

Abstract
BACKGROUND AND PURPOSE
mTOR inhibitors are currently used as immunosuppressants in transplanted patients and as promising anti-cancer agents. However, new-onset diabetes is a frequent complication occurring in patients treated with mTOR inhibitors such as rapamycin (Sirolimus). Here, we investigated the mechanisms associated with the diabetogenic effects of chronic Sirolimus administration in rats and in in vitro cell cultures.
EXPERIMENTAL APPROACH
Sirolimus was administered to rats fed either a standard or high-fat diet for 21 days. Metabolic parameters were measured in vivo and in ex vivo tissues. Insulin sensitivity was assessed by glucose tolerance tests and euglycaemic hyperinsulinaemic clamps. Rapamycin effects on glucose metabolism and insulin signalling were further evaluated in cultured myotubes.
KEY RESULTS
Sirolimus induced a decrease in food intake and concomitant weight loss. It also induced specific fat mass loss that was independent of changes in food intake. Despite these beneficial effects, Sirolimus-treated rats were glucose intolerant, hyperinsulinaemic and hyperglycaemic, but not hyperlipidaemic. The euglycaemic hyperinsulinaemic clamp measurements showed skeletal muscle is a major site of Sirolimus-induced insulin resistance. At the molecular level, long-term Sirolimus administration attenuated glucose uptake and metabolism in skeletal muscle by preventing full insulin-induced Akt activation and altering the expression and translocation of glucose transporters to the plasma membrane. In rats fed a high-fat diet, these metabolic defects were exacerbated, although Sirolimus-treated animals were protected from diet-induced obesity.
CONCLUSIONS AND IMPLICATIONS
Taken together, our data demonstrate that the diabetogenic effect of chronic rapamycin administration is due to an impaired insulin action on glucose metabolism in skeletal muscles.
Keywords: Sirolimus, rapamycin, mTOR, PTEN, IRS, GLUT, skeletal muscle, insulin resistance, diabetes, euglycaemic hyperinsulinaemic clamps
Introduction
The protein kinase mTOR (mammalian target of rapamycin) is the catalytic subunit of the protein complexes mTORC1 and mTORC2. These complexes integrate inputs from multiple pathways, including those activated by insulin, growth factors, nutrients and mitogens, thereby acting as key regulators of cell growth and metabolism (Wullschleger et al., 2006; Polak and Hall, 2009). Since deregulation of mTOR activity has been linked to the development of various human cancers and metabolic diseases, mTOR inhibitors are predicted to represent powerful therapeutic agents (Manning, 2004; Dann et al., 2007; Menon and Manning, 2008). In this respect, the anti-fungal macrolide rapamycin (also known in clinics as Sirolimus or Rapamune) is a potent and specific mTOR inhibitor (Tsang et al., 2007), which is currently used as an immunosuppressor to prevent rejection of transplanted organs (Gutierrez-Dalmau and Campistol, 2007). Due to their strong anti-proliferative effects, rapamycin and derivatives have also been approved for the treatment of both renal cell carcinoma and mantle cell lymphoma and are presently tested in clinical trials as therapeutic alternatives to cure other cancer types (Konings et al., 2009; Dancey, 2010).
However, and although holding promise either as an immunosupressor or to treat specific tumours, chronic use of rapamycin has been associated with metabolic, haematological and kidney dysfunctions (Stallone et al., 2009). Importantly, new-onset diabetes, an important risk factor for graft failure and mortality, frequently occurs with rapamycin-based immunosuppressive therapy (Teutonico et al., 2005; Romagnoli et al., 2006; Johnston et al., 2008). This is particularly intriguing since acute administration of rapamycin was shown to improve insulin signalling and glucose uptake in muscle and adipose cells exposed to an excess of nutrients (Tzatsos and Kandror, 2006; Tremblay et al., 2007). This effect was attributed to the inhibition of a downstream effector of the mTORC1 complex, the ribosomal protein S6 kinase (S6K) that phosphorylates insulin receptor substrate (IRS)1 on serine residues in response to insulin, thereby triggering its degradation (Um et al., 2006). A decreased expression of IRS proteins as well as phosphorylation of their negative regulatory sites represent critical mechanisms leading to insulin resistance in peripheral insulin-sensitive tissues (Thirone et al., 2006). On the other hand, recent reports indicate that prolonged in vitro exposure to rapamycin inhibits mTOR within the mTORC2 complex (also known as PDK2) (Sarbassov et al., 2006), which was previously thought to be rapamycin-insensitive (Jacinto et al., 2004). Since mTORC2 phosphorylates and activates Akt, a crucial downstream insulin effector mediating most of the metabolic effects of the hormone (Sarbassov et al., 2006), inhibition of this complex is expected to have a major impact on insulin sensitivity.
Although a central role of mTOR in the control of insulin signalling, as well as in glucose and lipid metabolism, is now well accepted, the precise site of action and the mechanisms involved in the diabetogenic effect of rapamycin are still poorly defined. In this regard, Houde et al. (2010) recently examined the liver sensitivity to rapamycin and reported that hepatic gluconeogenesis was enhanced by chronically inhibiting mTOR in rats. Chronic rapamycin administration also affected insulin-dependent signalling in skeletal muscles of diabetic Psammomys obesus, suggesting an impaired insulin sensitivity of these tissues in this animal model (Fraenkel et al., 2008).
Based on these results, as well as on the fact that defective glucose metabolism in skeletal muscles is a major cause of impaired glucose homeostasis in type 2 diabetes, the aim of the present study was to investigate the effect of systemic and chronic administration in rats of the mTOR inhibitor rapamycin on glucose metabolism, in particular at the level of skeletal muscles. To this end, euglycaemic hyperinsulinaemic clamps, the gold standard method to evaluate overall insulin sensitivity, were performed in association with the labelled 2-deoxy-glucose procedure to measure muscle insulin sensitivity. The mechanisms underlying the metabolic defects induced by rapamycin were further investigated in vitro using rat L6 myotubes. Finally, we determined the effect of rapamycin treatment in a rat model of high fat diet-induced obesity.
Methods
Reagents and antibodies
All reagents and antibodies are described in Table S1.
Animals
Male Wistar rats (Charles River, Arbresle, France) were housed individually (23°C; light on: 07.00–19.00 h) and allowed free access to water and diet (RM1; metabolized energy 2.61 kcal·g−1). Food intake and body weight were measured daily (09:00 h). Rats were killed using isoflurane anaesthesia and rapid decapitation. Blood was collected, and tissues were freeze-clamped and stored at −80°C for further analyses. All animal care and experimental procedures were in accordance with the Swiss guidelines for animal experimentation and were ethically approved by the Geneva health head office.
Treatments
An initial study was performed on rats fed a standard diet. In this experiment, 10 week-old animals (325 g ± 5 g) were randomly divided into three groups: an ad libitum fed control group; a Sirolimus-treated group and a pair-fed (PF) control group fed the same amount of food as that consumed by Sirolimus-treated rats. Sirolimus is a clinically formulated injectable form of rapamycin, which contains, in addition to rapamycin, other inactive components (0.1% sodium CMC, 0.25% Polysobate 80) and was kindly provided by Wyeth Pharma GmbH (Munster, Germany). Animals received daily i.p. injections of either vehicle (0.1% sodium CMC, 0.25% Polysobate 80 in sterile water) for the control and the PF groups, or Sirolimus at a dose of 2 mg·kg−1·day−1.
A second study was carried out on rats fed a high fat (HF) diet. Here, four randomly divided subgroups of 7 week-old male Wistar rats (225 g ± 5 g) were fed either the standard (one subgroup) or a 45% HF diet (Ssniff® EF R/M; metabolized energy 5.42 kcal·g−1, three subgroups) for 6 weeks. After 3 weeks of the different diets (i.e. at the age of 10 weeks), rats fed the standard diet received a daily i.p. injection of vehicle, whereas the three subgroups of animals fed the HF diet were treated as follows: i.p. injection of vehicle for the control and the pair-fed groups, or Sirolimus at a dose of 2 mg·kg−1·day−1.
Respiratory exchange ratio and locomotor activity
Analyses were performed at the end of the 3 week i.p. injection in rats fed a HF diet. We used the 12-cage LabMaster system (TSE Systems GmbH, Berlin, Germany) of the Small Animal Phenotyping Core Facility (CMU, University of Geneva, Geneva), under controlled temperature (22 ± 1°C) and lighting (12 h light–dark cycle). Before the recording, animals were allowed a 4 day acclimatization period in training cages.
Glucose tolerance test (GTT)
Rats were food-deprived for 4 h (08.30–12.30 h), and a glucose load of 1.5 g·kg−1 was administered i.p. Blood samples were collected for glycaemia measurements using Glucotrend® Active (Roche, Basel, Switzerland) and for further analyses of insulin concentrations. The GTT was performed in both standard and HF-diet-fed animals after 10 days of treatment. The last injection of vehicle or Sirolimus was administered 4 h before the GTT.
Euglycaemic hyperinsulinaemic clamps
Overnight-fasted rats were anaesthetized with i.p. sodium pentobarbital (75 mg·kg−1). Euglycaemic hyperinsulinaemic clamps were performed using an insulin infusion of 18 mU·kg−1·min−1 known to completely suppress hepatic glucose production (Terrettaz et al., 1986), and the glucose infusion rate (GIR) was measured (Vettor et al., 1994). Once in steady state, a bolus of 2-deoxy-d-[1-3H]-glucose (30 µCi) was injected to determine the in vivo glucose utilization index of insulin-sensitive tissues, such as skeletal muscles and adipose tissue (Vettor et al., 1994). Rats were then killed by rapid decapitation, and tissues were rapidly removed, frozen and stored at −80°C. Tissue concentrations of 2-deoxy-d-[1-3H]-glucose-6-phosphate were used to calculate the in vivo glucose utilization index, expressed in ng·mg−1·min−1. These experiments were performed in animals fed the standard diet after 20 days of treatment. In that case, the last injection of vehicle or Sirolimus was administered 24 h before the beginning of the clamps.
