

Abstract
BACKGROUND AND PURPOSE
KB-R7943 is an isothiourea derivative that is used widely as a pharmacological inhibitor of sodium–calcium exchange (NCX) in experiments on cardiac and other tissue types. This study investigated KB-R7943 inhibition of hERG (human ether-à-go-go-related gene) K+ channels that underpin the cardiac rapid delayed rectifier potassium current, IKr.
EXPERIMENTAL APPROACH
Whole-cell patch-clamp measurements were made of hERG current (IhERG) carried by wild-type or mutant hERG channels and of native rabbit ventricular IKr. Docking simulations utilized a hERG homology model built on a MthK-based template.
KEY RESULTS
KB-R7943 inhibited both IhERG and native IKr rapidly on membrane depolarization with IC50 values of ∼89 and ∼120 nM, respectively, for current tails at −40 mV following depolarizing voltage commands to +20 mV. Marked IhERG inhibition also occurred under ventricular action potential voltage clamp. IhERG inhibition by KB-R7943 exhibited both time- and voltage-dependence but showed no preference for inactivated over activated channels. Results of alanine mutagenesis and docking simulations indicate that KB-R7943 can bind to a pocket formed of the side chains of aromatic residues Y652 and F656, with the compound's nitrobenzyl group orientated towards the cytoplasmic side of the channel pore. The structurally related NCX inhibitor SN-6 also inhibited IhERG, but with a markedly reduced potency.
CONCLUSIONS AND IMPLICATIONS
KB-R7943 inhibits IhERG/IKr with a potency that exceeds that reported previously for acute cardiac NCX inhibition. Our results also support the feasibility of benzyloxyphenyl-containing NCX inhibitors with reduced potential, in comparison with KB-R7943, to inhibit hERG.
Keywords: hERG, IhERG, IKr, KB-R7943, NCX, rapid delayed rectifier K+ current, sodium–calcium exchange
Introduction
Sodium–calcium exchange (NCX) proteins are expressed in many tissue types and are recognized to be important for cellular Ca2+ ion homeostasis (Dipolo and Beauge, 2006). In the heart, the cardiac NCX isoform (NCX1) contributes both to Ca2+ homeostasis and to electrogenesis due to its stoichiometry, and, in principle, partial NCX inhibition may be beneficial in some cardiac pathologies (Philipson and Nicoll, 2000; Shigekawa and Iwamoto, 2001; Watanabe et al., 2006; Toth et al., 2009; Zhang and Hancox, 2009). At present, however, there are no high-affinity selective NCX inhibitors available for clinical use.
KB-R7943 (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methane sulphonate) was developed as the first NCX-specific inhibitor (Iwamoto et al., 1996; Watanabe et al., 2006). It was initially reported to inhibit preferentially outward over inward cardiac NCX current (INCX) when the NCX was activated unidirectionally (Watano et al., 1996), but with bi-directional NCX activation, the compound was found to inhibit both NCX modes similarly, with IC50 of ∼1 µM (Kimura et al., 1999). KB-R7943 was reported also to affect cardiac Na+, Ca2+ and inwardly rectifying K+ currents with higher IC50 values (Watano et al., 1996; Tanaka et al., 2002). Although KB-R7943 transpires not to be entirely selective for the NCX, its ease of use and effectiveness as an NCX blocker means that it has been employed widely as a pharmacological tool for the study of the cardiac NCX in physiological and pathophysiological conditions (Amran et al., 2003; Toth et al., 2009).
The rapid delayed rectifier K+ current (IKr) plays an important role in cardiac action potential (AP) repolarization (Sanguinetti and Mitcheson, 2005; Sanguinetti and Tristani-Firouzi, 2006). KCNH2-encoded hERG (human Ether-à-go-go-Related Gene) protein forms the pore-forming subunit of IKr channels (Sanguinetti et al., 1995; Trudeau et al., 1995; Alexander et al., 2011). Due to structural features of the channel, hERG is able to interact with chemically and therapeutically diverse drugs that are associated with the acquired Long QT Syndrome (Sanguinetti and Mitcheson, 2005; Sanguinetti and Tristani-Firouzi, 2006). Consequently, it is customary for all promising drug candidates now to be tested for hERG channel inhibitory activity (ICH, 2005; Shah, 2005; Hancox et al., 2008). Despite the long use of KB-R7943 in experiments utilizing cardiac cell, tissue and intact heart preparations and the hERG channel's high susceptibility to pharmacological blockade, there is currently no information available regarding effects of the compound on hERG channel current (IhERG). One study has reported partial inhibition of guinea-pig ventricular outward delayed rectifier K+ current (IK) by a single concentration (10 µM) of KB-R7943 (Tanaka et al., 2002). However, guinea-pig IK is a composite current comprised of both IKr and the slow delayed rectifier current, IKs (Sanguinetti and Jurkiewicz, 1990), and the investigation by Tanaka et al. (2002) neither determined concentration-dependence of the compound's effect on composite IK, nor identified any specific inhibitory effect of KB-R7943 on IKr as opposed to IKs. In a very recent study of the role of NCX in spontaneous activity of cells from the rabbit atrioventricular (AV) node, KB-R7943 led to membrane potential depolarization at a concentration (5 µM) that inhibited completely inward NCX current (Cheng et al., 2011). This seemingly paradoxical effect might, in principle, be accounted for by an inhibitory action on an AV nodal IKr, as this is a major repolarizing current in these cells, which lack IKs (Mitcheson and Hancox, 1999; Sato et al., 2000). Given the lack of direct evidence either for or against KB-R7943 actions on hERG/IKr, the present investigation was conducted to determine whether or not KB-R7943 is able to inhibit hERG and native cardiac IKr and if so to characterize the underlying mechanism. The results: (i) establish KB-R7943 as a potent IhERG/IKr inhibitor; (ii) provide insight into the nature of interactions between the KB-R7943 molecule and the hERG channel pore; and (iii) demonstrate that the structurally related NCX inhibitor SN-6 inhibits IhERG much less potently than does KB-R7943.
Methods
Maintenance of cell lines stably expressing hERG
IhERG measurements were made from HEK 293 cells stably expressing wild-type (WT) hERG [donated by Professor Craig January; (Zhou et al., 1998)] or the hERG S6 mutations Y652A and F656A (Milnes et al., 2003a), or transiently expressing the hERG pore helix mutation S624A (transfections were conducted using lipofectamine LTX using CD8 as a transfection marker; see Zhang et al., 2010). Cells were passaged using enzyme-free cell dissociation solution (Millipore, Watford, UK) and were plated onto glass coverslip shards in 40 mm Petri dishes (Zhang et al., 2010). Dulbecco's minimum essential medium with Glutamax-1 (DMEM; Gibco, Paisley, UK) was supplemented with 10% fetal bovine serum, together with 50 µg·mL−1 gentamycin and 400 µg·mL−1 geneticin (G418; Gibco) (Zhang et al., 2010). Cells were incubated at 37°C (5% CO2) for a minimum of 1 day before electrophysiological recording.
Isolation of rabbit ventricular myocytes
Male New Zealand White rabbits (∼2–3 kg) were killed in accordance with the UK Home Office Animals (Scientific Procedures) Act, 1986; hearts were rapidly removed, and ventricular myocytes were isolated from the right ventricle using previously described methods (Hancox et al., 1993; Howarth et al., 1996). Before use, cells were kept at 4°C in Kraft–Brühe medium (Isenberg and Klockner, 1982). For the native IKr data presented in Figure 7, each drug concentration was tested on cells from two or more hearts.
Figure 7.
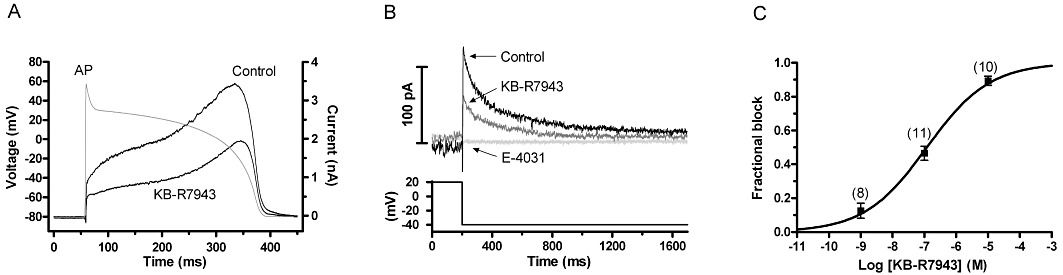
Effects of KB-R7943 on IhERG under AP clamp and on native IKr. (A) Representative records of IhERG under AP clamp conditions in control and in 100 nM KB-R7943, shown with the AP command waveform overlaid. AP commands were applied every 3 s, and online leak subtraction was performed using a P/4 protocol (Hancox et al., 1998). (B) Inhibition of native IKr by 100 nM KB-R7943. Membrane potential was stepped to −40 mV for 100 ms, +20 mV for 500 ms, then back to −40 mV for 1500 ms and finally back to the holding potential. The start-to-start interval for the protocol was 10 s. The figure focuses on the repolarization step, which elicited IKr tails. Representative traces are shown in control, following exposure to 100 nM KB-R7943 and following 10 µM E-4031. (C) Concentration–response relation for the action of KB-R7943 on IKr tails. Tail current amplitude was measured as the difference between peak tail current and the current level attained during the 100 ms to −40 mV. Numbers of replicates at each concentration are given in parentheses. Data were fitted with Equation 1 to give IC50 and nH values mentioned in the Results text.
