

Abstract
BACKGROUND AND PURPOSE
Graves' disease (GD) is an autoimmune disease in which the thyroid is overactive, producing excessive amounts of thyroid hormones, caused by thyroid-stimulating hormone (TSH) receptor-stimulating immunoglobulins (TSIs). Many GD patients also suffer from thyroid eye disease (Graves' ophthalmopathy or GO), as TSIs also activate TSH receptors in orbital tissue. We recently developed low molecular weight (LMW) TSH receptor antagonists as a novel therapeutic strategy for the treatment of GD and GO. Here, we determined the molecular pharmacology of a prototypic, nanomolar potent LMW TSH receptor antagonist, Org 274179-0.
EXPERIMENTAL APPROACH
Using CHO cells heterogeneously expressing human TSH receptors and rat FRTL-5 cells endogenously expressing rat TSH receptors, we determined the potency and efficacy of Org 274179-0 at antagonizing TSH- and TSI-induced TSH receptor signalling and its cross-reactivity at related follicle-stimulating hormone and luteinizing hormone receptors. We analysed the allosteric mode of interaction of Org 274179-0 and determined whether it is an inverse agonist at five naturally occurring, constitutively active TSH receptor mutants.
KEY RESULTS
Nanomolar concentrations of Org 274179-0 completely inhibited TSH (and TSI)-mediated TSH receptor activation with little effect on the potency of TSH, in accordance with an allosteric mechanism of action. Conversely, increasing levels of TSH receptor stimulation only marginally reduced the antagonist potency of Org 274179-0. Org 274179-0 fully blocked the increased basal activity of all the constitutively active TSH receptor mutants tested with nanomolar potencies.
CONCLUSIONS AND IMPLICATIONS
Nanomolar potent TSH receptor antagonists like Org 274179-0 have therapeutic potential for the treatment of GD and GO.
Keywords: TSH, thyroid, Graves' disease, Graves' ophthalmopathy, G protein-coupled receptor, allosterism, antagonism
Introduction
The thyroid-stimulating hormone (TSH) receptor is an essential regulator of the thyroid gland. This GPCR is responsible for the synthesis and release of the thyroid hormones thyroxine (T4) and triiodothyronine (T3) from the thyroid and is also required for thyrocyte growth and proliferation. The TSH receptor couples to AC and PLC via Gs and Gq/11 proteins respectively. Both signalling pathways are essential for thyroid hormone synthesis and release: the AC pathway is required for iodide uptake and secretion of T4 and T3 while the PLC pathway is responsible for synthesis of thyroid hormones (Corvilain et al., 2001; Song et al., 2010).
Inappropriate overstimulation of the TSH receptor leads to hyperthyroidism. In Graves' disease (GD), which afflicts ∼1.5% of the human population, in particular females, circulating TSH receptor-stimulating immunoglobulins (TSIs) potently activate TSH receptors in the thyroid and raise T3 and T4 serum levels (Rapoport et al., 1998; Prabhakar et al., 2003; Ando et al., 2004). Hyperthyroidism can also be caused by somatic, ‘gain-of-function’ mutations in the TSH receptor which confer TSH-independent, constitutive activity to the receptor in autonomous thyroid adenomas. These mutations are found in the extracellular N-terminus, the extracellular loops, the transmembrane domains and the third cytoplasmic loop of the receptor (Duprez et al., 1998; Corvilain et al., 2001). In addition to thyroid hormone synthesis and release, TSH receptor activation also stimulates thyrocyte proliferation; a signalling pathway which is of significant clinical relevance. TSH suppression with high doses of T4 is used to treat patients with well-differentiated thyroid cancer after thyroidectomy. This results in decreased recurrence rates and cancer-related mortality (Fiore et al., 2009). In addition, large epidemiological studies have shown that higher serum TSH levels are associated with an increased risk of thyroid cancer in patients with thyroid nodules (Boelaert et al., 2006; Haymart et al., 2008; Fiore et al., 2009).
In addition to expression in the thyroid gland, TSH receptors are also present in a number of extrathyroidal tissues, in particular in orbital fibroblasts and adipocytes of patients with GD. A large proportion (∼50–80%) of GD patients suffer from Graves' ophthalmopathy (GO), in which expansion of the orbital contents by adipogenesis, overproduction of glycosaminoglycans and oedema lead to proptosis and other eye complaints (Mourits et al., 1997). It is current belief that the TSIs also stimulate orbital TSH receptors and participate in the pathogenesis and pathophysiology of GO (Garrity and Bahn, 2006; Kumar et al., 2011; van Zeijl et al., 2011). This hypothesis is based on a number of observations. Firstly, expression of functional TSH receptors is significantly increased in orbital tissue of GO patients in the active phase (Valyasevi et al., 1999; Wakelkamp et al., 2003). Secondly, TSI levels correlate with the severity of GO (Gerding et al., 2000; Eckstein et al., 2006; Lytton et al., 2010). Thirdly, hypothyroidism with elevated serum TSH levels (Wiersinga, 2007) as well as radioiodine therapy (raising serum TSH and TSI levels) aggravates GO (Kung et al., 1994). Inhibition of TSH receptor signalling by LMW TSH receptor antagonists has therefore been advocated as a novel therapeutic strategy for GD and GO. The attractiveness of the LMW TSH receptor antagonist approach is that LMW TSH receptor antagonists will not only treat hyperthyroidism but may also treat and/or prevent GO.
In the past few years, LMW TSH receptor antagonists of two compound classes have been reported in the scientific literature (Neumann et al., 2008; 2010; 2011). These antagonists block TSH and TSI signalling, but display only high micromolar potencies (i.e. IC50 values ranging from 1 to 30 µM as measured in cAMP assays). Recently, we identified novel LMW TSH receptor antagonists that are about 1000-fold more potent, having the potential of oral bioavailability (Karstens et al., 2009). Here we present the allosteric mechanism of action of Org 274179-0, a representative of this tetrahydroquinoline compound series of LMW TSH receptor antagonists, with a molecular weight of 480.5 (Figure 1). Because of the limited bioavailability of human thyroid and orbital tissue, we performed these studies in CHO cells heterogeneously expressing hTSH receptors and in rat FRTL-5 cells endogenously expressing rat TSH receptors. Org 274179-0 inhibited TSH- and TSI-mediated cAMP formation and PLC activation at nanomolar concentrations. Org 274179-0 also fully blocked the increased basal activity of constitutively active TSH receptor mutants with comparable potencies.
Figure 1.

Chemical structure of Org 274179-0 and its stereoisomer, Org 274178-0.
Methods
Cell culture
CHO cells stably expressing CRE-luciferase with or without human (h) TSH receptors, hLH receptors or hFSH receptors were grown in DME/F12 medium containing 5% bovine calf serum and supplemented with penicillin G (100 U·mL−1) and streptomycin sulphate (0.1 mg·mL−1) in a humidified atmosphere in 5% CO2 at 37°C. FRTL-5 cells (ECACC) were grown in DMEM Ham's F12 with somatostatin (10 ng·mL−1), apo-transferrin (5 µg·mL−1), glycyl-L-histidyl-L-lysine acetate (0.1 µg·mL−1), insulin (10 µg·mL−1), hydrocortisone (10 nM), bovine TSH (10 U·L−1), penicillin G (100 U·mL−1), streptomycin sulphate (100 µg·mL−1) and 5% fetal calf serum. Before FRTL-5 cells were used in the cAMP assay, cells were cultured 3–5 days without bovine TSH to restore TSH responsiveness. All cell culture supplies were obtained from Gibco/BRL unless indicated otherwise.