Body composition
In both standard and HF-fed animals, an EchoMRI-700 quantitative NMR analyser (Echo Medical Systems, Houston, TX, USA) was used to measure total fat mass and lean body mass at the beginning and at the end of treatments (days 0 and 20).
Faeces and skeletal muscle triglyceride content
For the determination of TG content in faeces and skeletal muscles, weighted quantities of frozen tissue (≍100 mg) were powdered under liquid N2 and extracted overnight at 4°C in chloroform–methanol, as previously described (Veyrat-Durebex et al., 2009). The TG content was determined by colorimetric enzymatic analysis as in plasma samples.
Plasma measurements
Plasma glucose was measured by the glucose oxidase method (GLU kit, Roche Diagnostic GmbH, Rotkreuz, Switzerland). Plasma non-esterified fatty acid (NEFA) and triglyceride (TG) levels were determined using Wako Chemicals GmbH (Neuss, Germany) and Biomérieux kits (Marcy l'Etoile, France), respectively. Plasma insulin concentrations were measured by radioimmunoassay, as previously described (Herbert et al., 1965). Whole-blood rapamycin levels were determined by liquid chromatography-electrospray mass spectrometry, as previously described (Ansermot et al., 2008).
Cell cultures
Rat L6 muscle cells were grown in α-MEM/10% FBS and transferred to α-MEM/2% FBS to differentiate into myotubes as previously described (Mitsumoto and Klip, 1992). Rat L6-GLUT4myc myoblasts were kindly provided by A Klip (Toronto, Canada) and cultured as previously described (Wang et al., 1999). Myoblast differentiation into multinucleated myotubes (>85%) was monitored by phase contrast microscopy.
Western analyses
Tissues were homogenized, and cells were lysed in ice-cold RIPA buffer containing phosphatase and protease inhibitors. Equal amounts of proteins were resolved by 10% SDS-PAGE and blotted to nitrocellulose membranes. Proteins were detected with specific primary antibodies and HRP-conjugated secondary antibodies using an ECL kit. Western blots analyses were performed using the ChemiDoc™ XRS from Bio-Rad (Hercules, CA, USA) and the Quantity One™ Software.
Akt activity
Akt (PKB) activity was measured using an Akt Kinase Assay Kit (Nonradioactive) from Cell Signalling Tech. (Danvers, MA, USA) according to the manufacturer's instructions.
Glut4 translocation assay
GLUT4 translocation to the plasma membrane in response to insulin was determined as described in Ishikura et al. (2010) using L6 myoblasts expressing a chimeric GLUT4 transporter bearing a myc epitope in the exofacial portion of the transporter (GLUT4myc). In brief, after a 3 h period in serum-free medium, cells were incubated with or without 10−7 M insulin for 20 min at 37°C. Cells were washed in ice-cold PBS, fixed with 3% (v/v) paraformaldehyde and blocked with 5% (v/v) milk. Surface GLUT4myc was stained by incubating the cells for 1 h with an anti-myc primary antibody (9E10, 2 µg·mL−1) followed by a HRP-conjugated goat anti-mouse IgG secondary antibody. The amount of GLUT4myc expressed at the plasma membrane was then quantitatively determined using an OPD (Sigma, St. Louis, MO, USA) colorimetric assay.
Glucose uptake
Measurements of 2-deoxy-d-[2,6-3H]-glucose uptake by L6 myotubes were performed as previously described (Niu et al., 2003), with minor modifications. Briefly, cells were deprived of serum for 12 h, stimulated with 10−7 M insulin for 45 min before the addition of 5 µM radiolabelled 2-deoxyglucose (0.8 µCi·µL−1). 2-Deoxyglucose uptake was measured for 10 min at 37°C. Uptake was terminated by washing the cells three times with ice-cold PBS, and cells were lysed in 0.1N NaOH. Non-specific glucose uptake was determined in the presence of 10 µM cytochalasin B. Cell-associated radioactivity was measured by liquid scintillation counting in a β-counter (Wallac 1409, Perkin Elmer, Gaithersburg, MD, USA).
Glycogen synthesis
Glycogen synthesis was assessed by measuring d-[3-H3]-glucose incorporation into glycogen, as previously described (Huang et al., 2002), with minor modifications. Briefly, cells were deprived of serum for 12 h, stimulated with 10−7 M insulin for 30 min before the addition of radiolabelled d-glucose (1 µCi·µL−1) for an additional 90 min period. Cells were then washed in ice-cold PBS, lysed in NaOH 2N and boiled for 30 min. Glycogen was precipitated at 4°C, washed, and incorporation of radiolabelled glucose was determined by liquid scintillation counting in a β-counter.
Real-time PCR
Real-time PCR analyses were performed as previously described (Vinciguerra et al., 2008). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ribosomal protein large Po and β-actin transcripts were used as internal controls. Primer sequences are listed in Table S2.
Lipid staining (Oil red O) and triglyceride measurements
Liver sections were fixed in 4% buffered formaldehyde for 5 min at room temperature. Staining of intracellular neutral lipids was performed with Oil red O, and sections were further stained with Harris' haematoxylin. Intracellular triglyceride levels were quantified with a commercially available kit (GPO-PAP).
Statistical analysis
Results are expressed as means ± SEM. For in vivo experiments, statistical analyses using Student's t-test were performed when two groups were considered. An anova test followed by the post hoc Bonferroni test was used when more than two groups were analysed. For in vitro experiments, comparisons were made using the Student's t test. Differences were considered significant when *P < 0.05, **P < 0.01 or ***P < 0.001.
Results
Chronic mTOR inhibition by Sirolimus treatment in rats causes weight loss and a decreased fat mass
To investigate the effects of chronic mTOR inhibition on insulin sensitivity and glucose metabolism in vivo, Wistar rats were chronically administered for 3 weeks with either Sirolimus (2 mg·kg−1·day−1) or vehicle. After 20 days of such treatments, blood Sirolimus levels reached concentrations of 15.6 ± 0.8 µg·L−1, which are in the therapeutic range (5–15 µg·L−1) recommended for patients undergoing Sirolimus-based therapies (French et al., 2001). Sirolimus treatment of Wistar rats fed a standard diet resulted in a significant decrease in cumulative body weight (BW) gain from the third day to the end of the experiment (Figure 1A,B). This was associated with a significantly decreased food intake (Figure 1C). To delineate the effects of Sirolimus that were independent of changes in food intake, a group of vehicle-treated rats was pair-fed (PF) to the amount of food consumed by Sirolimus-treated animals. Cumulative BW gain of the PF control group was lower than that of ad libitum fed controls (Figure 1A,B), suggesting that the BW change of the Sirolimus-treated group was partly mediated by the anorexigenic effect of the drug. Accordingly, compared with the control group, food efficiency was decreased in both Sirolimus-treated and PF control animals (Figure 1D). However, and although not statistically significant, overall BW loss and decreased food efficiency tended to be higher in Sirolimus-treated than in PF rats. This suggested that Sirolimus may exert additional effects on BW that could be independent of changes in food intake. This prompted us to determine the nature of BW loss in the three experimental groups. To this end, body composition was measured before and at the end of the treatments, using an EchoMRI-700 analyser (Echo Medical Systems, Houston, TX, USA). We observed that Sirolimus induced a significant decrease in fat mass independently of changes in food intake (Figure 1E), while the % lean mass of Sirolimus-treated rats relative to total BW was increased compared with both control groups (Figure 1E).
Figure 1.
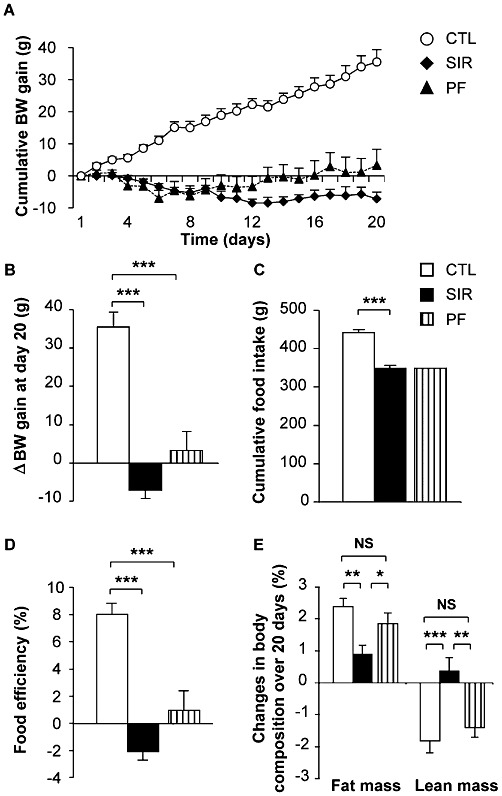
Chronic Sirolimus administration decreases body weight gain, food intake and fat mass of Wistar rats fed a standard diet. Wistar rats were chronically administered for 3 weeks with either vehicle or Sirolimus (2 mg·kg−1·day−1). A third group of rats was pair-fed (PF) with Sirolimus-treated animals. (A) Cumulative body weight (BW) gain over 3 weeks for control (CTL), Sirolimus-treated (SIR) and PF control rats (PF) (Initial BW of animals was of 329.8 ± 4.4 g, 331.2 ± 5.2 g and 334.3 ± 2.9 g; final BW was 365.3 ± 6.4 g, 324.0 ± 5.2 g and 337.6 ± 3.6 g for CTL, SIR and PF groups, respectively). (B) changes in BW, (C) cumulative food intake, (D) food efficiency [(BW gain/cumulative food intake) × 100] over 3 weeks and (E) changes in body composition determined by EchoMRI analysis over 20 days of treatment for control (CTL), Sirolimus-treated (SIR) and PF control rats. Values are mean ± SEM of seven to nine rats per group. NS: not significant, *P < 0.05, **P < 0.01 ***P < 0.001 using anova followed by the post hoc Bonferroni test.
