% and 40% of HKG, respectively).
Figure 6.
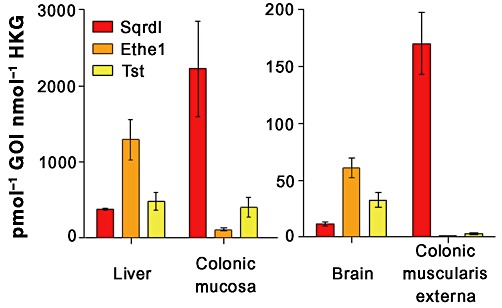
Expression of mRNA encoding SQR in the mouse liver, colonic mucosa and colonic muscularis externa, mRNA encoding Ethe1 in the mouse liver, colonic mucosa and brain, and mRNA encoding Tst in the mouse liver and brain. SQR, Ethe1 and Tst were expressed at low levels in brain tissue. Likewise, Ethe1 and Tst mRNA levels were at the detection limit in the colonic muscularis externa. Data are the mean ± SEM molar content of RNA encoding each gene of interest (GOI) in pmol, normalized to the average molar expression of RNA encoding β-actin and GAPDH as HKG in nmol for n= 3–4 animals.
Mouse liver and colon demonstrate SQR and Ethe1 immunoreactivity
Using two distinct antisera for SQR resulted in different immunoreactive structures in the mouse brain. The polyclonal antiserum available from Sigma identified immunoreactivity in epithelial cells of the choroid plexus and the subfornical organ. In addition, this antiserum identified diffuse immunoreactivity in the molecular layer of the cerebellum and glomeruli in the olfactory bulb. The polyclonal antiserum available from Proteintech identified diffuse immunoreactivity in the subfornical organ, but distinct immunoreactivity in the cell bodies of cortical neurons, cell bodies, dendrites and axons of Purkinji cells of the cerebellum, and cell bodies of mitrial cells, but not glomeruli in the olfactory bulb. The mostly non-overlapping immunoreactivity for SQR in the brain by these two antisera raises doubt regarding specificity of each antiserum and, together with the mRNA data, suggests that SQR expression might be quite limited in the brain. Of interest is that the subfornical organ, which exhibited overlapping immunoreactivity by both antisera, is a region of the brain outside the blood–brain barrier. In contrast to the CNS, both antisera identified similar immunoreactive structures in the liver and in the colon. Both antisera stained hepatocytes in the liver (Figure 7A and B). In the colon, both antisera-stained epithelial cells in the mucosa as well as numerous fine structures throughout the musculature and cells within the myenteric plexus (Figure 7C and D). Immunoreactivity for Ethe1 was not detected in the mouse brain. Conversely, Ethe1 immunoreactivity was detected in the epithelium of the mouse colonic mucosa (Figure 8A) as well as hepatocytes in the liver (Figure 8B).
Figure 7.
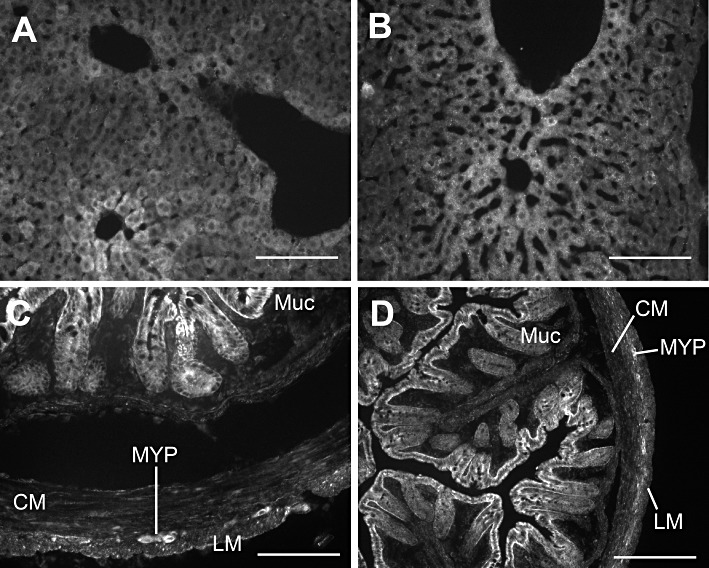
Representative micrographs illustrating immunoreactivity for SQR in the mouse liver (A and B) and colon (C and D). In the liver, both the Sigma antiserum (A) and Proteintech antiserum (B) produced immunoreactivity in hepatocytes. Likewise staining for both antisera were similar in the colon with strong immunoreactivity in epithelial cells and cells within the muscle layers and myenteric plexus (C and D) (Sigma: C; Proteintech: D). (Muc: colonic mucosa; CM: circular muscle layer of the colon; LM: longitudinal muscle layer of the colon; MYP: myenteric plexus of the colon.) Scale bars illustrate 100 µm.