Site-directed mutagenesis of TSH receptor
The TSH receptor substitution mutants, Ile568Thr, Asp619Gly, Ala623Val, Thr632Ile and Asp633Glu, were made by using the QuikChange II site-directed mutagenesis kit from Stratagene, according to the manufacturer's protocol with the appropriate primers. The complete coding sequence of each TSH receptor mutant was verified. All mutant and wild-type TSH receptor coding sequences were subcloned in the same eukaryotic expression vector (pKCR). The sources of the human TSH receptor and human LH receptor cDNAs, the construction of the chimeric cDNAs in which the extracellular N-terminus of the human TSH receptor and human LH receptor were exchanged (LTR and TLR), and the corresponding control TSH receptor and LH receptor cDNAs have been described previously (van Koppen et al., 2008).
Measurement of CRE-luciferase expression
For the measurement of luciferase activity, wells from a 384-well plate received 10 µL of DME/F12 modified medium supplemented with bovine insulin (1 µg·mL−1), penicillin G (100 U·mL−1), streptomycin sulphate (100 µg·mL−1) (assay medium) with or without agonist (TSH, M22, FSH, LH, serum IgG preparation) and 10 µL of assay medium containing 3% dimethylsulphoxide (DMSO) with or without Org 274179-0. Then, 10 µL of CHO cells stably expressing hTSH, hFSH and hLH receptors (7500 cells) were added per well. After incubation for 4 h in 5% CO2 at 37°C, plates were left to adapt to room temperature for 1 h followed by the addition of 15 µL SteadyLite (Packard, Groningen, the Netherlands). Luciferase activity was measured in an Envision (Perkin Elmer, Norwalk, CT, USA) after lysis of the cells for 1 h at room temperature. The concentrations of TSH, M22, TSIs, LH and FSH in the antagonist assays amounted to 80% of those used to achieve the maximum effect on the agonist response. Data points of the concentration–effect curves were fitted using GraphPad Prism version 4.03 (GraphPad Software).
Measurement of cAMP
For the measurement of cAMP synthesis in CHO.hTSH receptor and FRTL-5 cells, the AlphaScreen kit of Perkin Elmer was used. Org 274179-0 was diluted to the desired concentration in assay buffer, containing Hanks balanced salt solution (HBSS) supplemented with 5 mM HEPES, pH 7.4, 20 µM rolipram and 0.1% BSA. Ten microlitres of TSH or M22, and 10 µL assay buffer with or without Org 274179-0 (final DMSO concentration of 1%), were added to 10 µL of cell suspension (5000 cells per well) in Perkin Elmer 384 wells white culture plates. The cells were diluted in assay medium (DMEM Ham F12 supplemented with 1 µg·mL−1 bovine insulin, 5 µg·mL−1 human apo-transferrin, penicillin G (100 U·mL−1) and streptomycin sulphate (100 µg·mL−1). A cAMP standard curve was made to convert the signal into molar cAMP concentrations. The assay plates were incubated for 1 h at 37°C in 5% CO2. The reaction was stopped by adding 15 µL acceptor beads and 15 µL donor beads diluted in lysis buffer, consisting of 0.1% BSA, 0.3% Tween-20 and 5 mM HEPES, pH 7.4 in water. After 1 h at room temperature in the dark, luminescence was measured on an Envision. For FRTL-5 cells, cells were incubated for 60 min at 37°C in 5% CO2. Data points of concentration–effect curves were fitted using GraphPad Prism version 4.03.
Measurement of PLC
For the measurement of PLC activity in CHO.hTSH receptor cells, cells on six well plates were pre-labelled in inositol-free DME medium (custom-made by Gibco) with 1 µC·mL−1myo-[3H]-inositol at 5% CO2 in a humidified atmosphere at 37°C. After 48 h, cells were washed twice with HBSS (118 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 15 mM HEPES, 0.9 g·L−1 glucose, pH 7.4) + 0.1% BSA (37°C) to remove free [3H]-myo-inositol. Then, cells were pre-incubated for 15 min with 2 mL 10 mM LiCl in HBSS + 0.1% BSA in a dry incubator at 37°C. Stimulation was started by the addition of bovine TSH or M22, in the presence or absence of Org 274179-0 in a volume of 20 µL medium (final DMSO concentration of 0.1%). After 60 min, the medium was removed from the plates and phospholipids were isolated by chloroform/methanol extraction. [3H]-inositol phosphates in the methanol fractions were isolated by ion exchange chromatography (van Koppen et al., 2008).
Isolation of IgG from patients with Graves' disease
IgG was isolated from sera of GD patients by protein G Sepharose 4 Fast Flow chromatography (ProtG; Amersham Pharmacia Biotech Benelux, Roosendaal, the Netherlands) after obtaining informed consent, as described previously (van Zeijl et al., 2011). The TSH-binding inhibitory immunoglobulin (TBII) serum levels of the GD patients ranged from 51 to 256 U·L−1.
[125I]-TSH dissociation assays
CHO.hTSH receptor cells were collected from the tissue culture flasks by a rubber policeman in ice-cold 10 mM Tris-HCl containing 5 mM MgCl2 and 250 mM saccharose, pH 7.4 containing protease inhibitor cocktail (EDTA-free; Roche, Almere, the Netherlands). Following Potter homogenization, the cell homogenates were centrifuged at 48,200×g for 30 min at 4°C. Then, cell pellets were resuspended in ice-cold 10 mM Tris-HCl buffer containing 5 mM MgCl2 with protease inhibitor cocktail (EDTA-free, Roche) and aliquots were stored at −80°C. Protein concentration was determined by the Bradford assay. For measuring [125I]-TSH dissociation, 150 µL buffer (10 mM Tris-HCl + 5 mM MgCl2, 0.1% BSA) with or without 200 nM bovine TSH, 100 µL cell homogenate (15 µg of membrane protein, diluted 1:24 in buffer) and 50 µL [125I]-TSH (16 000–30 000 cpm) in buffer were incubated at room temperature. After 16 h, 5 µL of buffer with or without 6.2 µM bovine TSH (100 nM final) + 5 µL of vehicle (6.2% DMSO in buffer) with or without 62 µM Org 274179-0 (1 µM final) were added to the incubation medium. The [125I]-TSH dissociation reaction was stopped after 1, 2 and 4 h by addition of 500 µL ice-cold 10 mM Tris-HCl, 5 mM MgCl2, 0.1% BSA. Following centrifugation at 15 000×g for 5 min at room temperature and aspiration of the supernatant, centrifuge tubes were cut and radioactivity in the membrane pellet was determined in a Cobra II (Packard) γ counter.
Operational model of allosterism – fitting
The functional interaction between Org 274179-0 and TSH or M22 in the CRE-luciferase assays was also fitted according to the following operational model of allosterism (Leach et al., 2007):
where Em is the maximum possible tissue response, [A] and [B] are the concentrations of orthosteric ligand (TSH or M22) and Org 274179-0, respectively, KA and KB are the equilibrium dissociation constant of the orthosteric ligand and Org 274179-0, respectively, τA and τB are the operational measures for the efficacies of the orthosteric ligand and Org 274179-0, respectively, n denotes the slope factor of the curve, α is the binding co-operativity parameter between the orthosteric ligand and Org 274179-0, and β denotes the allosteric effect of Org 274179-0 on the efficacy of the orthosteric agonist.