Chronic mTOR inhibition by Sirolimus induces hyperglycaemia, glucose intolerance and insulin resistance
Glucose tolerance was assessed in animals fed a standard diet after 10 days of treatment. Sirolimus induced a significant impairment of glucose tolerance compared with control rats (Figure 2A), despite the fact that Sirolimus-treated animals had a lower BW gain and decreased caloric intake, two factors which usually tend to increase insulin sensitivity (Wing et al., 1994). We also observed a higher basal insulinaemia in Sirolimus-treated animals compared with controls (Figure 2B), that, in view of their normal basal glycaemia (Figure 2A), represents the first evidence of insulin resistance. This was confirmed by the respective homeostatic model assessment (HOMA) values (Table 1). The Sirolimus treatment also induced a delayed but enhanced glucose-induced insulin response during the GTT (Figure 2B).
Figure 2.
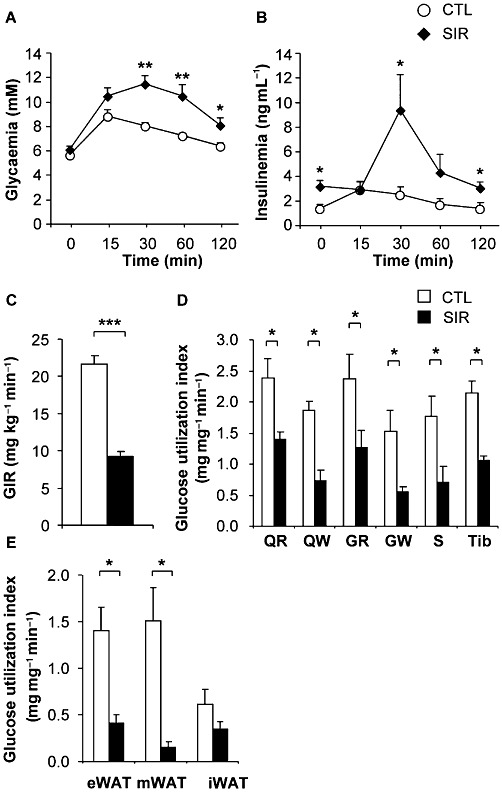
Chronic mTOR inhibition induces glucose intolerance and muscle insulin resistance in Wistar rats fed a standard diet. Wistar rats were chronically administered with either vehicle or Sirolimus (2 mg·kg−1·day−1). (A) Glycaemia and (B) insulinaemia during glucose tolerance tests (1.5 g glucose·kg−1) after 10 days of treatment in vehicle- (CTL) or Sirolimus- (SIR) treated rats. Values are mean ± SEM of seven to eight rats per group. *P < 0.05, **P < 0.01 compared with controls using one-way anova. Euglycaemic hyperinsulinaemic clamps were performed after 3 weeks of chronic administration of either vehicle or Sirolimus (2 mg·kg−1·day−1). (C) GIR in vehicle- (CTL), or Sirolimus-treated (SIR) rats. (D) Insulin-stimulated glucose utilization index in skeletal muscles: red quadriceps (QR), white quadriceps (QW), red gastrocnemius (GR), white gastrocnemius (GW), soleus (S), tibialis (Tib). (E) Insulin-stimulated glucose utilization in white adipose tissue (WAT) depots: epididymal (eWAT), mesenteric (mWAT) and inguinal (iWAT). Values are mean ± SEM of seven to eight rats per group. *P < 0.05, ***P < 0.001 compared with controls using Student's t-test.
Table 1.
Plasma TG, NEFA, insulin and glucose levels in rats fed a standard diet
| Vehicle | Sirolimus | |
|---|---|---|
| TG (mM) | 1.65 ± 0.19 | 1.17 ± 0.07* |
| NEFA (mM) | 0.15 ± 0.02 | 0.17 ± 0.02 |
| Fasting insulinaemia (ng·mL−1) | 1.4 ± 0.2 | 3.5 ± 0.6* |
| Fasting glycaemia (mM) | 4.6 ± 0.1 | 6.4 ± 0.5* |
| HOMA-IR | 8.4 ± 1.4 | 28.0 ± 4.9* |
| Final insulinaemia (ng·mL−1) | 44.4 ± 3.2 | 65.3 ± 3.6* |
| Final glycaemia (mM) | 5.5 ± 0.2 | 5.4 ± 0.1 |
Values are mean ± SEM.
P < 0.05 using the Student's t test.
Surprisingly and contrasting with the fact that a subset of patients under Sirolimus therapy displays hyperlipidaemia, plasma triglyceride (TG) and NEFA levels of Sirolimus-treated rats were either unchanged or decreased compared to the vehicle-treated control group (Table 1).
Chronic mTOR inhibition by Sirolimus triggers skeletal muscle insulin resistance
To investigate the effect of mTOR inhibition on insulin sensitivity, euglycaemic hyperinsulinaemic clamps were performed in rats fed a standard diet and treated or not with Sirolimus. These experiments were designed to specifically focus on insulin-induced glucose metabolism in skeletal muscles and adipose tissue by infusing insulin at a rate known to suppress hepatic glucose production in all groups (Terrettaz et al., 1986). Following an overnight fast, glycaemia and insulinaemia were higher in Sirolimus- than in vehicle-treated rats (Table 1). Glycaemia was similar in both groups at the end of the clamps, while the hyperinsulinaemia reached was higher in the Sirolimus- than in the vehicle-treated group (Table 1). This resulted from the higher basal values brought about by the Sirolimus treatment, together with the fact that the same rate of insulin was infused in both groups. Despite this higher insulinaemia, Sirolimus-treated animals displayed a marked decrease in the glucose infusion rate (GIR) compared with the vehicle-infused controls during the euglycaemic hyperinsulinaemic clamps (Figure 2C). In keeping with such a decreased GIR in Sirolimus-treated animals, the glucose utilization index measured in both red and white skeletal muscles was strongly reduced by the treatment (Figure 2D). Although the low amount of fat mass is unlikely to significantly contribute to the observed overall insulin resistance, Sirolimus treatment also significantly decreased the glucose utilization index of the epididymal and mesenteric, but not the inguinal fat depot (Figure 2E).
Chronic mTOR inhibition by Sirolimus impairs Akt signalling and glucose transporter expression in skeletal muscle
To gain insight into the molecular mechanisms by which Sirolimus triggers muscle insulin resistance, we analysed the expression and phosphorylation of critical insulin signalling effectors in skeletal muscle. As muscles were collected from animals immediately at the end of the 2-DG procedure during euglycaemic hyperinsulinaemic clamps, they were stimulated with insulin before ex vivo tissue analyses. As shown in Figure 3A, the mTOR pathway was effectively blocked by Sirolimus, as evidenced by the lack of phosphorylation of S6, a substrate of the S6K, which is directly activated by mTORC1. Although phosphorylation of the insulin receptor and of Akt on Thr308 was unaffected, Akt phosphorylation on Ser473, which depends on the mTORC2 complex, was completely inhibited in Sirolimus-treated animals, in contrast to what has previously been observed in the liver of rapamycin-treated animals (Houde et al., 2010). This impaired Akt activation was further reflected by a decreased phosphorylation of AS160/TBCD4 and the glycogen synthase kinase 3 (GSK3) (Figure 3A), two important substrates of Akt controlling translocation of glucose transporters to the plasma membrane and glycogen synthesis. These data indicate that chronic Sirolimus exposure prevents full activation of Akt in response to insulin, thereby affecting glucose metabolism in skeletal muscles.
Figure 3.