Electrophysiological recording
Cells were superfused in an experimental chamber mounted on the stage of an inverted microscope (Eclipse TE2000-U, Nikon) with a Tyrode's solution containing (mM) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 5 HEPES (titrated to pH 7.45 with NaOH). For experiments in which a high [K+]e (94 mM) was used, KCl was substituted for NaCl in this solution to attain the total desired [K+]. Experimental solutions were applied using a home-built, warmed solution delivery system capable of changing the bathing solution surrounding a cell in <1 s (Levi et al., 1996). Patch pipettes (A-M Systems, Sequim, WA, USA) were pulled using a Narishige vertical electrode puller (Narishige PP-83) and heat-polished to a final resistance of 2–3 MΩ (Narishige MF-83). The pipette solution contained (mM) 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, 10 HEPES (titrated to pH of 7.2 with KOH). The pipette and external solutions were identical for IhERG and IKr measurements. Series resistance values typically lay between 4 and 7 MΩ and were compensated by ∼70% or more. Measurements were made at 35–37°C. Action potential voltage clamp (AP clamp) experiments employed a ventricular AP waveform identical to that used in prior experiments from our laboratory (e.g. Zhang et al., 2010).
KB-R7943 and SN-6
KB-R7943 and SN-6 (Tocris, Bristol, UK) were dissolved in dimethyl sulphoxide (DMSO) to produce stock solutions of 1 µM–100 mM, which were kept at −20°C. The stock solutions were diluted, as necessary, with Tyrode's solution to give a final DMSO concentration in experimental superfusate of 1/1000.
Docking simulations
KB-R7943 was docked into open state hERG pore homology models constructed on a MthK-based template (Witchel et al., 2004). Docking was performed using the Flexidock module of Sybyl. Docking runs were set up with at least 40 different configurations of the drug within the hERG pore binding pocket comprising the amino acid residues T623, S624, V625, G648, Y652, F656, S660 and all atoms within 5 Å. Rotation of side chain bonds within the residues listed was allowed during the docking. 60 000 generations of the genetic algorithm were calculated in each docking run.
Analysis
Data are presented as mean ± SEM. Statistical analyses were performed using Microsoft Excel (Microsoft Corporation) and Prism (GraphPad Software Inc.), whilst fits to particular data sets were performed either using Prism or Clampfit of pClamp 10.0 (Axon instruments, Molecular Devices). Comparisons were made using one-way anova, paired t-test or unpaired t-test as appropriate; P < 0.05 was taken as significant.
Concentration–response data were fitted with a Hill equation of the form:
| (1) |
where Fractional block refers to the degree of inhibition of IhERG or IKr by a given concentration of KB-R7943 (X, the logarithm of concentration); IC50 is [KB-R7943] producing half-maximal inhibition of IhERG or IKr, and nH is the Hill coefficient for the fit.
For voltage-dependent activation and inactivation, the half-maximal activation or inactivation voltage (V0.5) and slope factor (k) values were derived from fits to the relevant data with standard Boltzmann functions. Time courses of current activation and deactivation were determined respectively by fitting data with single or bi-exponential functions.
Results
Concentration-dependent inhibition of IhERG by KB-R7943
A standard voltage protocol (lower trace of Figure 1A), as used in previous studies of IhERG pharmacology from our laboratory (e.g. McPate et al., 2008; Zhang et al., 2010), was applied from a holding potential of −80 mV in initial experiments to investigate IhERG inhibition by KB-R7943. The protocol incorporated a brief (50 ms) pre-pulse from −80 to −40 mV before the +20 mV test command, in order to quantify the instantaneous current at −40 mV. Comparison between this instantaneous current and the peak outward IhERG tail amplitude on repolarization to −40 mV enabled the accurate measurement of IhERG tail amplitude (McPate et al., 2008; Zhang et al., 2010). Figure 1A shows representative traces illustrating the effect of 100 nM KB-R7943. The compound produced a rapid reduction in both end-pulse current and tail current amplitudes, with inhibition complete within ∼1–2 min (current amplitudes in KB-R7943 were similar at 1 and 2 min of drug exposure). The extent of inhibition of IhERG tails recorded at −40 mV was quantified for a range of concentrations between 1 nM and 30 µM in order to construct a concentration–response relation (Figure 1B). A fit to the resulting data with Equation 1 yielded an estimated IC50 value of 88.6 ± 25.7 nM and an nH value for the fit of 0.50 ± 0.06. However, the IC50 derived from the line of best fit to the data may slightly overestimate potency, as the experimentally derived extent of IhERG tail inhibition by 100 nM KB-R7943 with this protocol was 43.6 ± 2.6% (n= 12), suggestive of an IC50 closer to 100 nM. The inhibitory effect of KB-R7943 was partially reversible, with recovery to 91.8 ± 4.1% of control amplitude following exposure to 1 nM KB-R7943 (n= 5) and to 40.5 ± 6.5% of control amplitude following exposure to 30 µM (n= 6) of the compound.
Figure 1.
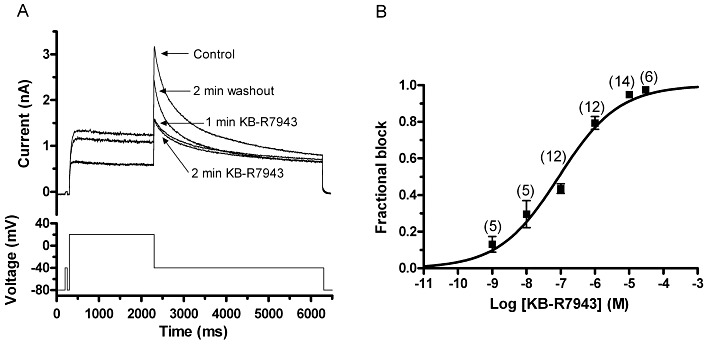
Concentration-dependent inhibition of IhERG by KB-R7943. (A) Upper panel shows representative IhERG traces in normal Tyrode's solution, after 1 and 2 min exposure to 100 nM KB-R7943 and after 2 min of washout. Lower panel shows voltage protocol (start-to-start interval of 12 s). (B) Concentration–response relation for the action of KB-R7943 on IhERG tails. Tail current amplitude was measured as the difference between peak tail current and the current level attained during the brief pre-pulse to −40 mV. Numbers of replicates at each concentration are given in parentheses. Data were fitted with Equation 1 to give IC50 and Hill coefficient (nH) values mentioned in the Results text.
Voltage-dependence of IhERG inhibition by KB-R7943
The protocol used to investigate voltage-dependence of inhibition of IhERG by KB-R7943 is shown in the lower traces of Figure 2A (control) and 2B (in 100 nM KB-R7943). From a holding potential of −80 mV, 2 s depolarizing voltage commands were applied to a range of test potentials between −40 and +40 mV, and tail currents were then recorded during a 4 s repolarization step to −40 mV (McPate et al., 2008; Zhang et al., 2010); 100 nM KB-R7943 appeared to produce a dual effect on IhERG. At potentials of approximately −10 mV and more positive to this, the compound produced a marked inhibition of IhERG both during and following the depolarizing voltage command (Figure 2A and B); however, at more negative potentials, this was not evident, and at −30 and −40 mV, an increase in elicited IhERG was seen (Figure 2A and B). Figure 2Ci and Di shows mean I–V relations for end-pulse (Figure 2Ci) and tail (Figure 2Di) currents. The data in Figure 2Di were used to derive half-maximal activation parameters: for the pooled mean data plot shown, the derived V0.5 and k values were, respectively, in control −15.0 and 6.9 mV, and in KB-R7943 these were −24.6 and 6.6 mV. When fits were made to data from individual cells, the mean V0.5 and k values then obtained were: control −14.1 ± 2.3 and 6.7 ± 0.5 mV (n= 6), and following KB-R7943, these were −24.9 ± 2.6 mV (P < 0.01 vs. control) and 6.2 ± 0.5 mV (P > 0.05 vs. control). Figure 2Cii and Dii shows, respectively, plots of mean fractional block of end-pulse current (Figure 2Cii) and tail current (Figure 2Dii). Both plots indicate marked voltage-dependence of the observed effect (P < 0.01 for each; one-way anova across the potential range from −40 to +40 mV). In Figure 2Dii, activation curves for IhERG are also plotted. The range of steepest change in fractional inhibition coincides with the steep part of the activation curves. The leftward shift in activation with KB-R7943 is likely to account for the increase in current seen at negative voltages in the membrane potential range examined.
Figure 2.