Source of reagents
Org 274179-0 ((S)-N-(1-acetyl-4-methyl-4-phenyl-1,2,3,4-tetrahydro-quinolin-6-yl)-3-(3-trifluoromethyl-phenyl)-propionamide; MW of 480.5) and its (R)-stereoisomer, Org 274178-0, were synthesized in house, as described previously (Karstens et al., 2009). Chemical purity was ≥99.3% by LC-MS; enantiomeric purity by chiral HPLC was ≥99.7%. [125I]-bovine TSH was obtained as part of the TSH receptor Autoantibody Coated Tube kit from RSR Ltd (Cardiff, UK). The purified, monoclonal TSI M22 was purchased from AV7 Ltd, FIRS Laboratories (Cardiff, UK). Recombinant human TSH (Thyrogen®) was purchased from Genzyme. Recombinant human FSH and LH were synthesized in house. Bovine TSH was purchased from Sigma, myo-[2-3H]-inositol (spec. act. 21 Ci·mmol−1) was obtained from Gibco/BRL.
Results
Adenylyl cyclase signalling
In the human thyroid, the AC pathway stimulates T4 and T3 secretion as well as thyroid proliferation in response to TSH receptor stimulation. To investigate the inhibitory potency of Org 274179-0, we determined the ability of Org 274179-0 to inhibit cAMP-responsive element (CRE)-mediated luciferase expression stimulated by human and bovine TSH in our CHO.hTSH receptor screening cell line stably expressing a CRE-luciferase reporter. In addition, we stimulated the TSH receptor in this assay with the (purified) monoclonal TSI, M22. M22 is derived from a patient with GD and can be regarded as a prototypic TSI (Sanders et al., 2004; 2006; Morshed et al., 2009). The pEC50 values of bTSH, human recombinant TSH and M22 in stimulating CRE-luciferase in CHO.hTSH receptor cells were 8.46 ± 0.08, 8.50 ± 0.03 and 8.68 ± 0.15, respectively. The Emax value of M22 in CHO.hTSH receptor cells (CRE-luciferase read-out) was 83 ± 7% of TSH (data not shown). As shown in Figure 2A, left panel (and summarized in Table 1), Org 274179-0 completely inhibited bTSH-, hTSH- and M22-mediated CRE-luciferase transactivation in the CHO cell line with IC50 values of 30, 26 and 56 nM, respectively. We also determined the potencies of Org 274179-0 at inhibiting cAMP formation induced by bTSH, human recombinant TSH and M22 in the same cell line. The pEC50 values of bTSH, human recombinant TSH and M22 in CHO.hTSH receptor cells were 8.35 ± 0.04, 7.95 ± 0.06 and 8.60 ± 0.06, respectively. The Emax value of M22 was 53 ± 5% of TSH in CHO.hTSH receptor cells. The IC50 values of Org 274179-0 for inhibiting cAMP formation induced by bTSH, hTSH and M22 were 9, 11 and 5 nM. The higher IC50 values in the luciferase assays are probably related to a slight degree of ‘receptor reserve’ in the CRE-luciferase reporter assay (due to signal amplification). We also determined the potency of Org 274179-0 at inhibiting TSH receptor stimulation in rat FRTL-5 thyroid cells, which endogenously express rat TSH receptors, by bTSH and M22. The pEC50 values of bTSH and M22 in FRTL-5 cells were 8.34 ± 0.04 and 8.66 ± 0.08, respectively. The Emax value of M22 was 72 ± 7% of TSH in FRTL-5 cells (data not shown). Org 274179-0 was also found to be a potent and full antagonist at the rat TSH receptor. The IC50 values for inhibiting bTSH- and M22-mediated cAMP formation in FRTL-5 cells were 5 and 2 nM, respectively (Figure 2B). These values are very similar to IC50 values (cAMP read-out) found in the CHO cells expressing the human TSH receptor. To demonstrate stereoselectivity of the putative binding pocket of Org 274179-0, the potency of its (R)-stereoisomer, Org 274178-0, was also determined (Figure 2C). Org 274178-0 was 646-fold less potent than Org 274179-0. Hence, the eudismic ratio of Org 274179-0 for TSH receptor antagonist activity is 646.
Figure 2.
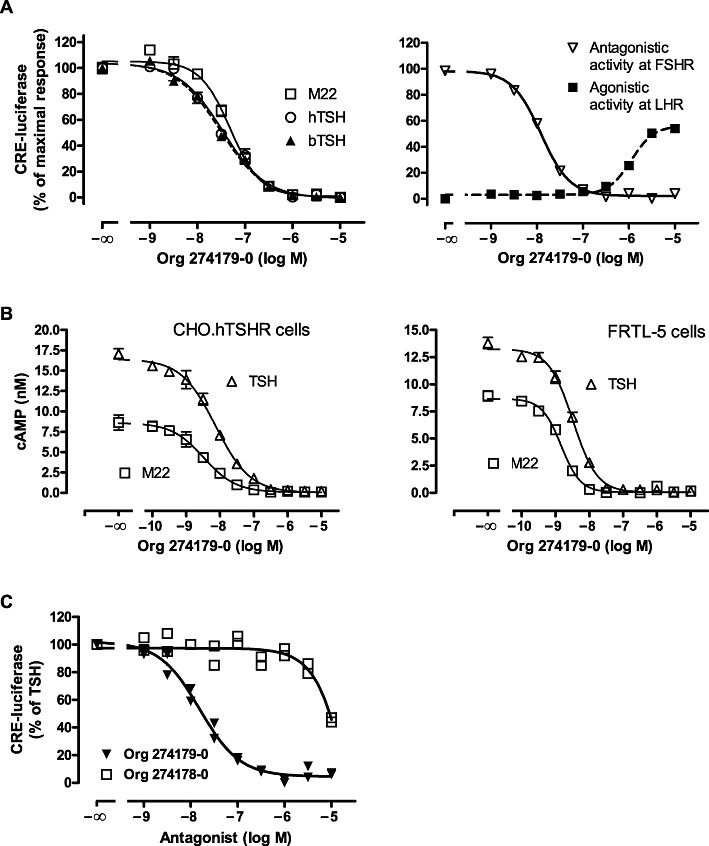
TSH receptor antagonism induced by Org 274179-0. (A, left panel) Inhibition of TSH- and M22-induced CRE-luciferase activity by Org 274179-0 in CHO.hTSH receptor cells stably expressing CRE-luciferase. CHO.hTSH receptor cells were incubated for 4 h at 37°C with 31.6 nM human recombinant TSH, 20 nM bovine TSH or 10 nM M22 in the presence or absence of the indicated concentrations of Org 274179-0. (A, right panel) Inhibition of FSH receptor-induced and stimulation of LH receptor-mediated CRE-luciferase activity in CHO.hFSH receptor and CHO.hLH receptor cells, respectively, by Org 274179-0. CHO.hFSH receptor and CHO.hLH receptor cells were incubated for 4 h at 37°C with 48 pM human recombinant FSH or buffer, respectively, in the presence or absence of the indicated concentrations of Org 274179-0. CRE-luciferase activity was determined after luminescence counting. Data are from a single experiment (mean ± SD, done in quadruplicate), representative of at least four experiments. Corresponding pIC50 values of Org 274179-0 are listed in Table 1. (B) Inhibition of TSH- and M22-induced cAMP synthesis in CHO.hTSH receptor and FRTL-5 cells by Org 274179-0. CHO.hTSH receptor and FRTL-5 cells were incubated for 60 min at 37°C with 20 or 50 nM bTSH (CHO.hTSHR or FRTL-5 cells, respectively) or 10 nM M22 (CHO.hTSHR and FRTL-5 cells) in the presence or absence of the indicated concentrations of Org 274179-0. Data are from a single experiment (mean ± SD, done in quadruplicate), representative of five and two experiments, respectively. Corresponding pIC50 values are listed in Table 1. (C) Stereoselectivity of Org 274179-0 at inhibiting TSH-induced CRE-luciferase activity in CHO.hTSH receptor cells stably expressing CRE-luciferase. Cells were stimulated with 20 nM bTSH. Org 274178-0 is the C-4 epimer of Org 274179-0 (Figure 1). Data are the duplicate measurements of a single experiment, representative of two experiments. Corresponding pIC50 values of Org 274179-0 and Org 274178-0 are 7.84 ± 0.02 and 5.03 ± 0.01, respectively.