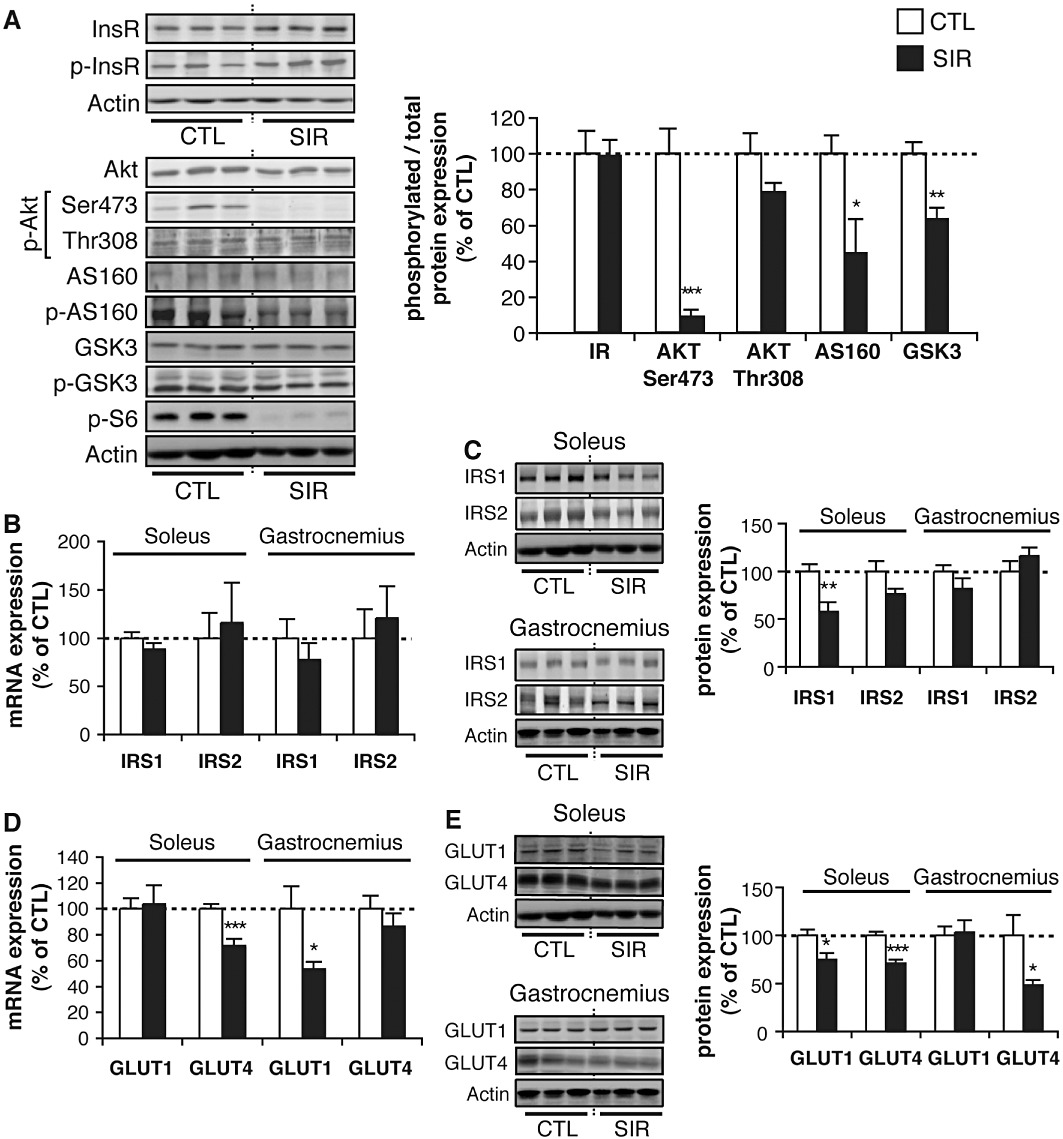
Chronic mTOR inhibition alters insulin signalling and glucose transporter expression in skeletal muscles. Wistar rats fed a standard diet were chronically administered for 3 weeks with either vehicle (CTL) or 2 mg·kg−1·day−1 Sirolimus (SIR). Western blot analyses were then performed on explanted skeletal muscle tissues (soleus or gastrocnemius) of insulin-stimulated rats (tissues sampled immediately at the end of the 2-DG procedure carried out under a 2 h constant insulin infusion at a rate of 18 mU·kg−1·min−1). (A) Representative Western blots and quantifications of phosphorylated over total insulin receptor (InsR), Akt, AS160, GSK3 and S6 ratios in soleus muscles of CTL and SIR rats. (B) mRNA and (C) protein expression of IRS1 and IRS2 in soleus and gastrocnemius muscles of CTL and SIR rats. (D) mRNA and (E) protein expression of GLUT1 and GLUT4 in soleus and gastrocnemius muscles of CTL and SIR rats. Actin detection was used as loading controls for the blots. Values are mean ± SEM of five to eight animals per group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle-treated rats. Representative blots are derived from three different animals per group.
Based on previous reports showing that S6K promotes IRS1 protein degradation (Um et al., 2006), Sirolimus treatment was expected to increase intracellular IRS protein content in rat muscles. On the contrary, depending on the muscle type, we found either a decrease or no change in IRS1 and IRS2 protein expression (Figure 3C), without any alteration in their mRNA levels (Figure 3B). However, it is unlikely that a partial decrease in IRS1 or IRS2 expression is responsible for the strong inhibition of Akt phosphorylation on Ser473, given that Akt phosphorylation on Thr308 was unaffected (Figure 3A).
In vitro analyses have previously shown that expression of glucose transporters may also be modulated by mTOR inhibition (Taha et al., 1995; 1999). Glucose uptake in skeletal muscle is mediated through insulin-dependent and -independent mechanisms, all requiring appropriate expression of specific glucose transporters. More specifically, GLUT1 mediates basal glucose transport, whereas GLUT4 is responsible for insulin-stimulated glucose uptake (Wood and Trayhurn, 2003). We thus investigated whether GLUT1 or GLUT4 expression was affected in vivo by chronic exposure to Sirolimus. As shown in Figure 3D,E, a decreased expression of either GLUT1 or GLUT4 was observed at the mRNA and/or the protein level in the soleus and the gastrocnemius muscles. These data suggest additional mechanisms by which muscle glucose uptake is impaired in response to chronic mTOR inhibition.
Rapamycin inhibits insulin-mediated Akt activation in rat L6 myotubes
Insulin signalling was further investigated in vitro in cultured L6 myotubes exposed to rapamycin for 48 h. In these conditions, viability of L6 myotubes was unaffected (data not shown). Control and rapamycin-treated L6 myotubes were deprived of serum and stimulated with insulin for 5–15 min. Activation of the mTOR/S6K, Akt and ERK1/2 pathways were then assessed by Western blot analyses (Figure 4A,B). As expected, insulin-induced S6K phosphorylation was inhibited by rapamycin. Furthermore, mTOR inhibition resulted in a lower phosphorylation of IRS1 on Ser636/639, with no change in the phosphorylation of Akt on Thr308. In contrast, Akt phosphorylation on Ser473 was significantly decreased, again supporting an inhibitory effect of chronic rapamycin treatment on mTORC2, as previously reported in myeloid cancer cells (Sarbassov et al., 2006). Consistent with a decreased phosphorylation of Akt on Ser473, Akt activity was strongly impaired (Figure 4C). Surprisingly however, analyses of phosphorylated Akt substrates with phosphospecific antibodies for Akt substrates revealed that rapamycin treatment prevented the Akt-dependent phosphorylation of only a subset of these potential substrates in either L6 myotubes (Figure 4D) or skeletal muscles (Figure S1).
Figure 4.
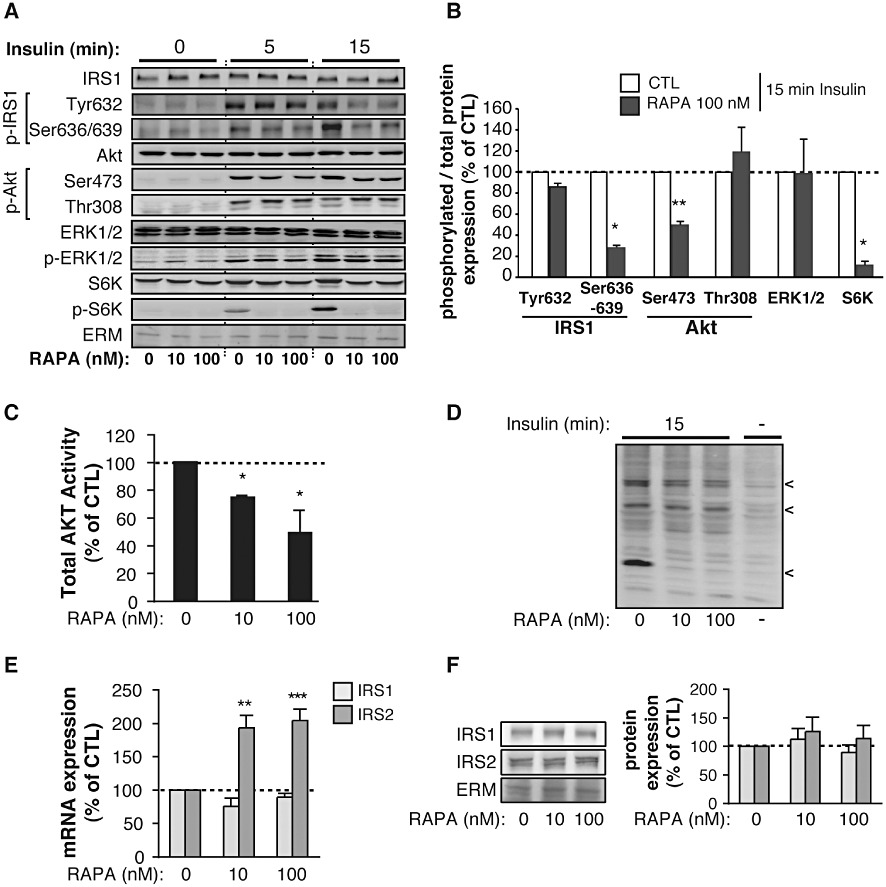
Chronic mTOR inhibition by rapamycin alters Akt activation by insulin in L6 myotubes. L6 myotubes were exposed for 48 h to either DMSO (CTL) or 10–100 nM rapamycin (RAPA) before analyses. (A) Representative Western blots of total and phosphorylated IRS1, Akt, ERK1/2 and S6K expression in rapamycin-treated L6 myotubes stimulated or not with 10−7 M insulin for 5 and 15 min. ERM detection was used as loading controls for the blots. (B) Quantification of IRS1, Akt, ERK1/2 and S6K phosphorylation in L6 myotubes treated (RAPA) or not (CTL) with 100 nM of rapamycin and stimulated with 10−7 M insulin for 15 min. Values are mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 compared with control cells. (C) Akt activity in L6 myotubes treated or not with 100 nM of rapamycin (RAPA) and stimulated with 10−7 M insulin for 15 min. Values are mean ± SEM of two to three independent experiments. *P < 0.05 compared with control cells. (D) Phosphorylated Akt substrates in L6 myotubes treated or not with 100 nM rapamycin (RAPA) and stimulated or not with 10−7 M insulin for 15 min. Representative blot is derived from four independent experiments. (E) mRNA and (F) protein expression of IRS1 and IRS2 in rapamycin-treated (RAPA) L6 myotubes. Values are mean ± SE of three to four independent experiments. **P < 0.01, ***P < 0.001 compared with control cells.
Finally and in contrast to Akt, ERK1/2 activation by insulin was unaffected by rapamycin. Although S6K has been shown to trigger IRS1 degradation through its phosphorylation on serine residues, we also did not observe a higher protein expression of IRS1 or IRS2 in L6 myotubes treated with rapamycin, despite the fact that IRS2 mRNA expression was significantly upregulated (Figure 4E,F).
