

Abstract
BACKGROUND AND PURPOSE
Inhalation of a β-adrenoceptor agonist (β-agonist) is first-line asthma therapy, used for both prophylaxis against, and acute relief of, bronchoconstriction. However, repeated clinical use of β-agonists leads to impaired bronchoprotection and, in some cases, adverse patient outcomes. Mechanisms underlying this β2-adrenoceptor dysfunction are not well understood, due largely to the lack of a comprehensive animal model and the uncertainty as to whether or not bronchorelaxation in mice is mediated by β2-adrenoceptors. Thus, we aimed to develop a mouse model that demonstrated functional β-agonist-induced β2-adrenoceptor desensitization in the context of allergic inflammatory airway disease.
EXPERIMENTAL APPROACH
We combined chronic allergen exposure with repeated β-agonist inhalation in allergen-treated BALB/C mice and examined the contribution of β2-adrenoceptors to albuterol-induced bronchoprotection using FVB/NJ mice with genetic deletion of β2-adrenoceptors (KO). Associated inflammatory changes – cytokines (ELISA), cells in bronchoalevolar lavage and airway remodelling (histology) and β2-adrenoceptor density (radioligand binding) – were also measured.
KEY RESULTS
β2-Adrenoceptors mediated albuterol-induced bronchoprotection in mice. Chronic treatment with albuterol induced loss of bronchoprotection, associated with exacerbation of the inflammatory components of the asthma phenotype.
CONCLUSIONS AND IMPLICATIONS
This animal model reproduced salient features of human asthma and linked loss of bronchoprotection with airway pathobiology. Accordingly, the model offers an advanced tool for understanding the mechanisms of the effects of chronic β- agonist treatment on β-adrenoceptor function in asthma. Such information may guide the clinical use of β-agonists and provide insight into development of novel β-adrenoceptor ligands for the treatment of asthma.
Keywords: β-adrenoceptor, β-agonist, receptor desensitization, airway remodelling, airway inflammation, asthma, loss of bronchoprotection, mouse
Introduction
Asthma is a chronic disease characterized by airway inflammation, hyperresponsiveness and remodelling. Arguably, the most debilitating symptoms associated with this disease are wheezing and shortness of breath, both of which ultimately result from increased bronchomotor tone. Agonists of the β-adrenoceptor (β-agonists) are the oldest and most commonly prescribed therapeutic agents for the management of asthma and are potent bronchodilators that relieve asthmatic bronchospasm and thereby resolve airway obstruction. In addition to mediating bronchodilation, β-agonists also confer bronchoprotection; that is, inhibition of induced bronchoconstriction (Abisheganaden and Boushey, 1998). Although β-agonists are very effective at improving lung function acutely, chronic use limits their therapeutic efficacy and, in some cases, leads to deleterious effects (Taylor, 2009).
Numerous clinical studies demonstrate that chronic use of β-agonists (both short- and long-acting) results in loss of bronchoprotection (Cheung, 1992; Bhagat et al., 1995; Drotar et al., 1998; Lipworth et al., 1998; Jokic et al., 2001). The clinical significance of this experimental observation is its association with a reduced ability to oppose allergen-mediated bronchconstriction and cross-tolerance to rescue β-agonists. Additionally, the mechanisms of desensitization of β2-adrenoceptors that underlie loss of bronchoprotection have been implicated in promoting asthma pathogenesis and worsening of asthma control (see Deshpande and Penn, 2006).
Although many clinical studies report that β-agonist treatment is efficacious and safe (Drazen et al., 1996; Dennis et al., 2000; Bateman et al., 2008), reports linking chronic β-agonism to worsening asthma control and asthma-related death are sufficient in number to raise legitimate questions about how best to clinically utilize this class of drugs to treat asthma (Stolley and Schinnar, 1978; Spitzer et al., 1992; Pearce et al., 1995; Sears, 2002; Salpeter et al., 2006). An important step towards addressing these concerns is to gain a mechanistic understanding of how, in a disease setting, β-adrenoceptor function and regulation are altered by chronic β-agonist activation and how this differs from what occurs during acute activation. It is difficult to imagine how a drug that is so beneficial acutely could lead to detrimental effects when administered chronically. However, this β-adrenoceptor paradox is not unique to asthma (Bond, 2001).