Figure 8.
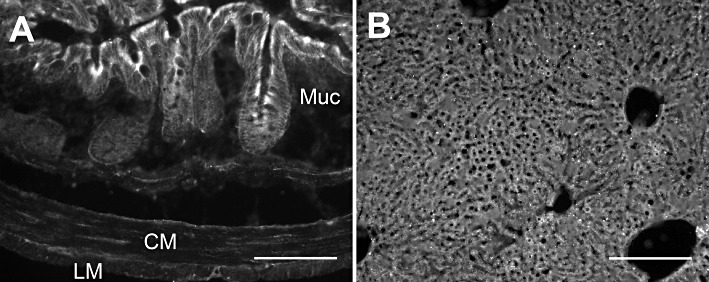
Representative micrographs illustrating immunoreactivity for Ethe1 in the mouse colon (A) and liver (B). Immunoreactivity was strong in the epithelial cells of the colonic mucosa while weak in the colonic muscularis externa. In the liver, there was strong immunoreactivity in hepatocytes. Scale bars illustrate 100 µm.
Discussion and conclusions
The results of this study support the concept that SQR contributes to the catabolism of H2S and suggest that robust catabolic pathways contribute to low resting tissue levels of H2S, thereby increasing the signalling capacity of H2S synthesized on demand. This study demonstrates for the first time that stigmatellin, an inhibitor of SQR, enhanced H2S release in the colonic muscularis externa and inhibited H2S catabolism in the colonic muscularis externa and liver. A related SQR inhibitor, antimycin A, also inhibited H2S catabolism in the colonic muscularis externa. These findings extend previous reports that SQR contributes to H2S catabolism in the rat liver (Hildebrandt and Grieshaber, 2008), and that recombinant expression of the human SQR gene oxidizes sulphide (Lagoutte et al., 2010). Furthermore, our findings in the present study suggest that catabolism of endogenous H2S through SQR contributes to H2S signal termination in the colonic muscularis externa and liver. The lack of effect of stigmatellin and antimycin A on H2S catabolism in brain tissue, which may be a reflection of the observed low levels of SQR mRNA and protein expression compared with colonic muscularis externa and liver tissues, suggest that SQR does not contribute to H2S signal termination in the brain. These results confirm a previous finding that mitochondria isolated from mouse brain lack SQR-like enzymatic activity (Lagoutte et al., 2010). In the brain, other unidentified enzymatic pathways of H2S catabolism to thiosulphate and sulphate and H2S sequestration probably contribute to H2S signal termination. This difference in H2S catabolism is not unlike different mechanisms of H2S production in the brain and periphery (Ishigami et al., 2009).