Rapamycin prevents insulin-induced glucose uptake and glycogen synthesis in L6 cells by altering the expression of GLUT transporters and GLUT4 translocation to the plasma membrane
To further delineate the molecular mechanisms by which mTOR inhibition affects skeletal muscle glucose metabolism in vivo, we investigated the effects of rapamycin on glucose uptake and glycogen synthesis in cultured rat L6 myotubes exposed for 48 h to 10–100 nM rapamycin. In control myotubes, basal glucose uptake was increased by 1.89 ± 0.22-fold following incubation with insulin (Figure 5A). In contrast, 48 h exposure to 10–100 nM rapamycin strongly decreased the basal, as well as the insulin-stimulated glucose uptake (Figure 5A). Similarly, insulin stimulation increased glycogen synthesis in control myotubes by 2.57 ± 0.33-fold (Figure 5B). However, following 48 h exposure to rapamycin, basal glycogen synthesis was reduced, and the insulin-induced glycogen synthesis was completely inhibited (Figure 5B).
Figure 5.
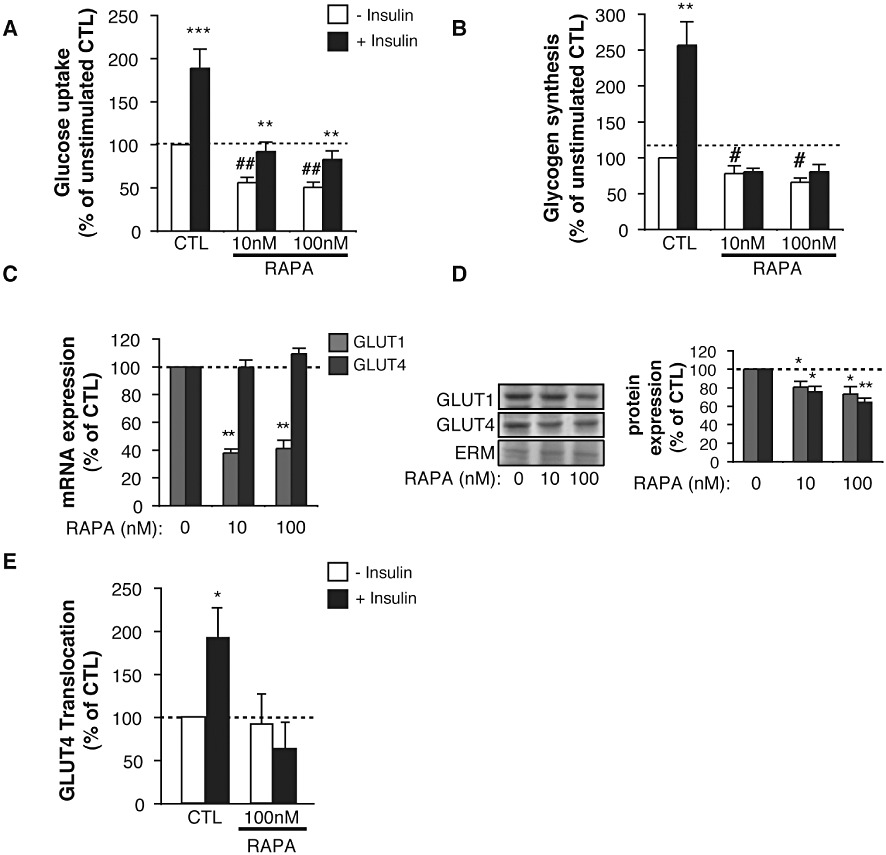
Chronic mTOR inhibition by rapamycin impairs glucose uptake, glycogen synthesis, GLUT transporters expression and translocation to the plasma membrane in response to insulin in L6 cells. L6 myotubes were exposed for 48 h to either DMSO (CTL) or 10–100 nM rapamycin (RAPA) before analyses. Basal (– Insulin) and insulin-stimulated (+Insulin) glucose uptake (A) and glycogen synthesis (B) in L6 myotubes. Results are mean ± SEM of three to nine independent experiments. **P < 0.01 and ***P < 0.001 compared with unstimulated cells for each condition. #P < 0.05 and ##P < 0.01 compared with unstimulated control cells. (C) mRNA and (D) protein expression of GLUT1 and GLUT4 in rapamycin-treated (RAPA) L6 myotubes. Values are mean ± SEM of three to four independent experiments. *P < 0.05, **P < 0.01 compared with control cells. (E) GLUT4myc translocation to the plasma membrane in L6 myoblasts following 20 min exposure to 10−7 M insulin. Values are mean ± SEM of four independent experiments. *P < 0.05 compared with unstimulated control cells.
We then assessed whether an altered expression or translocation to the plasma membrane of GLUT transporters could contribute to the inhibition of glucose uptake and glycogen synthesis in L6 cells. Consistent with our ex vivo observations (Figure 3D), rapamycin treatment downregulated the protein expression of both GLUT1 and GLUT4 in L6 myotubes. However, at the mRNA level, only GLUT1 was decreased, suggesting that rapamycin treatment down-regulates GLUT4 at a post-transcriptional level (Figure 5C,D). More importantly, rapamycin prevented almost completely the translocation of GLUT4 to the plasma membrane following insulin stimulation (Figure 5E).
Sirolimus prevents weight gain and decreases fat mass of rats fed a HF diet
Given that chronic exposure to Sirolimus induces weight loss and decreased fat mass in rats fed a standard diet, we hypothesized that mTOR inhibition could, at least partly, counteract the development of obesity and insulin resistance in response to high fat feeding. To address this question, we investigated the effects of Sirolimus in Wistar rats fed a high fat (HF) diet for 6 weeks and treated or not with Sirolimus or the vehicle for the last 3 weeks. A vehicle-treated control group was pair-fed (PF) to the amount of food consumed by Sirolimus-treated animals. An additional control group was fed a standard diet over the 6 week period and was infused with the vehicle for the last 3 weeks.
Rats fed a HF diet before the treatment with Sirolimus exhibited a significant increase in body weight gain as compared to rats fed a standard diet (Figure 6A,B). However, Sirolimus administration significantly reduced the body weight increase (Figure 6B), consistent with the data obtained in rats fed a standard diet (Figure 1). The decreased body weight in Sirolimus-treated rats partly resulted from reduced food intake (Figure 6C,D) but also from additional mechanisms, as it was significantly more pronounced in the Sirolimus-treated than in the PF control group (Figure 6B). Accordingly, food efficiency was lower in Sirolimus-treated rats independently of changes in food intake (Figure 6E). Similar to what we did in rats fed a standard diet, the effect of Sirolimus treatment on body composition was evaluated using an EchoMRI-700 analyser. We observed that Sirolimus induced a significant decrease in fat mass and a concomitant increase in the % of lean body mass (Figure 6F). Such effects of Sirolimus were unrelated to changes in food intake, as they were not observed in the PF group (Figure 6F). To delineate the mechanisms by which the Sirolimus reduces the fat mass in rats fed a HF diet, we measured the triglyceride (TG) content in the faeces and in skeletal muscles, as well as energy expenditure and locomotor activity of vehicle- and Sirolimus-treated rats. The effect of Sirolimus on the fat mass was not dependent on changes in intestinal absorption, as the faeces TG content did not reveal any difference between the groups (Figure 6G). However, we could not formally exclude the possibility that Sirolimus induces a redistribution of lipid storage from the adipose tissue to skeletal muscles. Indeed, the TG content in muscles tended to be more elevated in Sirolimus-treated animals than in control or PF groups, although the values measured were not significantly different (Figure 6H). In contrast, the respiratory exchange ratio (RER), measured by indirect calorimetry, was lower in Sirolimus- than in vehicle-treated and PF rats (Figure 6I), indicating a higher rate of lipid oxidation in the animals receiving the drug. Of note, locomotor activity and energy expenditure were unaffected by Sirolimus administration (data not shown).
Figure 6.
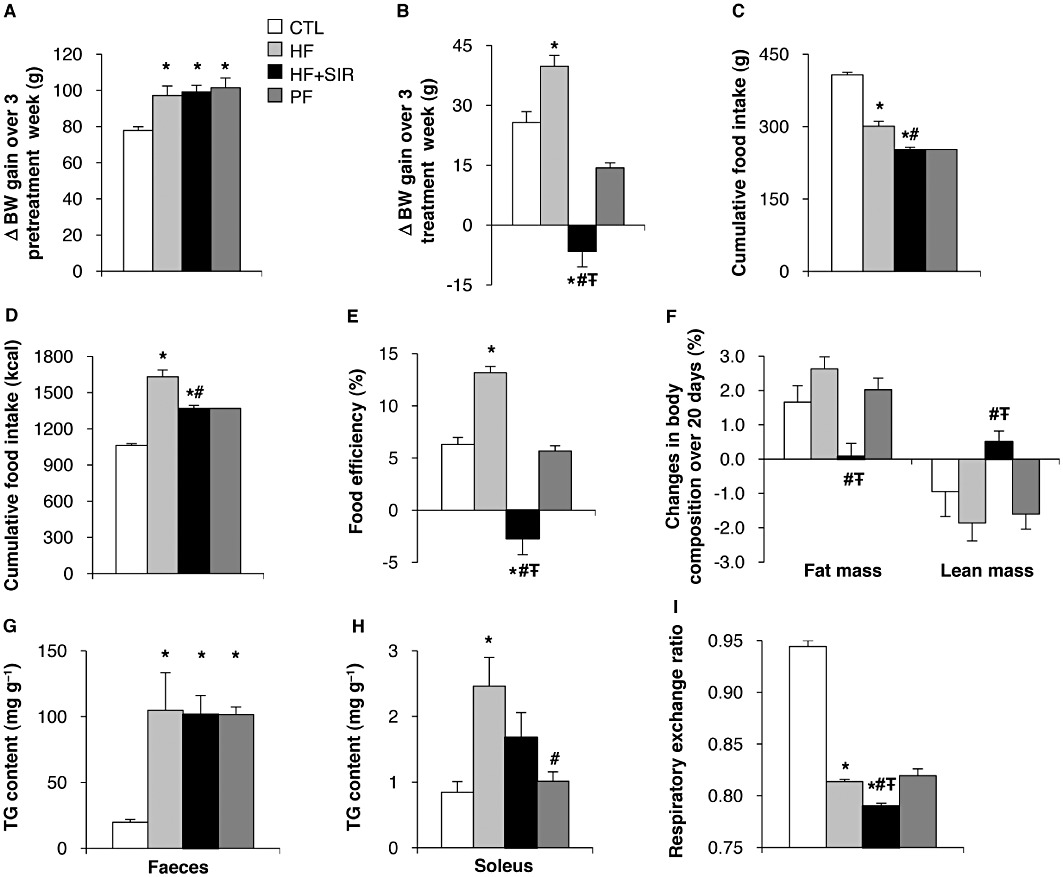
Effect of chronic Sirolimus administration on body weight (BW) gain and composition of Wistar rats fed a high fat (HF) diet. Wistar rats were fed a HF diet for 6 weeks. The last 3 weeks of the experiment, HF diet animals were chronically administered (for 3 weeks) either with vehicle (HF) or Sirolimus (HF + SIR). A third group of rats was pair-fed (PF) with Sirolimus-treated animals. An additional control group of rats (CTL) was fed a standard diet for 6 weeks. (A) Changes in BW of rats fed a standard or a HF diet during weeks 1 to 3 of the experiment. (B) Changes in BW of rats fed a standard or a HF diet with or without Sirolimus treatment during weeks 4 to 6 of the experiment. (C–D) Cumulative food intake over 3 weeks in g (C) and kcal (D). (E) Food efficiency [(BW gain/cumulative food intake) × 100] calculated over weeks 4 to 6 of the treatment period. (F) Changes in body composition determined by EchoMRI analysis over 20 days of treatment. (G) TG content in faeces, (H) TG content in soleus muscle and (I) respiratory exchange ratio (VCO2: VO2). Values are mean ± SEM of six rats per group. *P < 0.05 compared to standard diet fed controls. #P < 0.05 compared with HF diet-fed controls. ŦP < 0.05 compared with HF diet fed PF controls using anova followed by the post hoc Bonferroni test.