A classic example of the β-adrenoceptor paradox comes from the study of heart failure where chronic use of β-agonists causes not only desensitization to their inotropic effect but also leads to increased cardiac morbidity and mortality (Packer, 1989; Petrofski and Koch, 2003). An understanding of how excessive β-adrenoceptor activation in asthma can transform the beneficial effects of β-agonists into deleterious ones is currently lacking due to methodological and ethical constraints on human research and the lack of a comprehensive animal model.
Although animal models exist for studying the physiological effects of either chronic allergen exposure (Kamachi et al., 2001; Sugiura et al., 2007) or chronic β.-agonist administration (Finney et al., 2000; 2001; Tamaoki et al., 2004), few combine these two treatments. Moreover, of the few that do, the treatment duration is insufficient to demonstrate enhanced disease (Kamachi et al., 2001; Callaerts-Vegh et al., 2004; Riesenfeld et al., 2010; Lundblad et al., 2011), and only one has directly assessed whether or not β2-adrenoceptors become functionally desensitized (Callaerts-Vegh et al., 2004). Perhaps one reason why murine models of β2-adrenoceptor desensitization in the context of allergic airway inflammation are not widely reported is the speculation that murine airways have the ability to bronchorelax via β1-adrenoceptors.
β1-Adrenoceptors are expressed in greater abundance than are β2-adrenoceptors in murine tracheal smooth muscle and are functionally predominant in mediating tracheal smooth muscle relaxation (Henry and Goldie, 1990; Henry et al., 1990). However, the diameter of airways distal to the trachea is the main determinant of airway resistance during bronchoconstriction, and yet localization of β2-adrenoceptor subtypes in mouse bronchioles has not been examined. The density of β2-adrenoceptors increases with increasing airway generation in humans (see Johnson, 1998), and murine airways are not devoid of functional β2-adrenoceptors. Taken together, it is plausible that β2-adrenoceptors are the functionally predominant β-adrenoceptor in murine bronchioles.
The goal of our study was to demonstrate that β2-adrenoceptors mediated bronchorelaxation in mice and to develop an animal model that mimicked both the loss of bronchoprotection (an indicator of β2-adrenoceptor desensitization) and worsening of asthma (defined by enhanced inflammatory phenotype), observed in some human asthmatics. To this end, we tested the magnitude of β-agonist-mediated bronchoprotection in β2-adrenoceptor-KO mice and combined chronic allergen exposure with repeated β-agonist inhalation in allergen-sensitized BALB/C mice.
Methods
All animal care and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Duke University Medical Center and were carried out in accordance with the standards established by the US Animal Welfare Acts. Male BALB/C mice aged 6–8 weeks were purchased from The Jackson Laboratory (Bar Harbor, ME). β2-Adrenoceptor-KO male mice (Rohrer et al., 1999) aged 6–10 weeks and FVB/NJ WT mice aged 6–8 weeks (purchased from The Jackson Laboratory, stock # 1800) were kept in a pathogen-free barrier facility.