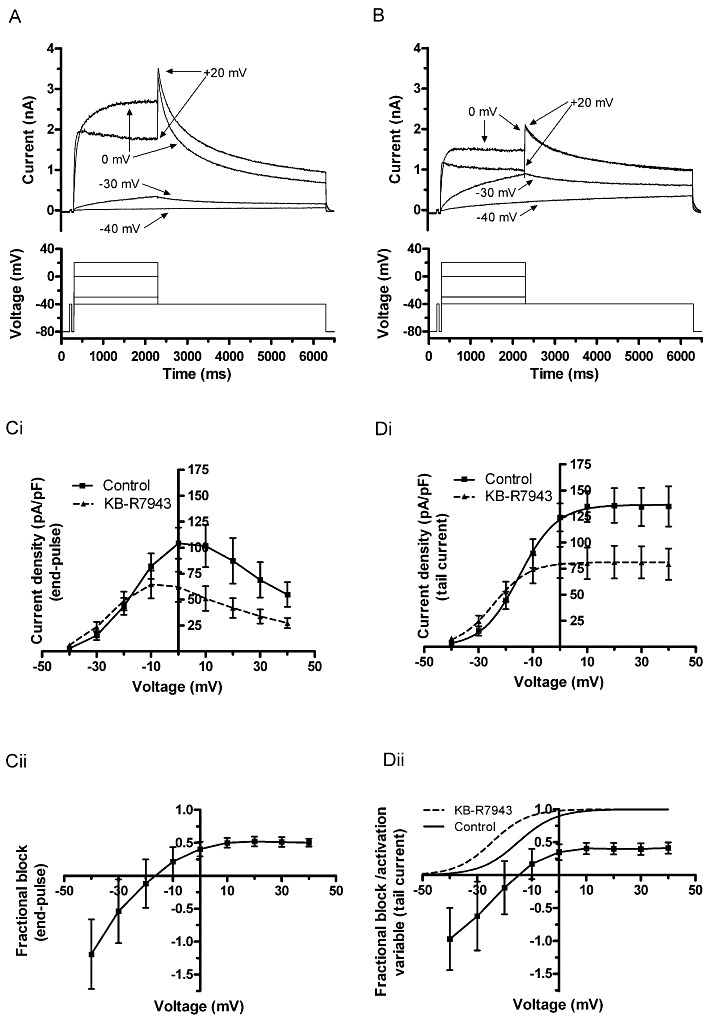
Voltage-dependence of IhERG inhibition by KB-R7943. Upper traces show representative IhERG records in control (A) and in the presence of 100 nM KB-R7943 (B). Lower traces show corresponding voltage steps in the experimental protocol. Currents were evoked by a series of 10 mV increments of step depolarizations between −40 and +40 mV from a holding potential of −80 mV. However, for clarity of display, only selected steps are shown. (Ci) Mean I–V relation for end-pulse currents in control and in the presence of 100 nM KB-R7943 (n= 6 cells). (Cii) Mean fractional block of end-pulse currents against voltage. (Di) Mean I–V relation for IhERG tails in control and in the presence of KB-R7943 (n= 6 cells). Data were fitted with a Boltzmann equation to give V0.5 and k values in the Results text. (Dii) Corresponding plot of mean fractional block of tail currents. Superimposed on this plot are continuous plots describing voltage-dependent activation of IhERG in control and KB-R7943.
Effects of KB-R7943 on time-dependent activation and deactivation of IhERG
An ‘envelope of tails’ protocol was used to investigate the development of inhibition of IhERG by KB-R7943 with time following membrane depolarization (e.g. Milnes et al., 2003a; Zhang et al. 2010). The protocol is shown as the lower set of traces in each of Figure 3A and B, with representative sets of current traces shown above the voltage protocol. As is characteristic of IhERG with this protocol (e.g. Milnes et al., 2003a; Zhang et al., 2010), in both control and drug-containing solutions, IhERG tail amplitude increased progressively as the duration of the depolarizing command to +20 mV increased, with current amplitude in 100 nM KB-R7943 smaller than that for the corresponding command pulse in control solution. Notably, even with relatively brief depolarizations, current was suppressed by KB-R7943. Figure 3C shows a plot of mean time course of development of the currents in control and KB-R7943, with monoexponential fitting to derive activation time constant (τ) values. Fits to the pooled mean data yielded activation τ values of ∼59 and ∼90 ms in control and KB-R7943, whilst the mean values derived from fits to data from each experiment yielded mean τ values of 60.7 ± 7.7 ms (control) and 99.7 ± 15.7 ms (KB-R7943; n= 8; P < 0.01 vs. control). Figure 3D shows a plot of fractional inhibition of IhERG against corresponding test pulse durations, focusing on the first ∼110 ms of the protocol. There was little difference in inhibition at the different time points [anova analysis across the full range of test pulse durations (up to 810 ms) showed no significant differences; P > 0.05; n= 8]. Thus, IhERG block by KB-R7943 was evident rapidly on membrane depolarization. Effects of the agent on current deactivation were quantified by assessing deactivation time-constant (τ) values for tail currents elicited in control and 100 nM KB-7943, by the protocol shown in Figure 1A: the derived τ values were τfast of 294 ± 24 ms and τslow of 1969 ± 147 ms in control and τfast of 350 ± 26 ms and τslow of 2332 ± 122 in KB-R7943 (n= 6; P < 0.05 for both τfast and τslow). In summary, KB-R7943 slowed the deactivation time course of IhERG; it also produced a modest slowing of IhERG activation at a test voltage (+20 mV) at which full IhERG activation could be attained in both control and drug conditions (Figure 2D).
Figure 3.
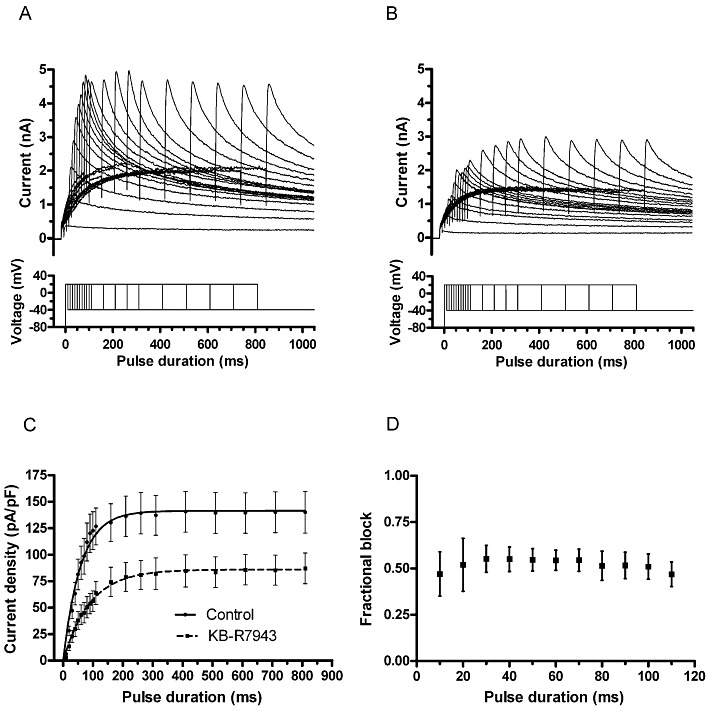
Time course of IhERG activation and KB-R7943. (A, B) Upper traces show representative currents elicited by envelope of tails protocol shown as lower traces in each panel: (A) currents in control and (B) during exposure to KB-R7943. (C) Mean plots of current amplitude against depolarizing pulse duration (n= 8), fitted with a single-exponential equation to yield τ values given in the Results text. (D) Plot of mean level of fractional block, focusing on the first 110 ms of the protocol. There was no statistically significant time-dependent difference in this (one-way anova).
KB-R7943 and IhERG inactivation
Effects of KB-R7943 (100 nM) on the voltage-dependence of IhERG inactivation were assessed using the ‘availability’ protocol shown in Figure 4A. This was comprised of an initial depolarizing step to +40 mV to activate and inactivate IhERG, followed by 2 ms repolarization steps to potentials ranging from +50 to −140 mV, to relieve channel inactivation to differing extents; the membrane potential was then stepped back to +40 mV for 100 ms, and the amplitude of resulting current transients was used to assess IhERG availability (cf. Milnes et al., 2003a; McPate et al., 2005). Figure 4B shows mean availability plots, from which inactivation V0.5 values were obtained for control solution and following exposure to KB-R7943. The mean V0.5 values derived from fits to data from each of eight cells were −48.4 ± 3.7 mV in control and −57.0 ± 6.1 mV in KB-R7943 (P > 0.05), whereas the k values were 26.9 ± 2.5 and 24.1 ± 2.3 mV, respectively (P > 0.05). Thus, there was no statistically significant effect of KB-R7943 on the voltage-dependence of inactivation. The effect of promoting increased IhERG inactivation on the inhibitory action of KB-R7943 was assessed using the protocol shown in Figure 4C (lower trace; cf. Ridley et al., 2003). From a holding potential of −80 mV, membrane potential was stepped first to 0 mV for 3 s, followed by 4 s of depolarization to +80 mV and then a return to 0 mV. In both control and 100 nM KB-R7943, depolarization to 0 mV elicited sizeable IhERG, which was reduced when membrane potential was further stepped to +80 mV (due to more channels becoming inactivated) and which then increased on the step back to 0 mV. Currents were sampled just before the end of the first step to 0 mV, the step to +80 mV and of the return to 0 mV. Measurements were made in control solution, during a single application of the protocol after a 2 min equilibration period in KB-R7943 and after application of a high concentration (5 µM) of the selective hERG/IKr inhibitor E-4031 to allow subtraction of residual leak current. Figure 4D compares mean levels of fractional block of IhERG at the three time points (n= 7), showing there to be no significant difference between these (P > 0.05; anova; cf. Ridley et al., 2003).