Finally, when rats were fed a HF diet, Sirolimus treatment normalized plasma TG levels (Table 2). This effect was attributed to the anorexigenic effect of rapamycin as it was also observed in the HF/PF control group (Table 2). In contrast, Sirolimus administration had no effect on circulating NEFA concentrations in HF-fed animals (Table 2), consistent with the data obtained in rats fed a standard diet (Table 1).
Table 2.
Plasma TG and NEFA in rats fed a high fat diet
| Vehicle | Vehicle | Sirolimus | PF | |
|---|---|---|---|---|
| Standard diet | HF diet | HF diet | HF diet | |
| TG (mM) | 1.26 ± 0.18 | 2.27 ± 0.14a | 1.25 ± 0.15b | 1.13 ± 0.08b |
| NEFA (mM) | 0.15 ± 0.02 | 0.25 ± 0.02a | 0.29 ± 0.01a | 0.32 ± 0.02a |
TG and NEFA levels were determined under basal conditions. Values are mean ± SEM of six to seven animals per group.
P < 0.05 using anova followed by the post hoc Bonferroni test, with significance compared with a: vehicle-treated standard-diet-fed group and b: vehicle-treated HF-fed group.
Sirolimus prevents hepatic steatosis but exacerbates glucose intolerance and insulin resistance in rats fed a HF diet
Interestingly, although NEFA levels were still elevated in Sirolimus-treated HF-fed rats (Table 2), hepatic steatosis, which developed with HF feeding, was totally prevented (Figure 7C). In addition, the TG content as well as the PTEN expression, which is down-regulated in the liver by mTOR-dependent mechanisms, thereby triggering the development of steatosis (Vinciguerra et al., 2008), were restored to normal levels in Sirolimus-treated rats (Figure 7A,B).
Figure 7.
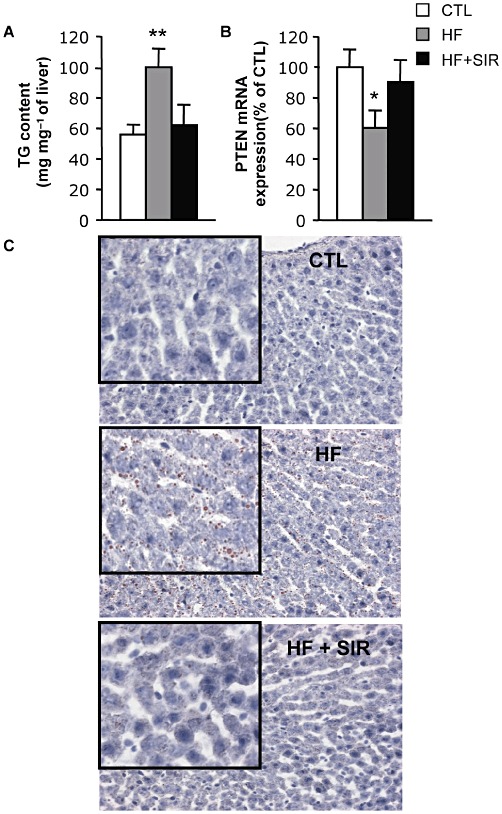
Sirolimus prevents triglyceride accumulation and PTEN down-regulation in the liver of HF-fed rats. Wistar rats were fed a HF diet for 6 weeks. During the last 3 weeks of the experiment, HF fed animals were divided into two groups either treated with 2 mg·kg−1·day−1 Sirolimus (HF + SIR) or vehicle (HF). An additional control group of rats (CTL) was fed a standard diet for 6 weeks. (A) Triglyceride content in the liver of CTL, HF and HF + SIR rats. (B) PTEN mRNA expression in the liver of CTL, HF and HF + SIR rats. Values are mean ± SEM of five to six animals per group. *P < 0.05, **P < 0.01 compared with controls. (C) Representative Oil red O staining of liver sections (200×) of CTL, HF and HF + SIR rats. Inserts are 400× magnifications of a representative area for each section.
However, despite the beneficial effects on body weight, plasma TG levels and hepatic steatosis, chronic Sirolimus administration further exacerbated the glucose intolerance brought about by HF feeding (Figure 8A). The treatment also resulted in a higher hyperinsulinaemia (Table 3) and a lack of glucose-induced insulin response during the GTT (Figure 8B), suggesting the occurrence of increased insulin resistance in peripheral tissues and/or a defective β-cell secretory response. This was further supported by a drastic increase in the respective calculated HOMA values (Table 3). Together, these data indicate that, although chronic mTOR inhibition in HF-fed rats has important beneficial effects on lipid metabolism (fat mass, liver steatosis and dyslipidaemia), it exacerbates the HF diet-induced overall insulin resistance and glucose intolerance.
Figure 8.
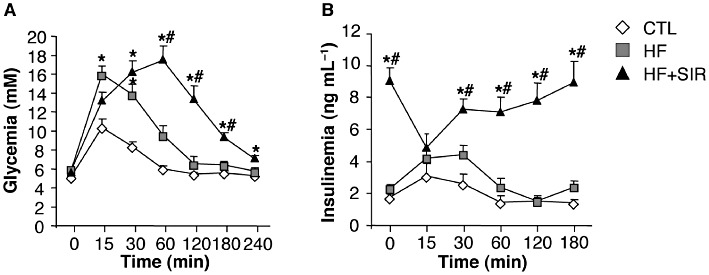
Chronic Sirolimus administration exacerbates glucose intolerance of rats fed a high-fat (HF) diet. Wistar rats were fed a HF diet for 6 weeks. During the last 3 weeks of the experiment, the animals fed the HF diet were divided into two groups, either treated with 2 mg·kg−1·day−1 Sirolimus (HF + SIR) or vehicle (HF). An additional control group of rats (CTL) was fed a standard diet for 6 weeks. (A) Glycaemia and (B) insulinaemia during glucose tolerance tests (1.5 g glucose·kg−1) after 10 days of treatment. Values are mean ± SEM of six to seven rats per group. *P < 0.05 compared with standard diet fed controls. #P < 0.05 compared with HF-fed controls.
Table 3.
Insulin and glucose levels in rats fed a high fat diet
| Vehicle | Vehicle | Sirolimus | |
|---|---|---|---|
| Standard diet | HF diet | HF diet | |
| Fasting insulinaemia (ng·mL−1) | 1.8 ± 0.2 | 2.3 ± 0.3 | 9.0 ± 0.8a,b |
| Fasting glycaemia (mM) | 5.2 ± 0.1 | 5.8 ± 0.2a | 5.8 ± 0.2a |
| HOMA-IR | 11.7 ± 1.3 | 17.4 ± 2.5 | 67.0 ± 6.9a,b |
Insulinaemia and glycaemia were measured after a 4 h period of fast. HOMA-IR was calculated as follows: (fasting plasma insulin in mU·L−1) × (fasting plasma glucose in mM)/22.5. Values are mean ± SEM of six to seven animals per group.
P < 0.05 using anova followed by the post hoc Bonferroni test, with significance compared with a: vehicle-treated standard-diet-fed group and b: vehicle-treated HF-fed group.
Discussion
Recently strong evidence has accumulated indicating that long-term rapamycin (Sirolimus) administration causes new-onset diabetes in organ-transplanted patients. However, the underlying mechanisms of the systemic metabolic effects of rapamycin are still poorly understood. Globally, our study indicates that, while rapamycin prevents excessive body weight gain, fat accumulation and hepatic steatosis, it also leads to hyperglycaemia, insulin resistance and glucose intolerance, phenomena that are exacerbated when associated with a HF diet. We also observed that skeletal muscle represents one of the major sites of insulin resistance in rats chronically exposed to rapamycin. At the molecular level, rapamycin-induced inhibition of insulin-dependent Akt activation, as well as expression/trafficking of glucose transporters, are important defects contributing to insulin resistance in muscle cells.