Mouse chronic ovalbumin (OVA) and albuterol treatment
Mice were sensitized on days 0 and 14 by i.p. injection of 10 µg ovalbumin (OVA) (Sigma, St. Louis, MO, USA) adsorbed to 2 mg of alum adjuvant (Pierce Biotechnology Inc., Rockford, IL, USA) diluted in saline, as previously described (Walker et al., 2003). Beginning on day 21 and continuing until day 62, mice were exposed for 60 min three times a week, with a rest day in between, to a 1% OVA-in-saline aerosol generated using a six-jet atomizer (TSI Inc., St. Paul, MN, USA). This OVA sensitization and challenge protocol is henceforth referred to as OVA treatment. Several hours after OVA aerosol exposure, mice were lightly anaesthetized with isoflurane and subjected to 50 µL oropharyngeal instillation of 1 mg·mL−1 albuterol sulphate (Sigma) or vehicle (0.9% saline). Animals were phenotyped on day 63, 24 h after the last albuterol or vehicle instillation. We administered the β-agonist by oropharyngeal instillation to mimic the route of delivery in humans and thereby negate potentially confounding effects of systemic β-agonist administration and control for the potential long-term effects of β-agonists on airway epithelial cells (McGraw et al., 2000; Nishimura et al., 2002).
Assessment of airway responsiveness: airway pressure time index (APTI) and impedance
Airway responsiveness was measured in a terminal experiment where mice were anaesthetized, paralysed and ventilated through a tracheal cannula (Walker et al., 1999). In brief, once mice were anaesthetized with i.p. 65 mg·kg−1 sodium pentobarbital, a jugular vein catheter and tracheal cannula were inserted and secured with sutures (Lundbeck, Inc., Deerfield, IL, USA). The animal was paralysed with 0.25 mg·kg−1 pancuronium bromide and ventilated with 100% oxygen (Sigma, St. Louis, MO, USA) at a constant volume of 7 mL·kg−1 and a frequency of 180 breaths·min−1. Mice were hyperinflated to 25 cmH2O 2 min before each injection of methacholine (MCh) (Sigma) to establish a constant volume history and respiratory mechanics. Drugs were injected at 5 min intervals. For APTI, peak tracheal pressure was continuously acquired from a tracheal cannula side port. APTI was calculated as the sum of the post MCh-induced changes in peak tracheal pressure (relative to pre-MCh peak tracheal pressure) integrated with respect to time (30 s). APTI is a measure that others have validated for its ability to provide a reasonable index of airway responsiveness (Levitt and Mitzner, 1988) as assessed by the more specific mechanical variables of resistance and compliance. For impedance measurements, mice were ventilated at 150 breaths·min−1 by a computer-controlled small animal ventilator (flexiVent, SCIREQ, Montreal, PQ, Canada) which generated sine wave volume perturbations (Schuessler and Bates, 1995). Following approximately 5 min of regular ventilation at a positive end-expiratory pressure (PEEP) of 3 cmH2O, a standard lung volume history was established followed by the acquisition of three baseline respiratory input impedance measurements. Bronchospasm was induced by aerosolizing 5 and 40 mg·mL−1 MCh (in saline) solutions using an ultrasonic nebulizer (flexiVent, SCIREQ). The aerosol was delivered to the airway opening by diverting the inspiratory ventilator flow through the aerosol chamber of the nebulizer for a total of 12 breaths (8 s aerosol) at a tidal volume of 10 mL·kg−1 at a rate of 150 breaths·min−1. Once the MCh had been delivered, the impedance measurements were made every 5–6 s, alternating between quickprime and snapshot measurements for a period of approximately 2 min. A 5 min washout period that included two deep sighs occurred before the next MCh challenge. The resultant total lung impedance signal contains information about the resistance and elastance properties of the lung from which Newtonian resistance can be calculated (Schuessler and Bates, 1995). In an idealized lung model, Newtonian resistance is a good indicator of the luminal diameter of the conducting airways (Proskocil and Fryer, 2005). APTI measurements were made on chronic OVA-treated BALB/C mice, whereas impedance measurements were made on β2-adrenoceptor-KO and FVB/NJ control mice.