Figure 4.
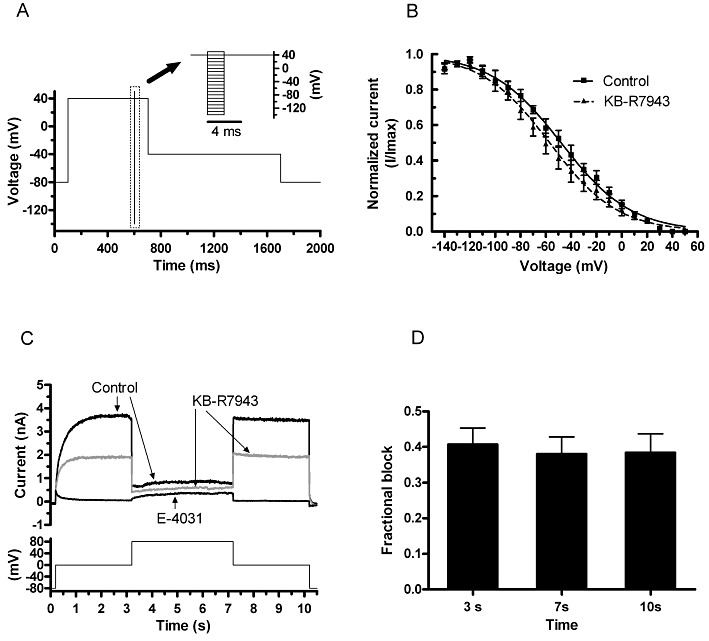
IhERG inactivation and KB-R7943. (A) Protocol used to investigate IhERG‘availability’. From −80 mV, the membrane potential was stepped to +40 mV for 500 ms; this was then followed by 2 ms repolarization steps to potentials ranging from −140 to +50 mV. Membrane potential was then stepped back to +40 mV for 100 ms (inset shows portion of protocol encompassing the transition to and from repolarizing steps). The amplitude of current transients elicited by the second step to +40 mV was used to assess IhERG availability. (B) Plots against voltage of normalized current transient amplitude in control and in 100 nM KB-R7943 (n= 8 cells); a Boltzmann equation was used to derive the V0.5 and k values shown in the Results text. (C) Three-step protocol used to assess the effect of promoting IhERG inactivation on the action of KB-R7943. Protocol is shown as lower trace; upper traces show representative currents in control, in the presence of 100 nM KB-R7943 and following exposure to 5 µM E-4031. (D) Mean level of fractional block at three time points during the protocol (n= 7). The three time points are given with reference to the start of the acquisition period (200 ms before the initial depolarization to 0 mV).
Effect of mutation of S6 aromatic amino acid residues on the action of KB-R7943
For a number of high-affinity hERG channel inhibitors, IhERG blockade involves drug binding within the inner cavity at a site involving aromatic residues (Y652 and F656) that are rendered accessible on channel gating (e.g. Mitcheson et al., 2000; Sanguinetti and Mitcheson, 2005; Sanguinetti and Tristani-Firouzi, 2006). Alanine mutants of these residues (Y652A and F656A) were therefore employed in order to explore further IhERG inhibition by KB-R7943 (cf.Milnes et al., 2003a; Ridley et al., 2004). Figure 5A shows data for Y652A-hERG. The same voltage protocol was employed as for WT IhERG in Figure 1. As indicated in Figure 5Ai and Aii, 100 nM KB-R7943 produced markedly less inhibition of Y652A-hERG in comparison with WT-hERG. Three KB-R7943 concentrations (100 nM, 1 µM and 10 µM) were tested on Y652A-hERG, and mean data from these experiments are shown in Figure 5B (with the concentration–response relation for WT IhERG shown overlaid). A fit to the data with Equation 1 gave an estimated IC50 for Y652A-hERG of 1.13 ± 0.14 µM, with an nH of 1.08 ± 0.16. Thus, the potency of IhERG inhibition by KB-R7943 was ∼13-fold lower for Y652A-hERG than for WT-hERG. The F656A-hERG mutant is comparatively poorly expressing (Mitcheson et al., 2000; Milnes et al., 2003a), and so, as in previous studies from our laboratory (e.g. Milnes et al., 2003a; Ridley et al., 2004), this mutant was studied using a high [K+]e (94 mM), employing the voltage protocol shown in Figure 5Ciii. Additional experiments were performed to assess inhibition of WT IhERG under these conditions (Figure 5Ci and D). The estimated IC50 for WT IhERG was 1.20 ± 0.02 µM, with an nH of 0.75 ± 0.01, indicating reduced potency of the compound against WT IhERG with raised [K+]e (cf. Wang et al., 1997; Ridley et al., 2004; Barrows et al., 2009); 1 µM KB-R7943 produced comparatively little inhibition of F656A IhERG under these conditions (compare Figure 5Ci and Cii); the estimated IC50 for inhibition of F656A-hERG was 11.09 ± 0.23 µM (with an nH of 0.84 ± 0.01), ∼9-fold higher than its WT control (Figure 5D). In order to investigate whether the compound may also be able to interact with the base of the hERG channel pore helix (cf.Mitcheson et al., 2000), additional experiments were performed using the S624A mutation, using the same recording conditions and protocol as used to study outward WT IhERG (Figure 6A and B). Figure 6A shows effects of 100 nM KB-R7943 on S624A IhERG; the compound reduced the tail current by ∼30%. Figure 6B shows concentration–response data for the S624A mutant: the derived IC50 was 216.7 ± 31 nM (nH= 1.13 ± 0.17), ∼2.4-fold that for WT IhERG. Considered together, these observations implicate inner cavity binding interactions in the IhERG inhibitory effect of KB-R7943.
Figure 5.
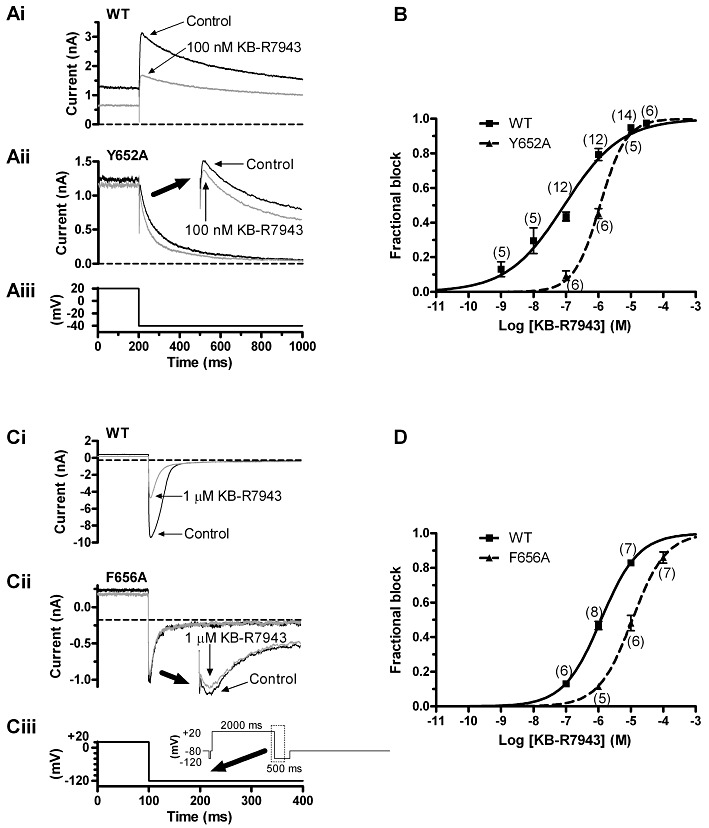
Effects of the Y652A and F656A mutations on the action of KB-R7943. (A) The traces in panels Ai and Aii show, respectively, WT IhERG and Y652A IhERG elicited in the absence and presence of 100 nM KB-R7943. The corresponding voltage protocol is shown in panel Aiii. (B) Mean data indicating effects of three concentrations of KB-R7943 on Y652A IhERG, fitted by Equation 1 to give the IC50 and nH values given in the text. The concentration–response relation for WT IhERG (identical to that shown in Figure 1) is shown overlain, for comparative purposes. (C) The traces in panels Ci and Cii show, respectively, WT IhERG and F656A IhERG elicited in the absence and presence of 1 µM KB-R7943. The corresponding voltage protocol is shown in panel Ciii and its inset. Experiments performed with high (94 mM) [K+]e. (D) Mean data indicating effects of three concentrations of KB-R7943 on each of WT and F656A IhERG tails, measured at −120 mV, fitted by Equation 1 to give the IC50 and nH values given in the text. For A and C, the horizontal dashed lines are drawn at the level of the current at the pre-pulse of −40 or −120 mV, against which peak tail current amplitudes were measured. For B and D, numbers in parentheses indicate numbers of replicates at each drug concentration tested on WT and mutant channels.
Figure 6.