Hyperlipidaemia, potentially resulting from increased adipose tissue lipolysis or hepatic TG synthesis, has been suggested to represent one of the factors contributing to peripheral insulin resistance following systemic mTOR inhibition (Morrisett et al., 2002; Chakrabarti et al., 2010; Houde et al., 2010; Kumar et al., 2010). However in our study, circulating NEFA and TG levels in Sirolimus-treated rats were not increased, but rather reduced, probably reflecting the marked rapamycin-induced decreases in body weight, food intake and fat mass, also observed in other rodent models (Chang et al., 2009; Houde et al., 2010; Kumar et al., 2010). In addition, when rats were fed a HF diet and treated with Sirolimus, ectopic deposition of lipids in peripheral tissues such as the liver and the skeletal muscles was decreased, although insulin resistance of these animals was exacerbated. Together, these data suggest that lipotoxicity in peripheral tissues is not the primary cause of the Sirolimus-induced insulin resistance in rats.
As regards the mechanisms by which the fat mass is reduced in animals treated with Sirolimus, previous studies have reported that rapamycin, or deletion of a component of the mTOR complexes, stimulates lipolysis and/or prevents lipogenesis in white adipose cells (Polak et al., 2008; Chakrabarti et al., 2010). In addition, our data indicated that the decrease in the fat mass was unrelated to a defect in lipid absorption, an increased energy expenditure or an ectopic redistribution of lipids in peripheral organs. In contrast, we observed a higher rate of lipid oxidation in peripheral tissues, which might be subsequent to a defective glucose uptake in animals treated with Sirolimus. Interestingly, alterations of the mitochondrial oxidative function has been previously reported in mice treated with rapamycin (Cunningham et al., 2007), but whether such defects affect insulin sensitivity and glucose tolerance remain controversial (Wredenberg et al., 2006; Cunningham et al., 2007; Pospisilik et al., 2007). Interestingly, taken together these data support the concept that although long-term systemic administration of rapamycin affects insulin sensitivity and the hepatic and muscle glucose metabolism, it might be paradoxically protective against the development of other metabolic disorders including the development of non-alcoholic fatty liver disease and diet-induced obesity.
A major finding of this study is that overall insulin resistance induced by chronic in vivo mTOR inhibition involves defective skeletal muscle glucose utilization. This was confirmed in vitro by showing that rapamycin decreases insulin-stimulated GLUT4 translocation to the plasma membrane, glucose uptake and glycogen synthesis in muscle L6 cells. Our data also indicate that rapamycin impairs muscle glucose metabolism by preventing the insulin-induced Akt activation, probably through an inhibition of the TORC2 complex (Sarbassov et al., 2006), and the phosphorylation of important Akt substrates, including AS160, which is critical to trigger GLUT4 translocation to the plasma membrane (Zaid et al., 2008). Controversial data have been reported about the effect of long-term exposure to rapamycin on Akt signalling in muscle. Houde et al. (2010) did not observe any difference in the ability of Akt immunoprecipitates coming from skeletal muscles of Sprague–Dawley rats to phosphorylate specific synthetic substrate peptides, but they did not analyse the phosphorylation status of Akt. In contrast, and similar to our observations in Wistar rats, Fraenkel et al. (2008) showed that chronic rapamycin administration affects insulin-dependent Akt and GSK3β phosphorylation in skeletal muscles of normoglycaemic and diabetic Psammomys obesus. Although the discrepancies between our results and the data from Houde et al. (2010) are still unclear, it is possible that the sensitivity of muscle Akt to rapamycin may vary among different animal models, or that a reduced Akt activity induced by rapamycin affects only a subset of Akt substrates. The latter hypothesis is supported by our analyses of various Akt substrate phosphorylation showing that only specific substrates are affected by a reduced Akt phosphorylation/activity in rapamycin-treated muscle cells or tissues. In addition, although it was reported that a small fraction of phosphorylated Akt is sufficient to induce GLUT4 translocation to the plasma membrane of wild-type L6 muscle cells (Hoehn et al., 2008), our data indicate that, in rapamycin-treated cells, fully activated Akt might be required to effectively incorporate glucose. This might be related to the lower expression of GLUT transporters in rapamycin-treated cells, the requirement for a timely and spatially specific activation of Akt to trigger GLUT4 translocation or negative cross-talks with other pathways altered by rapamycin. Along this line, it is likely that other effectors are involved in the chronic rapamycin effects on muscle glucose metabolism, since reduced glucose transport but increased basal glycogen synthase activity were reported in muscle-specific rictor knockout mice (Kumar et al., 2008).
Interestingly, we found that chronic rapamycin administration also down-regulates muscle GLUT4 expression in vivo and GLUT1/4 in L6 myotubes. Consistent with our data, insulin-induced GLUT1 expression was found to be attenuated by rapamycin in 3T3-L1 adipocytes and L6 cells (Taha et al., 1999). Acute inhibition of mTOR/S6K signalling by rapamycin was also reported to prevent IRS1 degradation, thus increasing insulin signalling and sensitivity (Um et al., 2006). However, we did not observe any increased IRS expression, or Akt phosphorylation on Thr308 in response to chronic rapamycin treatment in muscles ex vivo or in L6 myotubes. These data suggest that upon chronic mTOR inhibition, compensatory mechanisms may restore physiological signalling of IRS effectors.
In addition to the impressive muscle insulin resistance, defects in other tissues might also contribute to the diabetogenic effects of rapamycin. For example, chronic exposure to rapamycin was reported to enhance hepatic gluconeogenesis through mechanisms unrelated to a defective Akt signalling in the liver (Houde et al., 2010). Defects in hepatic insulin clearance were also reported in the same study (Houde et al., 2010), consistent with the increased insulinaemia that we observed in Sirolimus-treated rats compared with controls. Considering our data together with those reported in this previous study, it appears that systemic mTOR inhibition may lead to tissue-specific alterations of insulin signalling and glucose homeostasis. On the other hand, as regards the liver, we previously showed that free fatty acids trigger insulin resistance and steatosis in hepatocytes by down-regulating PTEN expression through mTOR-dependent mechanisms (Vinciguerra et al., 2008; 2009). In addition, a recent study also suggests that the mTORC1 complex controls diet-induced hepatic lipid accumulation (Kenerson et al., 2011). Consistent with these findings, we observed that mTOR inhibition in HF-fed rats prevented PTEN down-regulation and development of steatosis, although circulating NEFA levels were still high.
Finally, our data point to an effect of rapamycin on insulin secretion, as treated animals were hyperinsulinaemic in the basal state but had an impaired glucose-induced insulin response. Contradictory results have been reported on the presence or the absence of beneficial effects on the islet mass and function (Whiting et al., 1991; Kneteman et al., 1996; Marcelli-Tourvieille et al., 2007), while others described an impairment of beta-cell survival, insulin secretion and islets engraftment (Bell et al., 2003; Bussiere et al., 2006; Zhang et al., 2006; Fraenkel et al., 2008; Houde et al., 2010). Interestingly, adipose-specific rictor knockout mice have an enlarged pancreas and are hyperinsulinaemic, suggesting a potential for cross-talk between mTOR signalling in adipose tissue and pancreatic function (Cybulski et al., 2009). Although investigating the effects of rapamycin on the beta-cell is beyond the scope of this study, our data are consistent with an impairment of beta-cell function elicited by chronic mTOR inhibition.
In conclusion, rapamycin seems to decrease adiposity and to protect against diet-induced obesity, while promoting hyperglycaemia and hyperinsulinaemia, together with a strong muscle insulin resistance. Based on the present study, it is proposed that a decreased insulin-induced Akt activation and alterations of expression/trafficking of glucose transporters importantly contribute to the alterations of the glucose metabolism induced by rapamycin in skeletal muscles. Diabetes is an important independent risk factor for the development of several cancer types and a major cause of graft failure and mortality in transplanted patients. In light of such a spectrum of actions, caution should be exerted in the clinical use of mTOR inhibitors as immune suppressors or anti-cancer therapy.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grants N°310000–120147/1 and N°31003A-134919/1) to FRJ; the Swiss National Science Foundation (grant N°310030–135727/1), the Reuter Foundation, The Sir Jules Thorn Charitable Overseas Trust Reg., Schaan, The Fondation Romande pour la Recherche sur le Diabète and the EFSD Research Programme in Diabetes and Cancer to MF. The authors thank A Klip (Cell Biology Program, The Hospital for Sick Children, Toronto, Canada) for providing rat L6-GLUT4myc cells, C Wollheim, Department of Cell Physiology and Metabolism, University of Geneva, Switzerland for critically reading the manuscript and Wyeth Pharma GmbH (Germany) for providing the Sirolimus.
Glossary
- BW
body weight
- GIR
glucose infusion rate
- GLUT
glucose transporter
- GTT
glucose tolerance test
- HF
high fat
- IRS
insulin receptor substrate
- mTOR
mammalian target of rapamycin
- NEFA
non-esterified fatty acids
- PF
pair-fed
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- RAPA
rapamycin
- SIR
Sirolimus
- TG
triglycerides
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Chronic mTOR inhibition alters insulin-dependent phosphorylation of Akt substrates in skeletal muscles. Wistar rats fed a standard diet were chronically administered for 3 weeks with either vehicle (CTL) or 2 mg·kg−1·day−1 Sirolimus (SIR). Western analyses were then performed on explanted skeletal muscle tissues (gastrocnemius) of insulin-stimulated rats (tissues sampled immediately at the end of the 2-DG procedure carried out under 2 h constant insulin infusion at a rate of 18 mU·kg−1·min−1) to analyse the phosphorylation of Akt potential substrates in skeletal muscles. Representative blots are derived from three different animals per group.