β-Agonist-mediated bronchoprotection protocol
To assess β-agonist-mediated bronchoprotection, we first established (data not shown) that the dose of MCh administered was submaximal, and that after priming the bronchoconstrictor response with one low dose of MCh (50 µg·kg−1), repeated administration of MCh over 30 min resulted in similar magnitude bronchoconstrictor responses. In other words, the bronchoconstrictor response to MCh did not desensitize, a finding consistent with that of Finney et al. (2001). We then defined the bronchoconstrictor response to MCh by averaging the APTI response to two sequential doses of i.v. 100 µg·kg−1 MCh [a physiological dose (Walker et al., 1999)]. The APTI response to combined i.v.100 µg·kg−1 MCh and 30 µg·kg−1 albuterol was then measured. Bronchoprotection was calculated as % change in MCh-induced response by the following equation: [[(APTI response to MCh + albuterol) − (average APTI response to MCh)]/average APTI response to MCh × 100]. Whereas bronchoprotection in humans is calculated by comparing the protective effect of β-agonist against bronchoconstrictor stimuli measured at screening with that measured after several weeks of treatment (Cheung et al., 1992), repeat measures of lung mechanics in mice are rarely undertaken. Since inbred mice are nearly genetically identical, we compared the bronchoprotective effect of β-agonist in vehicle-treated mice with that measured in mice chronically treated with albuterol.
β-Adrenoceptor subtype mediating bronchoprotection
To assess the physiological importance of β-adrenoceptor subtypes in mediating airway bronchoprotection, we measured lung impedance and calculated airway resistance in wild-type and β2-adrenoceptor-KO mice using the flexiVent (SCIREQ). Baseline measurements were collected prior to each aerosol treatment. Mice were first exposed to a 20 s delivery of 15 mg·mL−1 albuterol aerosol or vehicle (0.9% saline) followed, sequentially, by 5 and 40 mg·mL−1 MCh aerosol exposures interspersed with another 15 mg·mL−1 albuterol or vehicle aerosol. Delivery of MCh and albuterol/control aerosols lasted 8 and 20 s respectively. The percent change in resistance was calculated as follows: [(pre-aerosol baseline resistance – MCh-induced resistance)/pre-aerosol baseline resistance × 100].
Whole lung lavage
At the termination of APTI measurements, mice were killed, and lungs were lavaged with saline. Cytokine concentrations and differential cell counts in lung lavage were determined as previously described (Walker et al., 2003). In brief, the percent of lavage cell types was calculated by differentiating, according to standard morphological criteria, 200 cells on slides stained using a Hema-3 staining kit (Fisher Scientific, Springfield, NJ, USA). Cytokine detection limits can be found at http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_3156.pdf.
Radioligand binding
Total β-adrenoceptor density expressed on plasma membranes of mouse lungs was measured by radioligand binding as previously described (Deshpande et al., 2008). In brief, whole lung plasma membranes were prepared in ice-cold homogenization buffer (75 mM Tris–HCl, 12.5 mM MgCl2, 2 mM EDTA, pH 7.4). Samples of lung plasma membranes (5 µg protein) were used for the radioligand binding assay where β-adrenoceptor total binding was determined by incubation with a saturating concentration (500 pM) of [125I]-iodocyanopindolol (ICYP; Perkin Elmer, Waltham, MA, USA) at 37°C for 40 min, a duration that achieves a steady state as verified by experiments in which longer incubations did not significantly alter specific binding. Inclusion of propranolol (20 µM; Sigma) defined non-specific binding and, although propranolol is a lipophilic antagonist, use of hydrophilic ligands such as isoprenaline (100 µM) and CGP 12 177 (1 µM) produced similar non-specific binding results (data not shown). Specific binding was calculated by subtracting non-specific binding from the total binding and maximal binding (Bmax) was estimated as specific binding per mg protein. As some treatment groups were chronically and/or acutely (just prior to lung collection) treated with albuterol, we first produced saturation binding curves using 5 to 750 pM ICYP to demonstrate that 500 pM is a saturating concentration for all treatment groups and represented approximately seven times the KD.