Table 1.
Antagonist and agonist activities of Org 274179-0 in CHO.hTSH receptor, FRTL-5, CHO.hFSH receptor, CHO.hLH receptor CRE-luciferase transactivation and cAMP assays
| Agonist | Cell line (read-out) | IC50 (pIC50) |
|---|---|---|
| bTSH | CHO.hTSH receptor (CRE-luciferase) | 30 nM (7.52 ± 0.22) |
| hTSH | CHO.hTSH receptor (CRE-luciferase) | 26 nM (7.58 ± 0.07) |
| M22 | CHO.hTSH receptor (CRE-luciferase) | 56 nM (7.25 ± 0.07) |
| bTSH | CHO.hTSH receptor (cAMP) | 9 nM (8.07 ± 0.08) |
| hTSH | CHO.hTSH receptor (cAMP) | 11 nM (7.95 ± 0.08) |
| M22 | CHO.hTSH receptor (cAMP) | 5 nM (8.34 ± 0.16) |
| bTSH | FRTL-5 (cAMP) | 5 nM (8.34 ± 0.14) |
| M22 | FRTL-5 (cAMP) | 2 nM (8.62 ± 0.18) |
| Agonist | Receptor cell line (read-out) | IC50 or EC50a (pIC50) |
|---|---|---|
| hFSH | CHO.hFSH receptor ant (CRE-luciferase) | 17 nM (7.77 ± 0.11) |
| – | CHO.hLH receptor ag (CRE-luciferase) | 1.1 µMa (5.94 ± 0.12) |
| hLH | CHO.hLH receptor ant (CRE-luciferase) | 24 ± 8 % inhibition at 10 µM |
Data are the mean ± SD of at least four experiments (except for ‘M22 on FRTL-5 cells’ where n= 2). Org 274179-0 showed full antagonist (ant), but no agonist (ag), activity on all TSH- and FSH-receptor-expressing cells.
Agonist Emax of Org 274179-0 on CHO.hLH receptor cells was 70 ± 15%.
The antagonist activity of Org 274179-0 was also determined against a panel of crude IgG preparations from patients with GD. These IgG preparations were isolated from (i) a serum pool of 20 GD patients with a final serum TBII value of 51 U·L−1 and (ii) from the individual sera of two GD patients with TBII values of 88 and 256 U·L−1. The in vitro TSH agonist Emax values of these TSI preparations tested at the maximally effective concentration of 10 mg·mL−1 IgG were 47, 72 and 100%, respectively of the maximal stimulation obtained with bTSH. The IC50 values of Org 274179-0 [determined in the presence of 3.16 mg·mL−1 IgG (CRE-luciferase read-out)] were 34, 39 and 41 nM, respectively, and Org 274179-0 displayed full antagonist activity (n= 2 sets of independent experiments, data not shown).
Org 274179-0 exhibited equipotent, full antagonist activity at the human FSH receptor (IC50 value of 17 nM, Figure 2A, right panel and Table 1). However, at the human LH receptor, Org 274179-0 showed cross-reactivity at concentrations that were ∼50-fold higher than required to inhibit TSH receptor activity. Org 274179-0 displayed partial agonist activity at the LH receptor (EC50 of 1.1 µM, Emax of 70%) (Figure 2A right panel, Table 1); it slightly inhibited LH receptor activity at high concentrations (24% at 10 µM, Table 1).
PLC signalling
We next tested whether Org 274179-0 also inhibits TSH receptor-mediated PLC activation in stably transfected CHO.hTSH receptor cells (Figure 3). The pEC50 values of bTSH and M22 for stimulating PLC were 7.21 ± 0.12 and 8.18 ± 0.08, respectively (n= 3 independent experiments). The Emax value of M22 was 92 ± 11% of bTSH. Org 274179-0 fully inhibited bTSH- and M22-mediated PLC activation with IC50 values of 5 nM (pIC50 of 8.43 and 8.14) and 4 nM (pIC50 of 8.32 and 8.58), respectively (n= 2 experiments each). These IC50 values are very similar to the IC50 values found for inhibiting cAMP formation. Org 274179-0 (1 µM) alone was ineffective at stimulating PLC, and Org 274179-0 (1 µM) had no effect on 1 mM ATP-induced PLC activation in these cells (data not shown).
Figure 3.
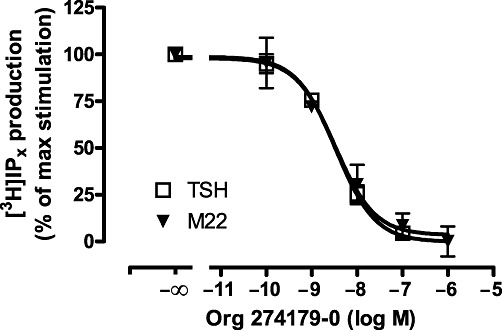
Inhibition of TSH- and M22-stimulated phospholipase C by Org 274179-0. CHO.hTSH receptor cells on six well plates and pre-labelled with myo-[3H]-inositol for 48 h were incubated with the indicated concentrations of Org 274179-0 in the presence of 500 nM bTSH or 10 nM M22 for 60 min followed by extraction of total [3H]-inositol phosphate production by chloroform/methanol extraction, liquid column chromatography and radioactivity counting. Data are mean ± range of two independent experiments, each done in duplicate. Total [3H]-inositol phosphate levels of TSH- and M22-stimulated and control cells were 1014, 780 and 330 cpm per well.
Allosteric action of Org 274179-0 at the TSH receptor
It is well known that the high molecular weight TSH and TSIs activate the TSH receptor by interacting with the N-terminus of the receptor (Rapoport et al., 1998; Sanders et al., 2003), while low molecular weight modulators of the glycoprotein hormone receptors, including the TSH receptor, are thought to interact with the transmembrane region of the receptors (e.g. van Koppen et al., 2008). We therefore investigated in more detail the allosteric action of Org 274179-0 at the TSH receptor. Firstly, we tested the inhibitory effect of Org 274179-0 on cAMP-CRE signalling of a hTSH receptor/hLH receptor chimera in which the N-termini of these receptors are exchanged. As shown in Figure 4, Org 274179-0 displayed high potency (nanomolar) in antagonizing the wild-type TSH receptor and the LT receptor chimera bearing the transmembrane domains of the TSH receptor but had negligible potency at inhibiting the wild-type LH receptor and the TL receptor chimera. These findings also indicate that Org 274179-0 does not inhibit TSH receptor activation by acting in a receptor-independent manner, downstream from the TSH receptor. Secondly, we tested whether Org 274179-0 affects [125I]-TSH binding and dissociation from the TSH receptor (Figure 5). Org 274179-0 did not affect either [125I]-TSH binding or the dissociation of [125I]-TSH from the TSH receptor. Thirdly, we investigated whether 1 µM Org 274179-0 affected the IC50 of bTSH for inhibiting [125I]-TSH binding; no effect on the IC50 of TSH was observed (IC50 of 3.0 and 3.6 nM in the absence and presence of 1 µM Org 274179-0; data not shown). Fourthly, we determined the effect of increasing concentrations of Org 274179-0 on the potency and efficacy of TSH at stimulating the cAMP signalling pathway. As shown in Figure 6, increasing concentrations of Org 274179-0 induced only a small rightward shift of the concentration–effect curves for TSH (as shown also by only a 2.5-fold increase in TSH's EC50) but significantly decreased the efficacy of TSH from 100% to ∼10%. Together, these results are fully commensurate with an allosteric action of Org 274179-0 at a binding site within the transmembrane domains of the receptor, thereby blocking signalling (but not binding) of TSH and TSI.