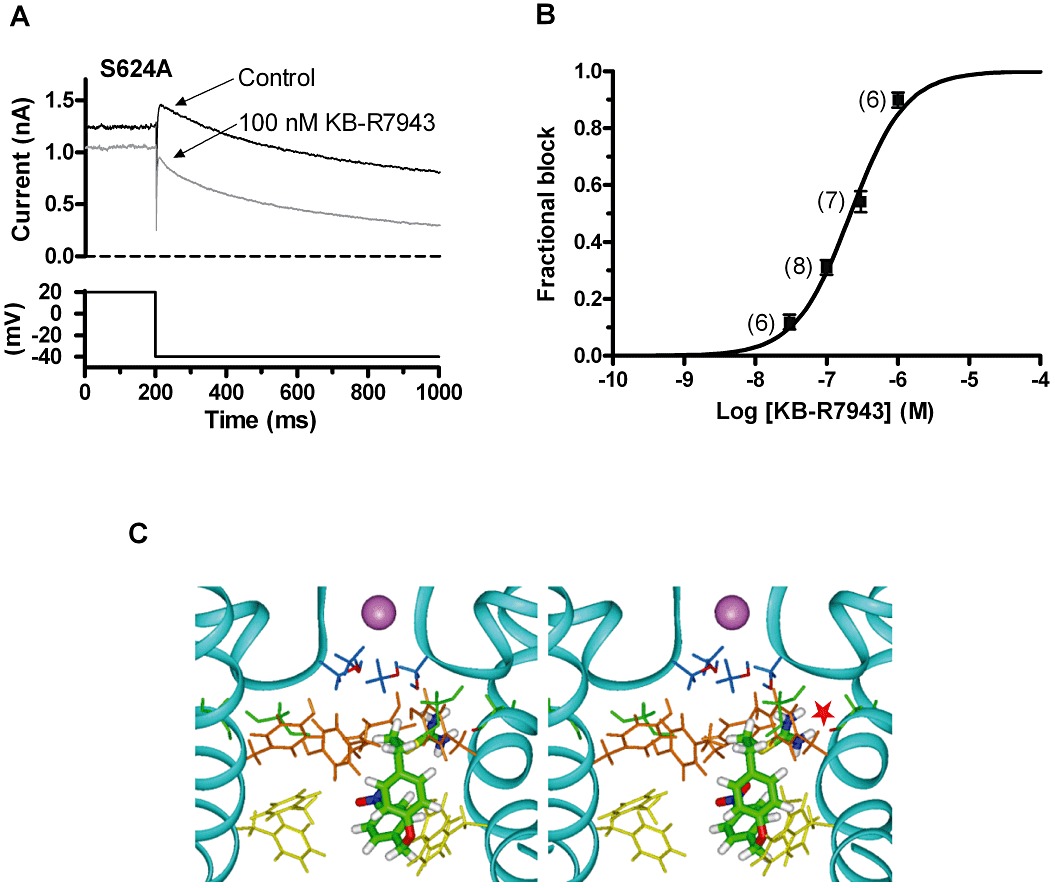
Effects of the S624A mutant on the action of KB-R7943 and simulated docking to the open hERG channel. (A) Representative records of S624A IhERG tails (upper traces) elicited on repolarization to −40 mV following a 2 s depolarization to +20 mV from −80 mV (lower trace shows corresponding portion of the voltage protocol) in control solution (standard Tyrode's solution) and following exposure to 100 nM KB-R7943. (B) Mean ± SEM fractional inhibition by SN-6 concentrations between 30 nM and 1 µM, fitted by Equation 1 to give the IC50 and nH values presented in the Results text. Numbers in parentheses indicate numbers of replicates at each drug concentration. (C) Stereoview of representative low energy score configuration for KB-R7943 docked into the open hERG channel pore homology model based on the MthK crystal structure. The protein backbone for two of the four subunits of the channel pore tetramer is indicated with a ribbon and a K+ ion (shown in mauve) occupies the S3 site in the selectivity filter. Gly648 (green) and the side chains of S624 (blue), Y652 (orange) and F656 (yellow) are shown as sticks. The red star marks a potential hydrogen bond between the thiourea group of KB-R7943 and the backbone carbonyl of G648 (green with red carbonyl oxygen).
In order to pursue further the nature of the interactions between KB-R7943 and hERG, ligand docking simulations were conducted in which interactions between the drug and channel were investigated in an open-channel configuration homology model (see Methods). Low energy binding configurations for KB-R7943 were dominated by extended conformations in which the drug lay approximately parallel to the pore axis with the positively charged thiourea group oriented towards the selectivity filter and the nitrobenzyl group oriented towards the cytoplasmic side of the pore (Figure 6C). The drug bound within a pocket comprising the side chains of Y652 and F656 enabling stacking interactions between aromatic rings on the drugs and the aromatic side chains of Y652 and F656; this is consistent with a significant contribution to binding with these residues and is in good agreement with the Y652A and F656A data shown in Figure 5. In low-energy configurations, the positively-charged thiourea group of KB-R7943 was also seen either to extend close to the ring of hydroxyl side chains of S624 or to locate near the C-terminus of a pore helix and to make a hydrogen bond with the backbone carbonyl group of G648. An additional thiourea hydrogen bond with the S624 side chain of the same subunit is possible in these poses, but would be anticipated to make only a small contribution to the binding energy, given the modest effect of the S624A mutation on IhERG block.
Effects of KB-R7943 under AP clamp and on native IKr
In order to evaluate the effect of KB-R7943 on IhERG elicited by a dynamic physiological waveform, action potential (AP) voltage-clamp experiments were performed. Current measurements were made first in control solution and then in the presence of 100 nM KB-R7943. IhERG throughout the repolarizing phase of the AP, was reduced in the presence of KB-R7943 (Figure 7A), with peak current reduced by 43.5 ± 1.9% (n= 7). The voltage at which maximal IhERG occurred was shifted by approximately −9.8 ± 1.2 mV in the presence of KB-R7943, which is similar to the extent of shift of IhERG activation V0.5 seen in Figure 2. In a subsequent set of experiments, inhibition by KB-R7943 of native ventricular IKr was assessed. From a holding potential of −80 mV, membrane potential was briefly stepped to −40 mV (for 100 ms; both to inactivate fast Na+ channels and provide a reference current level for tail current measurement), then to +20 mV (for 500 ms), then back to −40 mV for 1.5 s to elicit IKr tails. Figure 7B shows representative tail current traces in control solution, following 100 nM KB-R7943 and after subsequent application of a high concentration (10 µM) of E-4031. The IKr tail was markedly inhibited by KB-R7943, and the current remaining was abolished by E-4031 (thereby confirming identity of the tail current as IKr); 100 nM KB-R7943 inhibited native IKr by 46.5 ± 4.1% (n= 11), which is similar to the extent of WT IhERG inhibition produced by this concentration (P > 0.05; unpaired t-test). Figure 7C summarizes the effects of three concentrations of KB-R7943 on native IKr (1 nM, 100 nM and 10 µM). The data were fitted with Equation 1 to estimate an IC50 value for IKr inhibition: the derived value was 120.3 ± 27.5 nM, with an nH value for the fit of 0.44 ± 0.04. As observed for IhERG, deactivation of IKr was slowed by KB-R7943: with τfast of 81 ± 7 ms and τslow of 499 ± 65 ms in control and τfast of 144 ± 22 ms and τslow of 754 ± 192 ms in 100 nM KB-R7943 (P < 0.05 for each time constant; n= 6).
hERG inhibition by the structurally related NCX inhibitor SN-6
SN-6 (2-[4-(4-nitrobenzyloxy) benzyl] thiazolidine-4-carboxylic acid ethyl ester) is an NCX inhibitor that shares structural similarity to KB-R7943 (Figure 8A) and inhibits NCX1 (Iwamoto et al., 2004) and native cardiac INCX, apparently with improved selectivity (Niu et al., 2007). Therefore, in a final series of experiments, we investigated the propensity of this compound to inhibit IhERG. Figure 8B shows the effect of 10 µM SN-6 on the amplitude of IhERG tails on repolarization to −40 mV from +20 mV, whilst Figure 8C shows mean data across a range of concentrations from 1 nM to 100 µM. SN-6 produced a concentration-dependent inhibition of IhERG but was considerably less potent than KB-R7943 in this regard, with an estimated IC50 of 10.4 ± 3.3 µM and nH of 0.25 ± 0.03.
Figure 8.
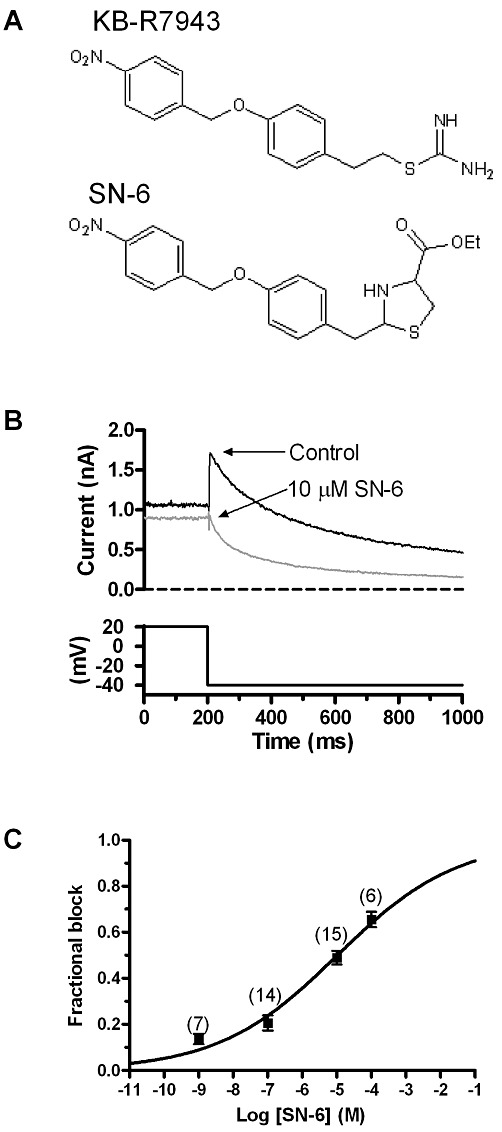
Effects of SN-6 on IhERG. (A) Structural formulae of KB-R7943 and SN-6 (structures from http://www.tocris.com). (B) Representative records of IhERG tails (upper traces) elicited on repolarization to −40 mV following a 2 s depolarization to +20 mV from −80 mV (lower trace shows corresponding portion of the voltage protocol) in control solution and following exposure to 10 µM SN-6. (C) Mean ± SEM fractional inhibition by SN-6 concentrations between 1 nM and 100 µM, fitted by Equation 1 to give the IC50 and nH values presented in the Results text. Numbers in parentheses indicate the numbers of replicates at each drug concentration.