SQR is a member of the disulphide oxidoreductase flavoprotein (DiSR) superfamily and is responsible for H2S oxidation by phototrophic and chemolithoautotrophic bacteria where H2S acts as an electron donor to the respiratory chain (Griesbeck et al., 2000). SQR was recently identified as the initial protein responsible for H2S oxidation in the rat liver (Hildebrandt and Grieshaber, 2008), and recombinant expression of the human SQR gene enhances sulphide oxidation of a mammalian cell line (Lagoutte et al., 2010). Stigmatellin and antimycin A are inhibitors of the cytochrome bc complex (complex III) involved in mitochondrial respiration (Potter and Reif, 1952; Kunze et al., 1984; Thierbach et al., 1984; von Jagow and Ohnishi, 1985; Zhang et al., 1998) as well as inhibitors of bacterial SQR (Griesbeck et al., 2000). It is possible that the actions of stigmatellin and antimycin A on H2S metabolism observed in the present study were through inhibition of complex III. However, several studies have been able to effectively separate sulphide oxidation activity from cytochrome bc oxidoreductase activity (Nubel et al., 2000; Kawamukai, 2002; Hildebrandt and Grieshaber, 2008; Lagoutte et al., 2010), suggesting that the enzymes are distinct proteins that share sensitivity to stigmatellin and antimycin A just as they share dependence on quinone as a cofactor. These enzymes are linked, however, as electrons donated to ubiquinone by SQR enter the electron transport chain through complex III and are eventually donated to oxygen (Griesbeck et al., 2000; Nubel et al., 2000; Hildebrandt and Grieshaber, 2008; Lagoutte et al., 2010). This biochemical process, without direct implication of SQR, has been proposed as the mechanism by which inhibition of H2S metabolism serves as a cellular oxygen sensor (Olson et al., 2008). While it is possible that stigmatellin and antimycin A inhibit H2S consumption via nonspecific effects downstream of blocking mitochondrial respiration (Thierbach et al., 1984; von Jagow and Ohnishi, 1985), some effects of inhibiting respiration, including the loss of mitochondrial voltage potential and reduced cell viability, do not occur with stigmatellin as it does with antimycin A (Armstrong et al., 2004). Furthermore, inhibition of cytochrome bc oxidoreductase by stigmatellin is not associated with an increase in reactive oxygen species often associated with other mitochondrial respiration inhibitors like antimycin A (Lai et al., 2005; Panee et al., 2007). In fact, stigmatellin has been shown to protect against the loss of mitochondrial voltage potential, mitochondrial damage and reduced cell viability caused by cellular models of oxidative stress (Armstrong et al., 2004; Belyaeva et al., 2006). These latter effects are probably due to the fact that stigmatellin binds to the Qo binding site of cytochrome b rather than the Qi site (site of action of antimycin A and related inhibitors), disabling the ability of the Rieske iron-sulphur protein (ISP) to move within the membrane and transfer electrons to the Qi site (von Jagow and Ohnishi, 1985; Zhang et al., 1998), so that electron transfer cannot take place. This type of respiratory chain inhibition is thus less damaging to the cell as ATP production can proceed, albeit at a lower efficiency, without the damaging effects of increased production of reactive oxygen species. We also used FCCP, an uncoupler of oxidative phosphorylation, and found that it did not inhibit H2S consumption in any tissues tested. These data provide the strongest support for our hypothesis that the action of stigmatellin- and antimycin A-mediated inhibition of H2S metabolism is distinct from inhibition of mitochondrial respiration. Therefore, we suggest that the effects of stigmatellin and antimycin A observed in our study were due to inhibition of SQR.
It has recently been proposed that protein cysteine residues act as a sink to remove free H2S by the formation of protein persulphides (Warenycia et al., 1990; Ishigami et al., 2009). While H2S can certainly be released from protein persulphides upon reduction, there is little existing direct evidence to suggest that appreciable free H2S, as supplied exogenously, or perhaps synthesized endogenously (Abe and Kimura, 1996; Wang, 2002; Furne et al., 2008; Linden et al., 2008), adds to the pool of persulphides or is retained as bound H2S. Ishigami et al. (2009) have suggested that the rapid disappearance of H2S exogenously applied to liver, brain and heart tissues resulted from absorption of sulphide because DTT-releasable H2S was enhanced following the exposure of tissue to 3.3 µM Na2S. However, they were not able to determine whether DTT-released H2S were the same molecules as the supplied Na2S. This limitation was circumvented in the present study by the use of H235S. Approximately 90% of H235S delivered to the mouse colon muscularis externa was recovered as either [35S]-sulphate, [35S]-thiosulphate or H235S. While it is possible that the unrecoverable label was bound to proteins in the tissue, the finding that samples without tissue had 15% loss of 35S suggests that 35S was lost in the experimental setup rather than being bound to tissue. The finding that the colonic muscularis externa catabolized H235S to only [35S]-sulphate or [35S]-thiosulphate is consistent with previous reports that nearly all H235S vascularly perfused or incubated with colonic mucosa, lung, kidney or liver tissues is recovered as [35S]-sulphate or [35S]-thiosulphate (Curtis et al., 1972; Bartholomew et al., 1980; Levitt et al., 1999). Our findings are also compatible with earlier and with more recent literature using non-radioactive technologies that found that systemically applied H2S is excreted as sulphate and does not remain as sulphide in the blood (Haggard, 1921; Denis and Reed, 1927; Whitfield et al., 2008). In the present study, only 65% of H235S delivered to mouse brain homogenates was recovered as either [35S]-sulphate, [35S]-thiosulphate or H235S. Samples without tissue had a 15% loss of 35S, similar to the results from experiments using colonic muscularis externa, suggesting that 20% of H235S was converted to a form that we did not assay. There was no evidence that the unidentified 35S-containing molecules were lipid soluble elemental sulphur. Rather, approximately half of the unidentified 35S-containing molecules were captured by a 30 kDa filter, while the other half remained unidentified on the HPLC column, that is not recovered as [35S]-sulphate or [35S]-thiosulphate. Although further studies are required to determine the molecular fate of the unrecovered H235S incubated with brain tissue, we cannot at the present time exclude the possibility that H2S becomes bound to protein sulphur pools in brain tissue as previously suggested (Ishigami et al., 2009). The brain may be unique in this regard.