Figure 4.
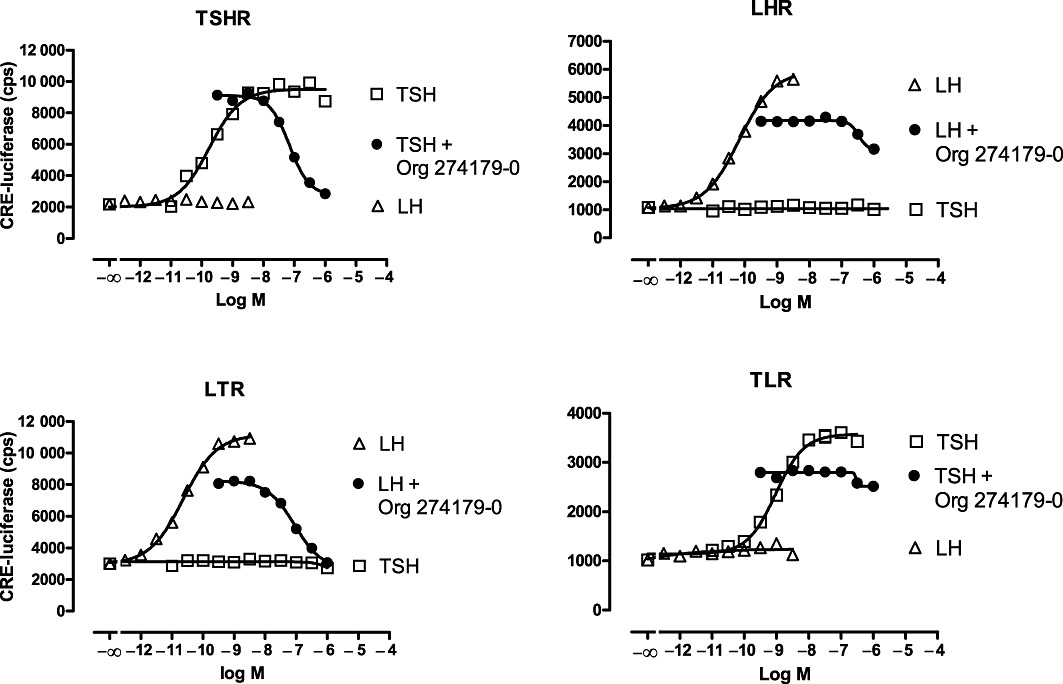
Effect of Org 274179-0 on TSH, LH or the chimeric LT and TL receptors transiently expressed in CHO cells together with luciferase. LT and TL receptors encode the TSH and LH receptor in which the extracellular N-terminus of the human TSH receptor and human LH receptor have been exchanged, respectively (van Koppen et al., 2008). CHO cells were incubated for 4 h at 37°C with recombinant human TSH or recombinant human LH in the presence or absence of Org 274179-0. Concentrations of TSH in the antagonist assays with TSH and TL receptors were 1 and 2 nM, respectively. Concentrations of hLH in the antagonist assays with LH and LT receptors were 150 and 70 pM, respectively. Data are from a single experiment (mean ± SEM, done fivefold), representative of two experiments. Corresponding IC50 values of Org 274179-0 in the two experiments were 70 and 35 nM (TSHR), and 90 and 59 nM (LTR).
Figure 5.

Effect of Org 274179-0 on [125I]-TSH dissociation from the TSH receptor. Crude membranes of CHO.hTSH receptor cells were incubated overnight with 16 000–30 000 cpm [125I]-TSH at room temperature with and without 100 nM bovine TSH for measurement of non-specific [125I]-TSH binding. After overnight incubation, bovine TSH (final concentration of 100 nM), Org 274179-0 (final concentration of 1 µM), TSH + Org 274179-0 (final concentration of 100 nM and 1.0 µM, respectively) or vehicle (buffer with DMSO; final concentration of 1%) were added. After incubation for 1, 2 or 4 h, cold incubation buffer was added, and tubes were centrifuged at 15 000×g. Bound [125I]-TSH was determined in the membrane pellet by γ counting. Data shown are the duplicates of a single experiment, representative of two independent experiments.
Figure 6.
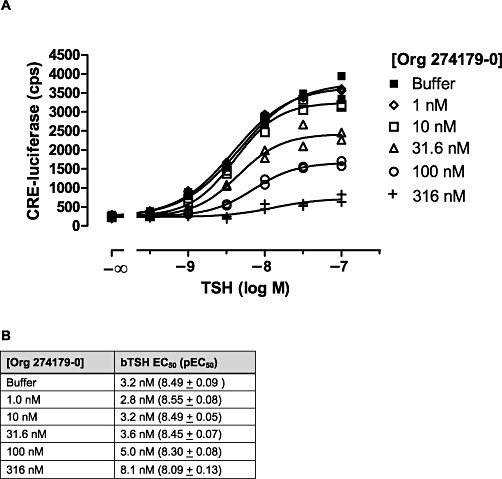
Effect of Org 274179-0 on the potency and efficacy of TSH at stimulating TSH receptor activation. (A) CHO.hTSH receptor cells were incubated with the indicated concentrations of bTSH in the absence and presence of Org 274179-0 for 4 h followed by lysis of the cells and measurement of luciferase activity. Data are the duplicates of a single experiment, representative of three experiments. (B) Summary of the potencies of bTSH in the absence or presence of the indicated fixed concentrations of Org 274179-0. Data are the mean EC50 or the mean pEC50± SEM of three experiments.
Analysis of the inhibitory potency of Org 274179-0 with increasing concentrations of M22
Serum TSI concentrations in GD patients can vary between patients and within patients over time (Eckstein et al., 2006). We therefore investigated how changes in the in vitro antagonist potency of Org 274179-0 are dependent on the M22 concentration in CHO.hTSH receptor cells. Increasing the level of TSH receptor stimulation from ∼10% to 100% (induced by 100 pM to 10 nM M22) led to only a relatively small, threefold, increase in the IC50 of Org 274179-0 (Figure 7).
Figure 7.