Discussion and conclusions
Although KB-R7943 is known not to be entirely selective for NCX (Amran et al., 2003; Watanabe et al., 2006), until now there has been no direct evidence for IhERG/IKr inhibition by this compound. Examination of previously published data by Tanaka et al. (2002) on the effect of 10 µM KB-R7943 on guinea-pig composite IK (both in respect of raw currents and ‘tail’ current–voltage relations in Figure 4 of that study) indicates that IKs is likely to have predominated in their recordings. Accordingly, the KB-R7943-sensitive current in that study is likely to have been comprised predominantly of IKs, although experimental data under selective recording conditions are not shown (Tanaka et al., 2002). Our data demonstrate unequivocally that KB-R7943 inhibits both hERG and native IKr at nanomolar to micromolar concentrations, with IC50 values of ∼100 nM. The majority of IhERG inhibitors exhibit IC50 values in the µM range (Shah, 2005), and in this context, KB-R7943 can fairly be considered to be a potent IhERG inhibitor. We observed that, in addition to inhibiting IhERG, KB-R7943 also modified voltage-dependent activation properties of the underlying channels. A similar dual effect has been observed for both low and high potency inhibitors that inhibit hERG in a gating-dependent fashion. For example, both desethylamiodarone and 4-aminopyridine inhibit IhERG at the majority of tested voltages, whilst producing an apparent augmentation of current at negative voltages – associated with a leftward voltage shift in current activation, as seen here for KB-R7943 (Ridley et al., 2003; Zhang et al., 2010). This feature of KB-R7943 inhibition is concordant with a gating-dependent channel blocking mechanism; however, our data from the envelope of tails protocol (Figure 3) indicate that the drug produces substantial hERG channel inhibition rapidly on membrane depolarization, and, on that basis, a possible contribution of closed channel block cannot entirely be excluded. The data in Figure 4 suggest that KB-R7943 exhibits little preference for inactivated over activated channels (cf. Ridley et al., 2003; Zhang et al., 2010), whilst the positive (as opposed to inverse) voltage-dependence and neutral time-dependence of inhibition seen here suggest that a preferential closed channel blocking mechanism is unlikely (cf.Milnes et al., 2003b). In addition, the fact that altering the fraction of inactivated WT IhERG channels did not significantly change the extent of observed IhERG inhibition (Figure 4) is concordant with the notion that decreased WT IhERG block with high [K+]e and inward K+ flux (Figure 5) may be accounted for by a direct interaction (electrostatic repulsion or ‘knock off’) between the permeant ion and KB-R7943 (Wang et al., 1997; Ridley et al., 2004; Barrows et al., 2009). It is notable that the Hill coefficient (nH) values for the concentration-dependence of KB-R7943 inhibition of WT IhERG (Figure 1) and native IKr (Figure 7) lay close to 0.5. This raises the possibility that KB-R7943 block of WT IhERG/IKr might involve negative co-operativity (cf. McPate et al., 2006). However, whilst this was seen for outward IhERG with normal [K+]e, when [K+]e was elevated, the nH was closer to 1 (0.75) for inward IhERG measurement in high [K+]e.
The impact of alanine substitution at Y652 and F656 seen here for KB-R7943 (Figure 5) is significant, though less marked than observed previously for some other high-affinity blockers (e.g. the Y652A mutation increased the IC50 for MK-499 block of IhERG by ∼94-fold, whilst F656A increased the IC50 by ∼650-fold, with both mutations also profoundly affecting the potency of cisapride and terfenadine; Mitcheson et al., 2000). Pore helix mutations, including S624A, also influence significantly the potency of IhERG inhibition by high-affinity inhibitors including methanesulphonanilides as well as that of cisapride and terfenadine (Mitcheson et al., 2000; Kamiya et al., 2006; Kamiya et al., 2008). Here, the S624A mutation exerted a relatively modest effect on blocking potency (Figure 6), suggesting that this residue may not be a key constituent of the drug binding site. It is notable, however, that although it was studied with an identical protocol and measurement conditions to WT IhERG, S624A hERG exhibited an nH value close to 1 for concentration-dependent IhERG inhibition, concordant with some influence of the residue on the manner of drug–channel interaction. Y652A-hERG also exhibited an nH for concentration-dependent inhibition close to 1. Thus, in electrophysiological experiments manipulation of the direction/magnitude of K+ flux and mutagenesis of S624 and Y652 residues influenced the steepness of observed inhibitory concentration-dependence, whilst Y652 and F656 were implicated as significant components of the KB-R7943 binding site. This was borne out by our docking simulations (Figure 6), which, additionally, demonstrate a likely orientation of the KB-R7943 molecule within the open channel pore. Stacking interactions between aromatic rings on the drugs and the aromatic side chains of Y652 and F656, together with potential interactions involving the positively charged thiourea group of KB-R7943 (electrostatic interactions with the pore helix dipole charge and hydrogen bond interactions with G648 carbonyl), may contribute to the relatively high affinity of this compound. Additional docking simulations (not shown) showed that SN-6 is unable to make equivalent hydrogen bond interactions to those seen for KB-R7943, and so these may contribute to the observed difference in inhibitory potency between KB-R7943 (Figures 1 and 7) and SN-6 (Figure 8).
In the first study of KB-R7943, the compound was found to inhibit Na+-dependent Ca2+ uptake into NCX1-transfected CC39 cells, rat aortic smooth muscle cells and cardiac myocytes, with IC50 values ranging between 1.6 and 2.4 µM (Iwamoto et al., 1996), whilst Na+i-dependent Ca2+ influx into Na+-loaded cardiac sarcolemmal vesicles was inhibited with an IC50 of 5.4 µM, and Na+o-dependent Ca2+ efflux was inhibited with an IC50 of 11–13 µM (Iwamoto et al., 1996). When INCX from guinea-pig ventricular myocytes was elicited in a unidirectional manner, outward and inward INCX were inhibited, respectively, with an IC50 of ∼320 nM and 17 µM (Watano et al., 1996). For bi-directional guinea-pig ventricular INCX, the reported IC50 was ∼1 µM (Kimura et al., 1999), whilst KB-R7943 was able to inhibit outward canine ventricular INCX more extensively than inward INCX, but with EC50 values, respectively, of ∼4.7 and 3.4 µM (Birinyi et al., 2005). As highlighted in the Introduction, the compound can also inhibit cardiac Na+ and Ca2+ and inwardly rectifying K+ currents at micromolar concentrations (Watano et al., 1996; Tanaka et al., 2002). There is evidence for additional off-target effects of the compound from experiments on non-cardiac preparations. For example, KB-R7943 inhibits recombinant Cav1.2 channels with an IC50 of ∼7.3 µM (Ouardouz et al., 2005) and canonical transient receptor potential channels with IC50 values of 460 nM (TRPC3), 710 nM (TRPC6) and 1.38 µM (TRPC5) (Kraft, 2007). At 10–20 µM, the compound can significantly inhibit store-operated Ca2+ entry into cultured neurons and astrocytes (Arakawa et al., 2000). In cultured hippocampal neurons, it inhibits NMDA receptor-mediated increases in cytosolic calcium with an IC50 of 13.4 µM (Brustovetsky et al., 2011), and it can inhibit mitochondrial Ca2+ uptake [with an IC50 of 5.5 µM in permeabilized HeLa cells (Santo-Domingo et al., 2007)] and mitochondrial complex I (Brustovetsky et al., 2011). Another study, focusing on acutely isolated CA1 hippocampal neurons, has reported inhibition of two populations of NMDA channels with high-affinity (IC50 of 0.8 µM) and low-affinity (IC50 of ∼11 µM) block by KB-R7943 (Sobolevsky and Khodorov, 1999). KB-R7943 has also been reported to inhibit a non-selective cation channel implicated in chemosensory transduction (with an IC50 of ∼11.7 µM; Pezier et al., 2009) and to activate vascular large conductance Ca2+-activated K+ channels (EC50 of ∼6.8 µM; Liang et al., 2008). Significantly, when the results of the present study are considered against the background of the compound's overall profile of effects, it is clear that (i) KB-R7943 inhibits hERG/IKr at least as potently has been reported previously for cardiac INCX, and (ii) none of the above-mentioned off-target effects is as potent as that seen in the present study for hERG/IKr. Thus, hERG/IKr channel inhibition appears to be the most potent off-target pharmacological action thus far identified for KB-R7943.