It is difficult to compare the rates of H2S synthesis and catabolism in the current study given the differences in experimental conditions. When we normalized the rate of synthesis (pmol·min−1) to the weight of the tissue (mg), we found that the rate of H2S catabolism in colonic muscularis externa (2.4 pmol·min−1·mg−1) exceeded the rate of H2S synthesis (1.7 pmol·min−1·mg−1). It should be noted that 10 mM of substrate (cysteine) was supplied in the H2S release experiments, while only 0.2 µM of substrate (H235S) was supplied in H2S consumption experiments, such that the latter might be an underestimate of physiological catabolism and the former may be an overestimate of physiological H2S production. Regardless, the difference in these rates may explain why the concentration of free H2S in the colonic muscularis externa is just above the lower detection limit (ambient H2S in room air) for the gas chromatograph. In the present study, the concentration of free H2S measured in the colonic muscularis externa was 76 fmol·mg−1. Previous studies indicate that the tissue levels of free H2S in the brain and liver are approximately 20 fmol·mg−1 (Furne et al., 2008) and <15 nM in the blood (Whitfield et al., 2008). Thus, the results of these previous studies, as well as the present study, support the concept that the level of free H2S in biological tissues is normally maintained very low by a robust catabolic system. Previous reports suggesting that tissue and blood levels of H2S are in the micromolar range (Abe and Kimura, 1996; Wang, 2002; Wallace et al., 2009) have recently been called into question (Furne et al., 2008; Whitfield et al., 2008; Linden et al., 2010). The high levels of H2S may have been due to the addition of cysteine, which can non-enzymatically produce H2S (Figure 1), tissue acidification with trichloroacetic acid and errors in standard curves constructed with impure NaHS used to quantify H2S using the methylene blue spectrophotometric assay (Ishigami et al., 2009).
It has generally been assumed that H2S conversion to sulphate occurs subsequent to thiosulphate (Levitt et al., 1999). In the present study, stigmatellin reduced the conversion of H235S to [35S]-thiosulphate by the colon muscularis externa but did not affect the conversion of H235S to [35S]-sulphate. One possible explanation may be that the enzymes responsible for the conversion of thiosulphate to sulphate are saturated such that reduced thiosulphate levels may still exceed the capacity of the enzymes and thus still have similar sulphate levels. While H2S has a low Km value of 2.9 µM for SQR (Hildebrandt and Grieshaber, 2008), thiosulphate has a Km value of 2.6 mM for thiosulphate:quinone oxidoreductase (Muller et al., 2004), the putative enzyme for thiosulphate oxidation, suggesting that saturation of thiosulphate catabolism is not likely to explain this observation. An alternative explanation is that another pathway for the production of sulphate from H2S exists in mouse colonic muscularis externa and liver. This idea is supported by the observation that stigmatellin did not inhibit H2S catabolism in the brain where sulphate and thiosulphate were still produced, and that it reduced but did not block H2S metabolism in the liver and colonic muscularis externa. A surprising finding of these studies was that while SQR is highly expressed in the liver, colonic mucosa and colonic muscularis externa, Ethe1, the putative sulphur dioxygenase responsible for SQR-persulphide oxidation and TST, the thiosulphate transferase responsible for thiosulphate production are highly expressed in the liver and colonic mucosa but negligible in the colonic muscularis externa. These results suggest that while the SQR-Ethe1-TST catabolism of H2S to thiosulphate previously described in the rat liver (Hildebrandt and Grieshaber, 2008) may be responsible for the catabolism of H2S in the mouse liver and colonic mucosa, other downstream acceptors of the SQR-persulphides (i.e. not Ethe1 and TST) may exist in colonic muscularis externa.