Table S1 Reagents and antibodies
Table S2 Primer sequences for real-time PCR
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ansermot N, Fathi M, Veuthey JL, Desmeules J, Rudaz S, Hochstrasser D. Simultaneous quantification of cyclosporine, tacrolimus, sirolimus and everolimus in whole blood by liquid chromatography-electrospray mass spectrometry. Clin Biochem. 2008;41:728–735. doi: 10.1016/j.clinbiochem.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Bell E, Cao X, Moibi JA, Greene SR, Young R, Trucco M, et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes. 2003;52:2731–2739. doi: 10.2337/diabetes.52.11.2731. [DOI] [PubMed] [Google Scholar]
- Bussiere CT, Lakey JR, Shapiro AM, Korbutt GS. The impact of the mTOR inhibitor sirolimus on the proliferation and function of pancreatic islets and ductal cells. Diabetologia. 2006;49:2341–2349. doi: 10.1007/s00125-006-0374-5. [DOI] [PubMed] [Google Scholar]
- Chakrabarti P, English T, Shi J, Smas CM, Kandror KV. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes. 2010;59:775–781. doi: 10.2337/db09-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GR, Wu YY, Chiu YS, Chen WY, Liao JW, Hsu HM, et al. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol. 2009;105:188–198. doi: 10.1111/j.1742-7843.2009.00427.x. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci U S A. 2009;106:9902–9907. doi: 10.1073/pnas.0811321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancey J. mTOR signaling and drug development in cancer. Nat Rev. Clin Oncol. 2010;7:209–219. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- French DC, Saltzgueber M, Hicks DR, Cowper AL, Holt DW. HPLC assay with ultraviolet detection for therapeutic drug monitoring of sirolimus. Clin Chem. 2001;47:1316–1319. [PubMed] [Google Scholar]
- Gutierrez-Dalmau A, Campistol JM. Immunosuppressive therapy and malignancy in organ transplant recipients: a systematic review. Drugs. 2007;67:1167–1198. doi: 10.2165/00003495-200767080-00006. [DOI] [PubMed] [Google Scholar]
- Herbert V, Lau KS, Gottlieb CW, Bleicher SJ. Coated charcoal immunoassay of insulin. J Clin Endocrinol Metab. 1965;25:1375–1384. doi: 10.1210/jcem-25-10-1375. [DOI] [PubMed] [Google Scholar]
- Hoehn KL, Hohnen-Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, et al. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008;7:421–433. doi: 10.1016/j.cmet.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Cheung AT, Parsons JT, Bryer-Ash M. Focal adhesion kinase (FAK) regulates insulin-stimulated glycogen synthesis in hepatocytes. J Biol Chem. 2002;277:18151–18160. doi: 10.1074/jbc.M104252200. [DOI] [PubMed] [Google Scholar]
- Ishikura S, Antonescu CN, Klip A. Documenting GLUT4 exocytosis and endocytosis in muscle cell monolayers. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1515s46. Chapter 15: Unit 15.15. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol. 2008;19:1411–1418. doi: 10.1681/ASN.2007111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenerson HL, Yeh MM, Yeung RS. Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PLoS ONE. 2011;6:e18075. doi: 10.1371/journal.pone.0018075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneteman NM, Lakey JR, Wagner T, Finegood D. The metabolic impact of rapamycin (sirolimus) in chronic canine islet graft recipients. Transplantation. 1996;61:1206–1210. doi: 10.1097/00007890-199604270-00015. [DOI] [PubMed] [Google Scholar]
- Konings IR, Verweij J, Wiemer EA, Sleijfer S. The applicability of mTOR inhibition in solid tumors. Curr Cancer Drug Targets. 2009;9:439–450. doi: 10.2174/156800909788166556. [DOI] [PubMed] [Google Scholar]
- Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC., Jr Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol. 2008;28:61–70. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Lawrence JC, Jr, Jung DY, Ko HJ, Keller SR, Kim JK, et al. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59:1397–1406. doi: 10.2337/db09-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelli-Tourvieille S, Hubert T, Moerman E, Gmyr V, Kerr-Conte J, Nunes B, et al. In vivo and in vitro effect of sirolimus on insulin secretion. Transplantation. 2007;83:532–538. doi: 10.1097/01.tp.0000255679.81792.dd. [DOI] [PubMed] [Google Scholar]
- Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;27(Suppl. 2):S43–S51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsumoto Y, Klip A. Development regulation of the subcellular distribution and glycosylation of GLUT1 and GLUT4 glucose transporters during myogenesis of L6 muscle cells. J Biol Chem. 1992;267:4957–4962. [PubMed] [Google Scholar]
- Morrisett JD, Abdel-Fattah G, Hoogeveen R, Mitchell E, Ballantyne CM, Pownall HJ, et al. Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J Lipid Res. 2002;43:1170–1180. [PubMed] [Google Scholar]
- Niu W, Huang C, Nawaz Z, Levy M, Somwar R, Li D, et al. Maturation of the regulation of GLUT4 activity by p38 MAPK during L6 cell myogenesis. J Biol Chem. 2003;278:17953–17962. doi: 10.1074/jbc.M211136200. [DOI] [PubMed] [Google Scholar]
- Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21:209–218. doi: 10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008;8:399–410. doi: 10.1016/j.cmet.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Romagnoli J, Citterio F, Nanni G, Favi E, Tondolo V, Spagnoletti G, et al. Incidence of posttransplant diabetes mellitus in kidney transplant recipients immunosuppressed with sirolimus in combination with cyclosporine. Transplant Proc. 2006;38:1034–1036. doi: 10.1016/j.transproceed.2006.03.072. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Stallone G, Infante B, Grandaliano G, Gesualdo L. Management of side effects of sirolimus therapy. Transplantation. 2009;87(8 Suppl.):S23–S26. doi: 10.1097/TP.0b013e3181a05b7a. [DOI] [PubMed] [Google Scholar]
- Taha C, Mitsumoto Y, Liu Z, Skolnik EY, Klip A. The insulin-dependent biosynthesis of GLUT1 and GLUT3 glucose transporters in L6 muscle cells is mediated by distinct pathways. Roles of p21ras and pp70 S6 kinase. J Biol Chem. 1995;270:24678–24681. doi: 10.1074/jbc.270.42.24678. [DOI] [PubMed] [Google Scholar]
- Taha C, Liu Z, Jin J, Al-Hasani H, Sonenberg N, Klip A. Opposite translational control of GLUT1 and GLUT4 glucose transporter mRNAs in response to insulin. Role of mammalian target of rapamycin, protein kinase b, and phosphatidylinositol 3-kinase in GLUT1 mRNA translation. J Biol Chem. 1999;274:33085–33091. doi: 10.1074/jbc.274.46.33085. [DOI] [PubMed] [Google Scholar]
- Terrettaz J, Assimacopoulos-Jeannet F, Jeanrenaud B. Severe hepatic and peripheral insulin resistance as evidenced by euglycemic clamps in genetically obese fa/fa rats. Endocrinology. 1986;118:674–678. doi: 10.1210/endo-118-2-674. [DOI] [PubMed] [Google Scholar]
- Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. 2005;16:3128–3135. doi: 10.1681/ASN.2005050487. [DOI] [PubMed] [Google Scholar]
- Thirone AC, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab. 2006;17:72–78. doi: 10.1016/j.tem.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26:63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, D'Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Vettor R, Zarjevski N, Cusin I, Rohner-Jeanrenaud F, Jeanrenaud B. Induction and reversibility of an obesity syndrome by intracerebroventricular neuropeptide Y administration to normal rats. Diabetologia. 1994;37:1202–1208. doi: 10.1007/BF00399793. [DOI] [PubMed] [Google Scholar]
- Veyrat-Durebex C, Montet X, Vinciguerra M, Gjinovci A, Meda P, Foti M, et al. The Lou/C rat: a model of spontaneous food restriction associated with improved insulin sensitivity and decreased lipid storage in adipose tissue. Am J Physiol Endocrinol Metab. 2009;296:E1120–E1132. doi: 10.1152/ajpendo.90592.2008. [DOI] [PubMed] [Google Scholar]
- Vinciguerra M, Veyrat-Durebex C, Moukil MA, Rubbia-Brandt L, Rohner-Jeanrenaud F, Foti M. PTEN down-regulation by unsaturated fatty acids triggers hepatic steatosis via an NF-kappaBp65/mTOR-dependent mechanism. Gastroenterology. 2008;134:268–280. doi: 10.1053/j.gastro.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Vinciguerra M, Sgroi A, Veyrat-Durebex C, Rubbia-Brandt L, Buhler LH, Foti M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology. 2009;49:1176–1184. doi: 10.1002/hep.22737. [DOI] [PubMed] [Google Scholar]
- Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, et al. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol. 1999;19:4008–4018. doi: 10.1128/mcb.19.6.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PH, Woo J, Adam BJ, Hasan NU, Davidson RJ, Thomson AW. Toxicity of rapamycin–a comparative and combination study with cyclosporine at immunotherapeutic dosage in the rat. Transplantation. 1991;52:203–208. doi: 10.1097/00007890-199108000-00004. [DOI] [PubMed] [Google Scholar]
- Wing RR, Blair EH, Bononi P, Marcus MD, Watanabe R, Bergman RN. Caloric restriction per se is a significant factor in improvements in glycemic control and insulin sensitivity during weight loss in obese NIDDM patients. Diabetes Care. 1994;17:30–36. doi: 10.2337/diacare.17.1.30. [DOI] [PubMed] [Google Scholar]
- Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89:3–9. doi: 10.1079/BJN2002763. [DOI] [PubMed] [Google Scholar]
- Wredenberg A, Freyer C, Sandstrom ME, Katz A, Wibom R, Westerblad H, et al. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun. 2006;350:202–207. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zaid H, Antonescu CN, Randhawa VK, Klip A. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem J. 2008;413:201–215. doi: 10.1042/BJ20080723. [DOI] [PubMed] [Google Scholar]
- Zhang N, Su D, Qu S, Tse T, Bottino R, Balamurugan AN, et al. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes. 2006;55:2429–2436. doi: 10.2337/db06-0173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.