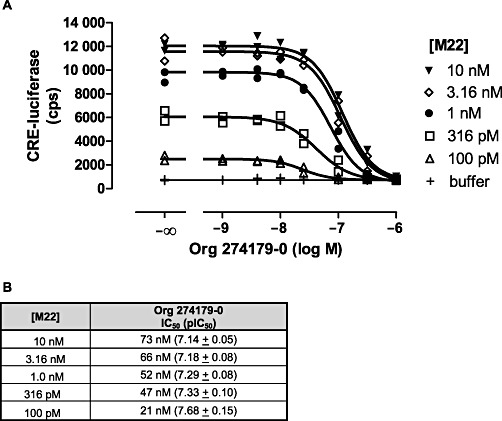
Analysis of the functional interaction between Org 274179-0 and M22 in regulating CRE-luciferase activity in CHO cells stably expressing hTSH receptors. (A) CHO.hTSH receptor cells were incubated with the indicated concentrations of M22 in the absence and presence of Org 274179-0 for 4 h followed by cell lysis and measurement of luciferase activity. Data are the duplicates of a single experiment, representative of three experiments. (B) Summary of the potencies of Org 274179-0 at inhibiting TSH receptor stimulation induced by fixed concentrations of M22. Data are the mean IC50 or the mean pIC50± SEM of three experiments.
To gain further insight in the mode of action of Org 274179-0 with the TSH receptor, the functional data in Figures 6 and 7 were fitted to the operational model of allosterism with the following assumptions. Firstly, as Org 274179-0 is a full antagonist at the TSH receptor with no agonist efficacy, logτB was constrained arbitrarily to −100. Secondly, as Org 274179-0 has no effect on the binding affinity of TSH (and presumably of M22 as well), log α was constrained to 0. Thirdly, as Org 274179-0 completely antagonized the TSH receptor, even in the presence of high concentrations of TSH or M22, log β was set to be −100. This quantitative analysis yielded log KB values of Org 274179-0 of −7.43 ± 0.12 (TSH) and −7.69 ± 0.22 (M22) (Table 2).
Table 2.
Operational model of allosterism parameters for the functional interaction of Org 274179-0 with TSH and M22 at the TSH receptor
| Parameter | TSH | M22 |
|---|---|---|
| logKA | −8.27 ± 0.08 | −8.65 ± 0.21 |
| logKB | −7.43 ± 0.12 | −7.69 ± 0.22 |
| logτA | 0.70 ± 0.03 | 0.59 ± 0.06 |
KA, KB and τA values were determined by fitting the data of Figure 6 (TSH) and Figure 7 (M22) to the operational model of allosterism. KA is the equilibrium dissociation constant of the agonist (TSH or M22). KB is the equilibrium dissociation constant of Org 274179-0. Log τA is the logarithm of the operational efficacy parameter of the agonist. For this analysis, log τB was fixed to −100, log α to 0, log β to −100, Emax of TSH to 100, and Emax of M22 to 120, because the Emax of M22 was 83% of that of TSH. Data are the mean ± SEM of three separate experiments.
Inverse agonist activity of Org 27419-0
Hyperthyroidism may not only be caused by TSH receptor-stimulating antibodies but also by somatic mutations in the TSH receptor, conferring significant constitutive activity to the receptor. These mutations invariably lead to an increase in basal cAMP production (Duprez et al., 1998; Corvilain et al., 2001). Because Org 274179-0 inhibits TSH signalling rather than TSH binding, we determined whether this TSH receptor antagonist also inhibits the increased basal activity of five selected, naturally occurring, constitutively active, TSH receptor mutants. These substitution mutants are Ile568Thr (mutation located in extracellular loop 2), Asp619Gly (intracellular loop 3), Ala623Val (intracellular loop 3), Thr632Ile and Asp633Glu (both in intracellular loop 3 and proximal to transmembrane domain 6). Transfection of the cells with increasing amounts of cDNA of mutant and wild-type TSH receptors significantly increased basal cAMP levels in the TSH receptor mutants compared with the wild-type receptor (Figure 8A). As shown in Figure 8B, Org 274179-0 fully inhibited the constitutive activity of all five TSH receptor mutants beyond the wild-type TSH receptor basal activity level. The IC50 values of Org 274179-0 at blocking the different receptor mutants were 8 nM for Ile568Thr, 74 nM for Asp619Gly, 22 nM for Ala623Val, 2 nM for Thr632Ile, and 19 nM for Asp633Glu TSH receptor. Importantly, Org 274179-0 was also found to suppress basal activity of the wild-type TSH receptor transiently expressed in the CHO cells. Org 274179-0 reduced basal cAMP synthesis of the wild-type TSH receptor by 65% with an IC50 value of 22 nM.
Figure 8.

Inverse agonism of Org 274179-0 at constitutively active mutant and wild-type (WT) TSH receptors. (A) DNA dose-dependent increase in basal cAMP synthesis in CHO cells transfected with WT and constitutively active mutant TSH receptors. CHO cells were transiently transfected with 10, 80 and 240 ng receptor cDNA in pKCR per 25 000 cells per well of 96 well plates and incubated for 60 min at 37°C. cAMP was measured using the AlphaScreen kit. (B) CHO cells were transiently transfected with 100 ng receptor cDNA per 15 000 cells and incubated with the indicated concentrations of Org 274179-0 for 60 min at 37°C. Basal cAMP concentrations of the I568T, D619G, A623V, T632I and D633E TSHR mutants were increased 3.0-, 2.3-, 2.6-, 2.9- and 2.0–fold over the basal activity of the WT TSH receptor, respectively. The maximum cAMP levels induced by 316 nM TSH at the mutant and WT TSH receptors were between 100 and 128 nM. Basal cAMP levels of the mutant TSH receptors were between 52% and 74% of the maximum cAMP levels induced by TSH (data not shown). Data are the mean ± SEM of a single experiment, done in quadruplicate, and representative of two independent experiments.
Discussion and conclusions
The aim of the present study was to investigate the mechanism of action of the LMW TSH receptor antagonist Org 274179-0 at the TSH receptor. Org 274179-0 is a representative of the newly described tetrahydroquinoline LMW TSH receptor antagonists that have nanomolar potencies at this receptor (Karstens et al., 2009). Org 274179-0 was found to be a full antagonist blocking the key signalling pathways of the TSH receptor (AC and PLC) with nanomolar potency. Analysis of the CRE-luciferase data according to the operational model of allosterism (Leach et al., 2007) also revealed nanomolar binding affinities of Org 274179-0 (i.e. 37 nM and 20 nM for the TSH- and M22-occupied TSH receptor, respectively). As both the AC and PLC pathways are present in human thyroid gland and in (differentiated) orbital fibroblasts of patients of GO (Atwa et al., 1995; van Zeijl et al., 2011), Org 274179-0 may be expected to act as a TSH receptor antagonist in the thyroid and orbital tissue of patients with GD and GO.
In the present study, we demonstrated that Org 274179-0 is an allosteric antagonist and most likely interacts with the transmembrane domain of the receptor. Firstly, we showed, using chimeric receptors of the TSH and LH receptors, that Org 274179-0 only antagonizes those receptors that contain the transmembrane region of the TSH receptor (Figure 4). Secondly, we demonstrated that Org 27419-0 does not change the dissociation of [125I]-TSH from the TSH receptor (Figure 5). Thirdly, we showed that increasing concentrations of Org 274179-0 induce only a small rightward shift of the concentration–effect curves of TSH (Figure 6). We therefore conclude that Org 274179-0 changes the efficacy of the orthosteric ligand-receptor complex at stimulating Gs and Gq proteins rather than modulating the binding affinity of the orthosteric ligand (TSH). We also observed that the potency of Org 274179-0 at inhibiting TSI signalling did not differ widely between the different TSI preparations and did not decrease significantly with increasing concentrations of M22 (Figure 7). These are important pharmacological properties of Org 274179-0 because TSIs are known to be heterogeneous within and between patients, and their concentrations may change during the course of the disease (Eckstein et al., 2006). It should be noted that, while the binding affinity of TSH was not affected by Org 274179-0, the EC50 value slightly increased with higher concentrations of Org 274179-0. This increase is likely to be the result of a reduction in receptor reserve with Org 274179-0 reducing the number of functional receptors.