H2S is gaining acceptance as a gaseous signal molecule involved in diverse cellular effects (Wang, 2002; Whiteman and Moore, 2009; Kimura, 2010; Linden et al., 2010). An important aspect of the biology of signal molecules is the termination of the signal. The results of this study support the concept that the mitochondrial protein SQR contributes to the rapid oxidation of H2S. As H2S appears to be anti-inflammatory (Wallace, 2010) and is beneficial in animal models of myocardial infarction (Ririe et al., 1997; Elrod et al., 2007; Zhu et al., 2007), it is reasonable to suggest that an inhibitor of a catabolic pathway for endogenous H2S may be an important target. New compounds that can inhibit H2S catabolic pathways will have to distinguish between SQR and cytochrome bc complex. Because crystal structures for bacterial SQR have recently been solved (Brito et al., 2009; Marcia et al., 2009), it is expected this development will be forthcoming.
Acknowledgments
We gratefully acknowledge the secretarial support of Ms Janice Applequist and Mr James Craighead and Mr Tyler King of the Mayo Clinic Division of Engineering for the design and fabrication of the apparatus used to capture released H2S. This work was supported by NIH grants DK17238 and DK76665 and a grant from the Minnesota Partnership for Biotechnology and Medical Genomics.
Glossary
- DiSR
disulphide oxidoreductase flavoprotein
- DTT
dithiothreitol
- Ethe1
ethylmalonic encephalopathy 1
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- GOI
gene of interest
- HKG
housekeeping genes
- SQR
sulphide quinone reductase
- Sqrdl
sulphide quinone reductase domain-like
- TRP
transient receptor potential
- TST
thiosulphate sulphur transferase
Conflicts of interest
No competing financial interests exist for the authors, DRL, JF, GJS, MA-R, MDL or JHS.
References
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JS, Yang H, Duan W, Whiteman M. Cytochrome bc(1) regulates the mitochondrial permeability transition by two distinct pathways. J Biol Chem. 2004;279:50420–50428. doi: 10.1074/jbc.M408882200. [DOI] [PubMed] [Google Scholar]
- Bartholomew TC, Powell GM, Dodgson KS, Curtis CG. Oxidation of sodium sulphide by rat liver, lungs and kidney. Biochem Pharmacol. 1980;29:2431–2437. doi: 10.1016/0006-2952(80)90346-9. [DOI] [PubMed] [Google Scholar]
- Belyaeva EA, Dymkowska D, Wieckowski MR, Wojtczak L. Reactive oxygen species produced by the mitochondrial respiratory chain are involved in Cd2+-induced injury of rat ascites hepatoma AS-30D cells. Biochim Biophys Acta. 2006;1757:1568–1574. doi: 10.1016/j.bbabio.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Brito JA, Sousa FL, Stelter M, Bandeiras TM, Vonrhein C, Teixeira M, et al. Structural and functional insights into sulfide: quinone oxidoreductase. Biochemistry. 2009;48:5613–5622. doi: 10.1021/bi9003827. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- Curtis CG, Bartholomew TC, Rose FA, Dodgson KS. Detoxication of sodium 35 S-sulphide in the rat. Biochem Pharmacol. 1972;21:2313–2321. doi: 10.1016/0006-2952(72)90382-6. [DOI] [PubMed] [Google Scholar]
- Denis W, Reed L. The action of blood on sulfides. J Biol Chem. 1927;72:385–394. [Google Scholar]
- Dunshee BR, Leben C, Keitt GW, Strong FM. The isolation and properties of Antimycin A. J Am Chem Soc. 1949;71:2436–2437. [Google Scholar]
- Ekundi-Valentim E, Santos KT, Camargo EA, adai-Souza A, Teixeira SA, Zanoni CI, et al. Differing effects of exogenous and endogenous hydrogen sulphide in carrageenan-induced knee joint synovitis in the rat. Br J Pharmacol. 2010;159:1463–1474. doi: 10.1111/j.1476-5381.2010.00640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esechie A, Enkhbaatar P, Traber DL, Jonkam C, Lange M, Hamahata A, et al. Beneficial effect of a hydrogen sulphide donor (sodium sulphide) in an ovine model of burn- and smoke-induced acute lung injury. Br J Pharmacol. 2009;158:1442–1453. doi: 10.1111/j.1476-5381.2009.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furne J, Saeed A, Levitt MD. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–R1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- Gallego D, Clave P, Donovan J, Rahmati R, Grundy D, Jimenez M, et al. The gaseous mediator, hydrogen sulphide, inhibits in vitro motor patterns in the human, rat and mouse colon and jejunum. Neurogastroenterol Motil. 2008;20:1306–1316. doi: 10.1111/j.1365-2982.2008.01201.x. [DOI] [PubMed] [Google Scholar]