Hyaluronan staining and BAL content
Slide-mounted lung sections were stained with biotinylated hyaluronan-binding protein (bHABP) (2 µg·mL−1) (Associates of Cape Cod Incorporated, East Falmouth, MA, USA) for 2 h and then developed using a Vectastain-Elite-ABC kit (Vector Laboratories, Burlingame, CA, USA). Hyaluronan concentrations in bronchoalveolar lavage (BAL) fluid of mice were measured with a competitive elisa-like assay using bHABP as described previously (Teder et al., 2002).
Histological staining and quantitative assessment
Inflated lungs were fixed in 4% paraformaldehyde and stored overnight at 4°C prior to cleaning. Lungs were subsequently embedded in paraffin and sectioned in 4 µm thick slices. After individual staining of slides, 10 images of randomly chosen variable size airways (but not terminal bronchioles) were photographed at 20× magnification. The method of slide analysis and semi-quantitation depended upon the structure being assessed and the stain used, as described below.
Haematoxylin and eosin (H&E) staining
Lung tissue inflammation was semi-quantitatively determined, without knowledge of the treatments, from H&E-stained sections using a six-tiered scoring system of inflammation severity (Hollingsworth et al., 2010). In brief, the inflammation score increased with increasing depth of perivascular or peribronchiolar inflammation and was further elevated when eosinophils were predominant.
Periodic acid Schiff (PAS) staining
To assess airway mucin production, lung sections were analysed using PAS staining (Hollingsworth et al., 2010). The scale ranged from 0 to 4, where 0 indicated no staining within the airway epithelium, 4 indicated greater than 75% airway epithelium staining, and scores 1, 2 and 3 represented up to 25%, 50% and 75% staining respectively.
Morphometric analysis of structural changes (collagen, α-smooth muscle actin (α-SMA), elastin)
Hart's method and Masson-trichrome stain were used to stain lung sections for elastin (Veness-Meehan, 2002) and collagen, respectively. To identify α-SMA, antibody clone 1A4 was used with a biotinylated horse-anti-mouse secondary antibody. α-SMA was quantitated by using the thresholding method. To semi-quantitate the thickness of airway wall collagen, the area between the outer extent of the total collagen layer in the submucosal region and the basement membrane was digitally traced. Using the colour segmentation function of the Image Pro ® software (Media Cybernetics, Inc., Bethesda, MD, USA), the area inside the tracing that was positively Masson trichrome-stained for collagen was quantitated. The basement membrane length was digitally calculated to normalize the ‘collagen area’. ImageJ software was used to semi-quantitatively assess α-SMA and elastin. α-SMA-positive cells in only lung bronchioles were counted. The surrounding tissue of the airways was erased in Adobe Photoshop, and a colour deconvolution module was employed to automatically threshold only the tissue expressing α-SMA. The volume percent of α-SMA-positive cells relative to total tissue volume was calculated and reported. An identical approach was taken to calculate the percentage of lung elastin except that the colour deconvolution profile was customized to account for the black colour of the Hart stain.
Statistics
Data are expressed as mean ± SEM. Statistical calculations were performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). Significant differences among groups were identified by anova and a Tukey honest significant difference post hoc test was used for individual comparisons. When variances were unequal, Welch's correction was applied. When only two groups were compared, a one- or two-tailed Student's t-test was applied as appropriate. A P-value of less than 0.05 was considered statistically significant.
Results
Airway responsiveness and bronchoprotection
As shown in Figure 1A, chronic OVA treatment resulted in a significant increase in airway response to MCh, relative to alum-treated mice. Although animals treated with OVA–albuterol showed a trend for increased responsiveness to MCh relative to OVA-treated animals, no statistically significant difference was measured. Thus, as is characteristic of human asthmatics, mice chronically treated with OVA demonstrate increased airway responsiveness.
Figure 1.