Given the results of the present study, it seems reasonable to conclude that in experiments on cardiac cells, tissues and intact hearts significant IhERG/IKr inhibition can be anticipated to occur at NCX-blocking concentrations of KB-R7943. It follows, therefore, that the propensity of the agent to inhibit hERG/IKr channels needs to be taken into account when interpreting experimental data with the compound obtained from cardiac preparations from species in which IKr participates in AP repolarization [these include human, dog, rabbit and guinea-pig (Sanguinetti and Jurkiewicz, 1990; Tamargo et al., 2004)] or pacemaking (Zaza et al., 1997; Mitcheson and Hancox, 1999; Sato et al., 2000). For example, it is noteworthy that a low concentration of KB-R7943 (i.v. injection to plasma levels of ∼31 nM) has been reported to prolong atrial effective refractory period (AERP) of anaesthetized dogs (Miyata et al., 2002), an effect that was attributed to action(s) of KB-R7943 other than NCX inhibition (Miyata et al., 2002), and that is concordant with hERG/IKr inhibition [IKr participates in atrial AP repolarization and pharmacological IKr blockade is known to exert an atrial class III anti-arrhythmic action (Torp-Pedersen et al., 2000; Tamargo et al., 2004; Doggrell and Hancox, 2005)]. The hERG/IKr-inhibition by KB-R7943 demonstrated here also explains recently reported membrane potential depolarization of rabbit AV node cells at compound concentrations sufficient to inhibit inward INCX (Cheng et al., 2011). This highlights the importance of using alternative/complementary interventions to KB-R7943 to alter NCX function in investigations of the role of the NCX in cardiac pacemaking (e.g. Sanders et al., 2006; Cheng et al., 2011). It is worth noting that in some experimental settings, at some drug concentrations, the ability of KB-R7943 to exert multiple actions may mean that overt AP or QT interval prolongation due to diminished IKr may not be evident, and hence, the consequences of IKr blockade may feasibly be overlooked, despite contributing to the overall actions of the compound. Indeed, if the overall experimental objectives of a cardiac study employing KB-R7943 do not require that IKr be present, it may be prudent to select a species in which IKr is not important for ventricular repolarization or to employ an alternative means of NCX inhibition. Considered in isolation, our KB-R7943 data might be interpreted to suggest that benzyloxyphenyl-based pharmacological strategies for NCX inhibition would be hampered by concurrent potent hERG inhibition. However, our findings with SN-6 (Figure 8) indicate that this need not necessarily occur to the extent that is evident here for KB-R7943. SN-6 has been reported to inhibit 45Ca2+ uptake in NCX1-transfected fibroblasts with an IC50 of 2.9 µM (Iwamoto et al., 2004) and to inhibit outward and inward components of bi-directional INCX from guinea-pig ventricular myocytes with IC50 values of 2.3 and 1.9 µM (Niu et al., 2007). Thus, the compound is comparable with KB-R7943 in its NCX-inhibitory potency, but the present study shows it to be ∼100-fold less potent as a hERG inhibitor; it also affects other currents less potently than KB-R7943 (Niu et al., 2007). It remains to be established whether benzyloxyphenyl-based NCX inhibitors entirely devoid of hERG activity can be produced. However, SN-6 may offer a useful complementary or alternative NCX inhibitor to KB-R7943 for use in experiments on cardiac preparations that contain IKr/hERG channels. An alternative to either compound is SEA0400, which inhibits INCX with greater potency than does KB-R7943 (Tanaka et al., 2002; Birinyi et al., 2005) and for which the available data suggest exerts little effect on cardiac composite delayed rectifier K+ current (Tanaka et al., 2002). Finally, it is worth noting that hERG channel expression is not restricted to the heart (Sanguinetti and Tristani-Firouzi, 2006; Larsen, 2010); consequently, the hERG-channel blocking action of KB-R7943 may also need to be taken into account when the compound is used in experimental studies of hERG-expressing non-cardiac tissue types.
Acknowledgments
The authors thank the British Heart Foundation for financial support (PG/08/036, PG/10/17) and Professor Clive Orchard for valuable discussion and support. The authors also thank Drs S Choisy and P Barman for assistance with cardiomyocyte isolation and Mrs Lesley Arberry for technical assistance.
Glossary
- AP
action potential
- AV
atrioventricular
- DMEM
Dulbecco's minimum essential medium
- nH
Hill coefficient
- hERG
human Ether-à-go-go-Related Gene
- IhERG
hERG potassium channel ionic current
- IKr
rapid delayed rectifier K+ current
- k
slope factor for voltage-dependent activation or inactivation relations
- KB-R7943
2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methane sulphonate
- NCX
sodium calcium exchange
- SEA0400
2-[4-[(2,5-difluorophenyl)methoxy]phenoxy]-5-ethoxyaniline
- SN-6
2-[[4-[(4-nitrophenyl)methoxy]phenyl]methyl]-4-thiazolidinecarboxylic acid ethyl ester
- TRPC
canonical transient receptor potential channel
- Vm
membrane potential
- V0.5
half maximal activation or inactivation voltage
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amran MS, Homma N, Hashimoto K. Pharmacology of KB-R7943: a Na+-Ca2+ exchange inhibitor. Cardiovasc Drug Rev. 2003;21:255–276. doi: 10.1111/j.1527-3466.2003.tb00121.x. [DOI] [PubMed] [Google Scholar]
- Arakawa N, Sakaue M, Yokoyama I, Hashimoto H, Koyama Y, Baba A, et al. KB-R7943 inhibits store-operated Ca2+ entry in cultured neurons and astrocytes. Biochem Biophys Res Commun. 2000;279:354–357. doi: 10.1006/bbrc.2000.3968. [DOI] [PubMed] [Google Scholar]
- Barrows B, Cheung K, Bialobrzeski T, Forster J, Schulze J, Miller A. Extracellular potassium dependency of block of HERG by quinidine and cisapride is primarily determined by the permeant ion and not by inactivation. Channels. 2009;3:240–249. [PubMed] [Google Scholar]
- Birinyi P, Acsai K, Banyasz T, Toth A, Horvath B, Virag L, et al. Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:63–70. doi: 10.1007/s00210-005-1079-x. [DOI] [PubMed] [Google Scholar]
- Brustovetsky T, Brittain MK, Sheets PL, Cummins TR, Pinelis V, Brustovetsky N. KB-R7943, an inhibitor of the reverse Na+/Ca2+ exchanger, blocks N-methyl-D-asparate receptor and inhibits mitochondrial complex I. Br J Pharmacol. 2011;162:255–270. doi: 10.1111/j.1476-5381.2010.01054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Smith GL, Hancox JC, Orchard CH. Inhibition of spontaneous activity of rabbit atrioventricular node cells by KB-R7943 and inhibitors of sarcoplasmic reticulum Ca2+ ATPase. Cell Calcium. 2011;49:56–65. doi: 10.1016/j.ceca.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipolo R, Beauge L. Sodium/calcium exchanger: influence of metabolic regulation on ion carrier interactions. Physiol Rev. 2006;86:155–203. doi: 10.1152/physrev.00018.2005. [DOI] [PubMed] [Google Scholar]
- Doggrell SA, Hancox JC. Ibutilide – recent molecular insights and accumulating evidence for use in atrial flutter and fibrillation. Expert Opin Investig Drugs. 2005;14:655–669. doi: 10.1517/13543784.14.5.655. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Levi AJ, Lee CO, Heap P. A method for isolating rabbit atrioventricular node myocytes which retain normal morphology and function. Am J Physiol. 1993;265:H755–H766. doi: 10.1152/ajpheart.1993.265.2.H755. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Levi AJ, Witchel HJ. Time course and voltage dependence of expressed HERG current compared with native ‘rapid’ delayed rectifier K current during the cardiac ventricular action potential. Pflugers Arch. 1998;436:843–853. doi: 10.1007/s004240050713. [DOI] [PubMed] [Google Scholar]
- Hancox JC, McPate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther. 2008;119:118–132. doi: 10.1016/j.pharmthera.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Howarth FC, Levi AJ, Hancox JC. Characteristics of the delayed rectifier potassium current (IK) compared in myocytes isolated from the atrioventricular node and ventricle of the rabbit heart. Pflugers Arch. 1996;431:713–722. doi: 10.1007/BF02253834. [DOI] [PubMed] [Google Scholar]
- ICH. 2005. ICH S7B Guideline (Step 5 Version): The Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002841.pdf (Accessed August 2011)