- Gil V, Gallego D, Jimenez M. Effects of inhibitors of hydrogen sulphide synthesis on rat colonic motility. Br J Pharmacol. 2011;164:485–498. doi: 10.1111/j.1476-5381.2011.01431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck C, Hauska G, Schutz M. Biological sulfide oxidation: sulfide-quinone reductase (SQR), the primary reaction. In: Pandalai SG, editor. Recent Research Developments in Microbiology. Trivadrum, India: Research Signpost; 2000. pp. 179–203. [Google Scholar]
- Haggard HW. The fate of sulfides in the blood. J Biol Chem. 1921;49:519–529. [Google Scholar]
- Hennig B, Diener M. Actions of hydrogen sulphide on ion transport across rat distal colon. Br J Pharmacol. 2009;158:1263–1275. doi: 10.1111/j.1476-5381.2009.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heytler PG, Prichard WW. A new class of uncoupling agents – carbonyl cyanide phenylhydrazones. Biochem Biophys Res Commun. 1962;7:272–275. doi: 10.1016/0006-291x(62)90189-4. [DOI] [PubMed] [Google Scholar]
- Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Ishigami M, Hiraki K, Umemura K, Ogasawara Y, Ishii K, Kimura H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid Redox Signal. 2009;11:205–214. doi: 10.1089/ars.2008.2132. [DOI] [PubMed] [Google Scholar]
- von Jagow G, Ohnishi T. The chromone inhibitor stigmatellin – binding to the ubiquinol oxidation center at the C-side of the mitochondrial membrane. FEBS Lett. 1985;185:311–315. doi: 10.1016/0014-5793(85)80929-7. [DOI] [PubMed] [Google Scholar]
- Kawamukai M. Biosynthesis, bioproduction and novel roles of ubiquinone. J Biosci Bioeng. 2002;94:511–517. doi: 10.1016/s1389-1723(02)80188-8. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: from brain to gut. Antioxid Redox Signal. 2010;12:1111–1123. doi: 10.1089/ars.2009.2919. [DOI] [PubMed] [Google Scholar]
- Krueger D, Foerster M, Mueller K, Zeller F, Slotta-Huspenina J, Donovan J, et al. Signaling mechanisms involved in the intestinal pro-secretory actions of hydrogen sulfide. Neurogastroenterol Motil. 2010;22:1224–1220. doi: 10.1111/j.1365-2982.2010.01571.x. [DOI] [PubMed] [Google Scholar]
- Kunze B, Kemmer T, Hofle G, Reichenbach H. Stigmatellin, a new antibiotic from Stigmatella aurantiaca (Myxobacterales). I. Production, physico-chemical and biological properties. J Antibiot (Tokyo) 1984;37:454–461. doi: 10.7164/antibiotics.37.454. [DOI] [PubMed] [Google Scholar]
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Lai B, Zhang L, Dong LY, Zhu YH, Sun FY, Zheng P. Inhibition of Qi site of mitochondrial complex III with antimycin A decreases persistent and transient sodium currents via reactive oxygen species and protein kinase C in rat hippocampal CA1 cells. Exp Neurol. 2005;194:484–494. doi: 10.1016/j.expneurol.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Levitt MD, Furne J, Springfield J, Suarez F, DeMaster E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J Clin Invest. 1999;104:1107–1114. doi: 10.1172/JCI7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt MD, Abdel-Rehim MS, Furne J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxid Redox Signal. 2011;15:373–378. doi: 10.1089/ars.2010.3525. [DOI] [PubMed] [Google Scholar]
- Linden DR, Sha L, Mazzone A, Stoltz GJ, Bernard CE, Furne JK, et al. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J Neurochem. 2008;106:1577–1585. doi: 10.1111/j.1471-4159.2008.05502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DR, Levitt MD, Farrugia G, Szurszewski JH. Endogenous production of H2S in the gastrointestinal tract: still in search of a physiologic function. Antioxid Redox Signal. 2010;12:1135–1146. doi: 10.1089/ars.2009.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcia M, Ermler U, Peng G, Michel H. The structure of Aquifex aeolicus sulfide : quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc Natl Acad Sci U S A. 2009;106:9625–9630. doi: 10.1073/pnas.0904165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FH, Bandeiras TM, Urich T, Teixeira M, Gomes CM, Kletzin A. Coupling of the pathway of sulphur oxidation to dioxygen reduction: characterization of a novel membrane-bound thiosulphate : quinone oxidoreductase. Mol Microbiol. 2004;53:1147–1160. doi: 10.1111/j.1365-2958.2004.04193.x. [DOI] [PubMed] [Google Scholar]