Constitutively active TSH receptor mutants are the cause of hyperthyroidism in patients with autonomously functioning thyroid adenomas. In correspondence with the literature, all TSH receptor mutants tested in the present study showed a significantly increased cAMP level in the absence of TSH. Org 274179-0 completely blocked the increased constitutive activity of the mutant TSH receptors with potencies between 8 and 74 nM. Org 274179-0 was also effective at suppressing basal cAMP synthesis in cells expressing the wild-type TSH receptor, with an IC50 value of 22 nM that is comparable to the IC50 value of 9 nM measured in the presence of a TSH concentration that induces 80% of the maximum attainable effect. Currently, patients with well-differentiated thyroid carcinoma are treated with supraphysiological doses of T4 after thyroidectomy to suppress endogenous TSH secretion thereby preventing TSH-mediated proliferation of residual thyroid carcinoma cells. This treatment, however, does not suppress basal, TSH-independent TSH receptor activity. Org 274179-0, in contrast, may well inhibit TSH-independent TSH receptor activity in vivo and therefore may show increased efficacy compared with TSH suppression with supraphysiological doses of T4. Also, long treatment with supraphysiological doses of T4 is associated with osteoporosis (Kung et al., 1993). Blocking the TSH receptor with antagonists like Org 274179-0 together with T4 replacement therapy may therefore keep thyroidectomized thyroid cancer patients euthyroid, thus preventing the adverse effects of long-term hyperthyroidism.
There is a high unmet need for novel approaches to treat and prevent GO. Current treatment regimes of active GO are aimed at reducing the volume of the orbital tissue by orbital decompression surgery, high doses of glucocorticoids or orbital radiotherapy (Wiersinga, 2007; 2009; Bartalena, 2011). The precise therapeutic strategy, however, is still a matter of debate. Glucocorticoids and orbital radiotherapy are effective in approximately only two-thirds of the patients and come with (substantial) side effects. Glucocorticoid therapy for GO is associated with transient Cushingoid features but also with adverse effects such as diabetes, depression, infections, hypertension, increased body weight, osteoporosis and cataracts. Orbital radiotherapy has limited side effects but proptosis, one of the main features of GO, often is barely affected by irradiation. As the TSH receptor is assumed to play a key role in the cascade of events leading to GO, blockade of this receptor with LMW TSH receptor antagonists could be a cause-directed approach to treat both hyperthyroidism and GO.
In our selectivity assays, Org 274179-0 not only antagonized the TSH receptor but also exhibited equipotent antagonistic activity at the FSH receptor. It is evident that this cross-reactivity at the FSH receptor is not optimal. In males, strong and lasting FSH receptor antagonism might lead to reduced spermatogenesis. In post-menopausal females, however, the presence of a FSH receptor antagonist would not have clinical consequences because FSH receptors are not expressed in their ovaries (or other tissues). Also in pre-menopausal women, who use hormonal contraceptives, the clinical consequences would be very limited. In pre-menopausal women, who do not use contraceptives, a circulating FSH receptor antagonist could lead to disturbances of the menstrual cycle. Having said that, it is current practice that all pre-menopausal female patients with GD treated with antithyroid drugs are strongly advised not to become pregnant during the course of the therapy. It seems therefore reasonable to assume that in the majority of patients, the benefits of Org 274179-0 in treating and preventing the signs of GO would outweigh the burden of a potential temporary impairment of fertility.
In conclusion, we report the molecular pharmacology of the first nanomolar potent TSH receptor antagonist, Org 274179-0, which opens the way for novel treatment paradigms in Graves' disease and Graves' ophthalmopathy.
Acknowledgments
We thank Mr. Maurice van Loosbroek, Mr. Robert Tan and Mr. Jeroen de Roos for expert technical assistance. We are very grateful to Drs. Clementine van Zeijl and Anita Boelen (Department of Endocrinology and Metabolic Diseases, AMC, University of Amsterdam) for providing sera of GD patients and healthy controls.
Glossary
- CRE
cyclic AMP-responsive element
- FSH
follicle-stimulating hormone
- FSHR
FSH receptor
- GD
Graves' disease
- GO
Graves' ophthalmopathy
- GPCR
G protein-coupled receptor
- LH
luteinizing hormone
- LMW
low molecular weight
- Org 274179-0
(S)-N-(1-acetyl-4-methyl-4-phenyl-1,2,3,4-tetrahydro-quinolin-6-yl)-3-(3-trifluoromethyl-phenyl)-propionamide
- TBII
TSH-binding inhibitory immunoglobulins
- TSH
thyroid-stimulating hormone
- TSI
TSH receptor-stimulating immunoglobulins
Conflict of interest
The authors, except Robert D Lane, are paid employees of MSD.
References
- Ando T, Latif R, Davies T. Concentration-dependent regulation of thyrotropin receptor function by thyroid-stimulating antibody. J Clin Invest. 2004;113:1589–1595. doi: 10.1172/JCI21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwa MA, Smallridge RC, Gist ID, Abo-Hashem EM, El-Kannishy HM, Burman KD. Effects of bradykinin and TSH on phospholipase C and A2 systems in retroocular fibroblasts obtained from patients with Graves' ophthalmopathy. Thyroid. 1995;5:S-13. [Google Scholar]
- Bartalena L. The dilemma of how to manage Graves' hyperthyroidism in patients with associated orbitopathy. J Clin Endocrinol Metab. 2011;96:592–599. doi: 10.1210/jc.2010-2329. [DOI] [PubMed] [Google Scholar]
- Boelaert K, Horacek J, Holder RL, Watkinson JC, Sheppard MC, Franklyn JA. Serum thyrotropin concentration as a novel predictor of malignancy in thyroid nodules investigated by fine-needle aspiration. J Clin Endocrinol Metab. 2006;91:4295–4301. doi: 10.1210/jc.2006-0527. [DOI] [PubMed] [Google Scholar]
- Corvilain B, Van Sande J, Dumont JE, Vassart G. Somatic and germline mutations of the TSH receptor and thyroid diseases. Clin Endocrinol (Oxf) 2001;55:143–158. [PubMed] [Google Scholar]
- Duprez L, Parma J, Van Sande J, Rodien P, Dumont JE, Vassart G, et al. TSH receptor mutations and thyroid disease. Trends Endocrinol Metab. 1998;9:133–140. doi: 10.1016/s1043-2760(98)00036-8. [DOI] [PubMed] [Google Scholar]
- Eckstein AK, Plicht M, Lax H, Neuhäuser M, Mann K, Lederbogen S, et al. Thyrotropin receptor autoantibodies are independent risk factors for Graves' ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab. 2006;91:3464–3470. doi: 10.1210/jc.2005-2813. [DOI] [PubMed] [Google Scholar]
- Fiore E, Rago T, Provenzale MA, Scutari M, Ugolini C, Basolo F, et al. Lower levels of TSH are associated with a lower risk of papillary thyroid cancer in patients with thyroid nodular disease: thyroid autonomy may play a protective role. Endocr Relat Cancer. 2009;16:1251–1260. doi: 10.1677/ERC-09-0036. [DOI] [PubMed] [Google Scholar]
- Garrity JA, Bahn RS. Perspective: pathogenesis of Graves' ophthalmopathy: implications for prediction, prevention, and treatment. Am J Ophthalmol. 2006;142:147–153. doi: 10.1016/j.ajo.2006.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MN, van der Meer JW, Broenink M, Bakker O, Wiersinga WM, Prummel MF. Association of thyrotrophin receptor antibodies with the clinical features of Graves' ophthalmopathy. Clin Endocrinol (Oxf) 2000;52:267–271. doi: 10.1046/j.1365-2265.2000.00959.x. [DOI] [PubMed] [Google Scholar]
- Haymart MR, Repplinger DJ, Leverson GE, Elson DF, Sippel RS, Jaume JC, et al. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J Clin Endocrinol Metab. 2008;93:809–814. doi: 10.1210/jc.2007-2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karstens WFJ, Conti PC, Timmers CM, Plate R, Van Koppen CJ. TSH receptor antagonizing tetrahydroquinoline compounds. 2009. WO2009/027482.