- Isenberg G, Klockner U. Calcium tolerant ventricular myocytes prepared by incubation in a ‘KB medium’. Pflugers Arch. 1982;395:6–18. doi: 10.1007/BF00584963. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Inoue Y, Ito K, Sakaue T, Kita S, Katsuragi T. The exchanger inhibitory peptide region-dependent inhibition of Na+/Ca2+ exchange by SN-6 [2-[4-(4-nitrobenzyloxy)benzyl]thiazolidine-4-carboxylic acid ethyl ester], a novel benzyloxyphenyl derivative. Mol Pharmacol. 2004;66:45–55. doi: 10.1124/mol.66.1.45. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of hERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Morishima M, Honjo H, Sanguinetti MC. Molecular determinants of hERG channel block by terfenadine and cisapride. J Pharmacol Sci. 2008;108:301–307. doi: 10.1254/jphs.08102fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J, Watano T, Kawahara M, Sakai E, Yatabe J. Direction-independent block of bi-directional Na+/Ca2+ exchange current by KB-R7943 in guinea-pig cardiac myocytes. Br J Pharmacol. 1999;128:969–974. doi: 10.1038/sj.bjp.0702869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem Biophys Res Commun. 2007;361:230–236. doi: 10.1016/j.bbrc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Larsen AP. Role of ERG1 isoforms in modulation of ERG1 trafficking and function. Pflugers Arch. 2010;460:803–812. doi: 10.1007/s00424-010-0855-8. [DOI] [PubMed] [Google Scholar]
- Levi AJ, Hancox JC, Howarth FC, Croker J, Vinnicombe J. A method for making rapid changes of superfusate whilst maintaining temperature at 37°C. Pflugers Arch. 1996;432:930–937. doi: 10.1007/s004240050217. [DOI] [PubMed] [Google Scholar]
- Liang GH, Kim JA, Seol GH, Choi S, Suh SH. The Na+/Ca2+ exchange inhibitor KB-R7943 activates large conductance Ca2+-activated K+ channels in endothelial and vascular smooth muscle cells. Eur J Pharmacol. 2008;582:35–41. doi: 10.1016/j.ejphar.2007.12.021. [DOI] [PubMed] [Google Scholar]
- McPate MJ, Duncan RS, Milnes JT, Witchel HJ, Hancox JC. The N588K-HERG K+ channel mutation in the ‘short QT syndrome’: mechanism of gain-in-function determined at 37°C. The N588K-HERG K+ channel mutation in the ‘short QT syndrome’: mechanism of gain-in-function determined at 37. Biochem Biophys Res Commun. 2005;334:441–449. doi: 10.1016/j.bbrc.2005.06.112. [DOI] [PubMed] [Google Scholar]
- McPate MJ, Duncan RS, Witchel HJ, Hancox JC. Disopyramide is an effective inhibitor of mutant hERG K+ channels involved in variant 1 short QT syndrome. J Mol Cell Cardiol. 2006;41:563–566. doi: 10.1016/j.yjmcc.2006.05.021. [DOI] [PubMed] [Google Scholar]
- McPate MJ, Duncan RS, Hancox JC, Witchel HJ. Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br J Pharmacol. 2008;155:957–966. doi: 10.1038/bjp.2008.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol. 2003a;139:887–898. doi: 10.1038/sj.bjp.0705335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnes JT, Dempsey CE, Ridley JM, Crociani O, Arcangeli A, Hancox JC, et al. Preferential closed channel blockade of HERG potassium currents by chemically synthesised BeKm-1 scorpion toxin. FEBS Lett. 2003b;547:20–26. doi: 10.1016/s0014-5793(03)00662-8. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC. An investigation of the role played by the E-4031-sensitive (rapid delayed rectifier) potassium current in isolated rabbit atrioventricular nodal and ventricular myocytes. Pflugers Arch. 1999;436:843–850. doi: 10.1007/s004249900118. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata A, Zipes DP, Hall S, Rubart M. KB-R7943 prevents acute, atrial fibrillation-induced shortening of atrial refractoriness in anesthetized dogs. Circulation. 2002;106:1410–1419. doi: 10.1161/01.cir.0000028587.85711.f6. [DOI] [PubMed] [Google Scholar]
- Niu C-F, Watanabe Y, Ono K, Iwamoto T, Yamashita K, Satoh H, et al. Characterization of SN-6, a novel Na+/Ca2+ exchange inhibitor in guinea-pig cardiac ventricular myocytes. Eur J Pharmacol. 2007;573:161–169. doi: 10.1016/j.ejphar.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Zamponi MW, Barr W, Kiedrowski L, Stys PK. Protection of ischemic rat spinal cord white matter: dual action of KB-R7943 on Na+/Ca2+exchange and L-type Ca2+ channels. Neuropharmacology. 2005;48:566–575. doi: 10.1016/j.neuropharm.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Pezier A, Bobkov YV, Ache BW. The Na+/Ca2+ exchanger, KB-R7943, blocks a nonselective cation channel implicated in chemosensory transduction. J Neurophysiol. 2009;101:1151–1159. doi: 10.1152/jn.90903.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- Ridley JM, Milnes JT, Zhang YH, Witchel HJ, Hancox JC. Inhibition of HERG K+ current and prolongation of the guinea-pig ventricular action potential by 4-aminopyridine. J Physiol. 2003;549:667–672. doi: 10.1113/jphysiol.2003.043976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley JM, Milnes JT, Witchel HJ, Hancox JC. High affinity HERG K+ channel blockade by the antiarrhythmic agent dronedarone: resistance to mutations of the S6 residues Y652 and F656. Biochem Biophys Res Commun. 2004;325:883–891. doi: 10.1016/j.bbrc.2004.10.127. [DOI] [PubMed] [Google Scholar]
- Sanders L, Rakovic S, Lowe M, Mattick PA, Terrar DA. Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sinoatrial node. J Physiol. 2006;571:639–649. doi: 10.1113/jphysiol.2005.100305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. J Gen Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. TIPS. 2005;26:119–124. doi: 10.1016/j.tips.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Santo-Domingo J, Vay L, Hernandez-SanMiguel E, Lobaton CD, Moreno A, Montero M, et al. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br J Pharmacol. 2007;151:647–654. doi: 10.1038/sj.bjp.0707260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Tanaka H, Habuchi Y, Giles WR. Electrophysiological effects of ibutilide on the delayed rectifier K+ current in rabbit sinoatrial and atrioventricular node cells. Eur J Pharmacol. 2000;404:281–288. doi: 10.1016/s0014-2999(00)00603-8. [DOI] [PubMed] [Google Scholar]
- Shah RR. Drug-induced QT interval prolongation – regulatory guidance and perspectives on hERG channel studies. Novartis Found Symp. 2005;266:251–280. [PubMed] [Google Scholar]
- Shigekawa M, Iwamoto T. Cardiac Na-Ca exchange: molecular and pharmacological aspects. Circ Res. 2001;88:864–876. doi: 10.1161/hh0901.090298. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AL, Khodorov BL. Blockade of NMDA channels in acutely isolated rat hippocampal neurons by the Na+/Ca2+exchange inhibitor KB-R7943. Neuropharmacology. 1999;38:1235–1242. doi: 10.1016/s0028-3908(99)00040-4. [DOI] [PubMed] [Google Scholar]
- Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9–33. doi: 10.1016/j.cardiores.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, Shigenobu K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol. 2002;135:1096–1100. doi: 10.1038/sj.bjp.0704574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torp-Pedersen C, Brendorp B, Kober L. Dofetilide: a Class III anti-arrhythmic drug for the treatment of atrial fibrillation. Expert Opin Investig Drugs. 2000;9:2695–2704. doi: 10.1517/13543784.9.11.2695. [DOI] [PubMed] [Google Scholar]
- Toth A, Kiss L, Varro A, Nanasi PP. Potential therapeutic effects of Na+/Ca2+ exchanger inhibition in cardiac diseases. Curr Med Chem. 2009;16:3294–3321. doi: 10.2174/092986709788803268. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, an inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett. 1997;417:41–47. doi: 10.1016/s0014-5793(97)01245-3. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Koide Y, Kimura J. Topics on the Na+/Ca2+ exchanger: pharmacological characterization of Na+/Ca2+ exchanger inhibitors. J Pharmacol Sci. 2006;102:7–16. doi: 10.1254/jphs.fmj06002x2. [DOI] [PubMed] [Google Scholar]
- Watano T, Kimura J, Morita T, Nakanishi H. A novel antagonist, No. 7943, of the Na+/Ca2+ exchange current in guinea-pig cardiac ventricular cells. Br J Pharmacol. 1996;119:555–563. doi: 10.1111/j.1476-5381.1996.tb15708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witchel HJ, Dempsey CE, Sessions RB, Perry M, Milnes JT, Hancox JC, et al. The low potency, voltage-dependent hERG blocker propafenone – molecular determinants and drug trapping. Mol Pharmacol. 2004;66:1201–1212. doi: 10.1124/mol.104.001743. [DOI] [PubMed] [Google Scholar]
- Zaza A, Micheletti M, Brioschi A, Rocchetti M. Ionic currents during sustained pacemaker activity in rabbit sino-atrial myocytes. J Physiol. 1997;505:677–688. doi: 10.1111/j.1469-7793.1997.677ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Hancox JC. Regulation of cardiac Na+-Ca2+ exchanger activity by protein kinase phosphorylation – still a paradox? Cell Calcium. 2009;45:1–10. doi: 10.1016/j.ceca.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Cheng H, Alexeenko VA, Dempsey CE, Hancox JC. Characterization of recombinant hERG K+ channel inhibition by the active metabolite of amiodarone desethyl-amiodarone. J Electrocardiol. 2010;43:440–448. doi: 10.1016/j.jelectrocard.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, et al. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]