- Nubel T, Klughammer C, Huber R, Hauska G, Schutz M. Sulfide : quinone oxidoreductase in membranes of the hyperthermophilic bacterium Aquifex aeolicus (VF5) Arch Microbiol. 2000;173:233–244. doi: 10.1007/s002030000135. [DOI] [PubMed] [Google Scholar]
- Olson KR, Healy MJ, Qin Z, Skovgaard N, Vulesevic B, Duff DW, et al. Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am J Physiol Regul Integr Comp Physiol. 2008;295:R669–R680. doi: 10.1152/ajpregu.00807.2007. [DOI] [PubMed] [Google Scholar]
- Panee J, Liu W, Nakamura K, Berry MJ. The responses of HT22 cells to the blockade of mitochondrial complexes and potential protective effect of selenium supplementation. Int J Biol Sci. 2007;3:335–341. doi: 10.7150/ijbs.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton R, Eggo MC, Merrill GA, Langman MJ, Singh S. Mucosal protection against sulphide: importance of the enzyme rhodanese. Gut. 2002;50:201–205. doi: 10.1136/gut.50.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter VR, Reif AE. Inhibition of an electron transport component by antimycin A. J Biol Chem. 1952;194:287–297. [PubMed] [Google Scholar]
- Pouokam E, Diener M. Mechanisms of actions of hydrogen sulphide on rat distal colonic epithelium. Br J Pharmacol. 2011;162:392–404. doi: 10.1111/j.1476-5381.2010.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- Schicho R, Krueger D, Zeller F, Von Weyhern CW, Frieling T, Kimura H, et al. Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology. 2006;131:1542–1552. doi: 10.1053/j.gastro.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Papapetropoulos A. Hydrogen sulfide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Kang L, Kimura T, Niki I. Hydrogen sulphide protects mouse pancreatic beta-cells from cell death induced by oxidative stress, but not by endoplasmic reticulum stress. Br J Pharmacol. 2011;162:1171–1178. doi: 10.1111/j.1476-5381.2010.01119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague B, Asiedu S, Moore PK. The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. Br J Pharmacol. 2002;137:139–145. doi: 10.1038/sj.bjp.0704858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierbach G, Kunze B, Reinchenbach H, Hofle G. The mode of action of stigmatellin, a new inhibitor of the cytochrome b-c1 segment of the respiratory chain. Biochimica Et Biophysica Acta (BBA) Bioenerg. 1984;765:227–235. [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxid Redox Signal. 2010;12:1125–1133. doi: 10.1089/ars.2009.2900. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009;137:569–578. doi: 10.1053/j.gastro.2009.04.012. 578. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346) Br J Pharmacol. 2010;159:1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- Warenycia MW, Goodwin LR, Francom DM, Dieken FP, Kombian SB, Reiffenstein RJ. Dithiothreitol liberates non-acid labile sulfide from brain tissue of H2S-poisoned animals. Arch Toxicol. 1990;64:650–655. doi: 10.1007/BF01974693. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Moore PK. Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability? J Cell Mol Med. 2009;13:488–507. doi: 10.1111/j.1582-4934.2009.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- Wilson K, Mudra M, Furne J, Levitt M. Differentiation of the roles of sulfide oxidase and rhodanese in the detoxification of sulfide by the colonic mucosa. Dig Dis Sci. 2008;53:277–283. doi: 10.1007/s10620-007-9854-9. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, et al. Electron transfer by domain movement in cytochrome bc1. Nature. 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC, Huang SH, et al. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J Appl Physiol. 2007;102:261–268. doi: 10.1152/japplphysiol.00096.2006. [DOI] [PubMed] [Google Scholar]