- Kumar S, Nadeem S, Stan MN, Coenen M, Bahn RS. A stimulatory thyrotropin receptor antibody enhances adipogenesis via phosphoinositide 3-kinase activation in orbital preadipocytes from patients with Graves' ophthalmopathy. J Mol Endocrinol. 2011;46:155–163. doi: 10.1530/JME-11-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AW, Lorentz T, Tam SC. Thyroxine suppressive therapy decreases bone mineral density in post-menopausal women. Clin Endocrinol (Oxf) 1993;39:535–540. doi: 10.1111/j.1365-2265.1993.tb02405.x. [DOI] [PubMed] [Google Scholar]
- Kung AW, Yau CC, Cheng A. The incidence of ophthalmopathy after radioiodine therapy for Graves' disease: prognostic factors and the role of methimazole. J Clin Endocrinol Metab. 1994;79:542–546. doi: 10.1210/jcem.79.2.7913934. [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taken advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Lytton SD, Ponto KA, Kanitz M, Matheis N, Kohn LD, Kahaly GJ. A novel thyroid stimulating immunoglobulin bioassay is a functional indicator of activity and severity of Graves' orbitopathy. J Clin Endocrinol Metab. 2010;95:2123–2131. doi: 10.1210/jc.2009-2470. [DOI] [PubMed] [Google Scholar]
- Morshed SA, Latif R, Davies TF. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology. 2009;150:519–529. doi: 10.1210/en.2008-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourits MP, Prummel MF, Wiersinga WM, Koornneef L. Clinical activity score as a guide in the management of patients with Graves' ophthalmopathy. Clin Endocrinol (Oxf) 1997;47:9–14. doi: 10.1046/j.1365-2265.1997.2331047.x. [DOI] [PubMed] [Google Scholar]
- Neumann S, Kleinau G, Costanzi S, Moore S, Jiang J, Raaka BM, et al. A low-molecular-weight antagonist for the human thyrotropin receptor with therapeutic potential for hyperthyroidism. Endocrinology. 2008;149:5945–5950. doi: 10.1210/en.2008-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Huang W, Eliseeva E, Titus S, Thomas CJ, Gershengorn MC. A small molecule inverse agonist for the human thyroid-stimulating hormone. Endocrinology. 2010;151:3454–3459. doi: 10.1210/en.2010-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Eliseeva E, McCoy JG, Napolitano G, Giuliani C, Monaco F, et al. A new small-molecule antagonist inhibits Graves' disease antibody activation of the TSH receptor. J Clin Endocrinol Metab. 2011;9:5945–5950. doi: 10.1210/jc.2010-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar BS, Bahn RS, Smith TJ. Current perspective on the pathogenesis of Graves' disease and ophthalmopathy. Endocr Rev. 2003;24:802–835. doi: 10.1210/er.2002-0020. [DOI] [PubMed] [Google Scholar]
- Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH)-receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19:673–716. doi: 10.1210/edrv.19.6.0352. [DOI] [PubMed] [Google Scholar]
- Sanders J, Evans M, Premawardhana LDKE, Depraetere H, Jeffreys J, Richards T, et al. Human monoclonal thyroid stimulating autoantibody. Lancet. 2003;362:126–128. doi: 10.1016/s0140-6736(03)13866-4. [DOI] [PubMed] [Google Scholar]
- Sanders J, Jeffreys J, Depraetere H, Evans M, Richards T, Kiddie A, et al. Characteristics of a human monoclonal autoantibody to the thyrotropin receptor: sequence, structure and function. Thyroid. 2004;14:560–570. doi: 10.1089/1050725041692918. [DOI] [PubMed] [Google Scholar]
- Sanders J, Bolton J, Sanders P, Jeffreys J, Nakata N, Richards T, et al. Effects of TSH receptor mutations on binding and biological activity of monoclonal antibodies and TSH. Thyroid. 2006;16:1195–1206. doi: 10.1089/thy.2006.16.1195. [DOI] [PubMed] [Google Scholar]
- Song Y, Massart C, Chico-Galdo V, Jin L, De Maertelaer V, Decoster C, et al. Species specific thyroid signal transduction: conserved physiology, divergent mechanisms. Mol Cell Endocrinol. 2010;319:56–62. doi: 10.1016/j.mce.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Valyasevi RW, Erickson DZ, Harteneck DA, Dutton CM, Heufelder AE, Jyonouchi SC, et al. Differentiation of human orbital preadipocyte fibroblasts induces expression of functional thyrotropin receptor. J Clin Endocrinol Metab. 1999;84:2557–2562. doi: 10.1210/jcem.84.7.5838. [DOI] [PubMed] [Google Scholar]
- van Koppen CJ, Zaman GJR, Timmers CM, Kelder J, Mosselman S, Van de Lagemaat R, et al. A signaling-selective, nanomolar potent allosteric low molecular weight agonist for the human luteinizing hormone receptor. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:503–514. doi: 10.1007/s00210-008-0318-3. [DOI] [PubMed] [Google Scholar]
- van Zeijl CJ, Fliers E, van Koppen CJ, Surovtseva OV, de Gooyer ME, Mourits MP, et al. Thyrotropin receptor-stimulating Graves' disease immunoglobulins induce hyaluronan synthesis by differentiated orbital fibroblasts from patients with Graves' ophthalmopathy not only via cyclic adenosine monophosphate signaling pathways. Thyroid. 2011;21:169–176. doi: 10.1089/thy.2010.0123. [DOI] [PubMed] [Google Scholar]
- Wakelkamp IM, Bakker O, Baldeshi L, Wiersinga WM, Prummel MF. TSH-R expression and cytokine profile in orbital tissue of active vs. inactive Graves' ophthalmopathy patients. Clin Endocrinol (Oxf) 2003;58:280–288. doi: 10.1046/j.1365-2265.2003.01708.x. [DOI] [PubMed] [Google Scholar]
- Wiersinga WM. Management of Graves' ophthalmopathy. Nat Clin Pract Endocrinol Metab. 2007;3:396–404. doi: 10.1038/ncpendmet0497. [DOI] [PubMed] [Google Scholar]
- Wiersinga WM. Therapy: evidence-based treatment of Graves' ophthalmopathy. Nat Rev Endocrinol. 2009;5:653–654. doi: 10.1038/nrendo.2009.222. [DOI] [PubMed] [Google Scholar]