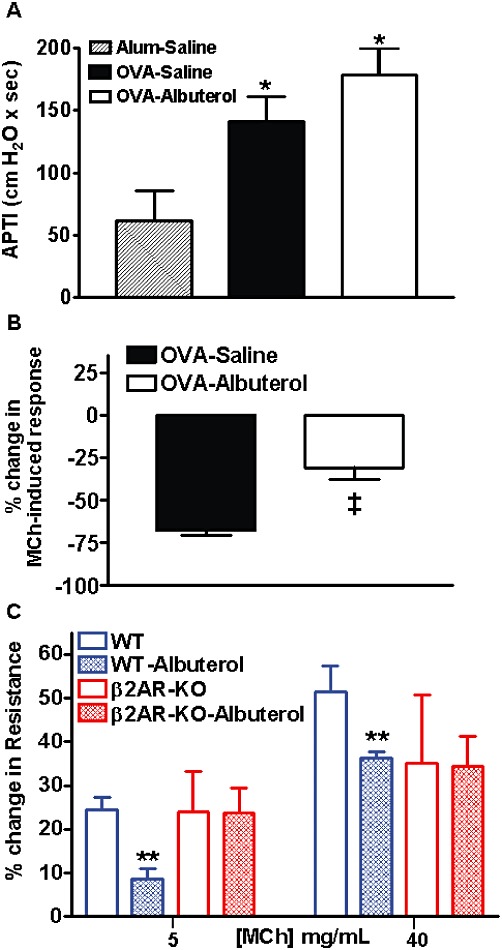
Assessment of airway responsiveness and bronchoprotection. (A) Relative to alum-treated mice, chronic OVA-treatment increased airway responsiveness to 100 µg·kg−1 MCh that is not significantly altered by additional chronic administration of albuterol (open bar). (B) The airway response to MCh in OVA-treated mice that are naïve to chronic oropharyngeal albuterol treatment is significantly abated by i.v administration of albuterol during MCh challenge. However, the effectiveness of i.v. 30 µg·kg−1 albuterol to functionally neutralize the airway response to MCh is significantly diminished in mice chronically treated with OVA and oropharyngeal albuterol. Values are mean ± SE from three to nine mice per group. (C) The percent change in airway resistance was significantly diminished by albuterol relative to vehicle at 5 and 40 mg·mL−1 aerosolized MCh in naïve wild type (WT), but not naïve β2-adrenoceptor KO (β2AR-KO) mice. Values are mean ± SE from three to four mice per group. *P < 0.05 compared with alum–saline; ‡P < 0.05 compared with OVA–saline; **P < 0.05 compared with WT using a one-tailed Student's t-test.
As shown in Figure 1B, i.v. albuterol substantially abated airway responsiveness to MCh in mice treated with OVA alone. However, mice chronically treated with OVA–albuterol were significantly refractory to the bronchoprotective effect of intravenous albuterol. Thus, our mouse model reproduced the association between chronic β-agonist use and β-adrenoceptor tolerance, or dysfunction, observed in human asthmatics.
Functional β-adrenoceptor subtype
Although studies show that the β1-adrenoceptor is the predominant adrenoceptor expressed in mouse trachea, localization of the β-adrenoceptor subtypes in more distal mouse airways has not been described. Since albuterol is only moderately selective for β2- over β1-adrenoceptors (Baker, 2005), we determined which β-adrenoceptor subtype mediated airway smooth muscle relaxation in our model. Thus, we compared the bronchoprotective effect of albuterol in β2-adrenoceptor-KO mice versus wild-type mice. As shown in Figure 1C, the percent change in airway resistance was significantly diminished by albuterol, relative to vehicle at 5 and 40 mg·mL−1 bronchoconstricting doses of MCh in naïve WT, but not naïve β2-adrenoceptor-KO, mice. These data indicate that, as in humans, bronchoprotection was primarily mediated by β2-adrenoceptors.
Lung inflammation
Our mouse model of chronic allergic airway inflammation demonstrates many other features of human chronic asthma. For example, chronically OVA-treated mice show increased numbers of eosinophils, lymphocytes and neutrophils in BAL (Figure 2A), increased airway mucin staining (Figure 3A–D) and enhanced peribronchiolar and perivascular inflammation (Figure 3E–H). The addition of albuterol to the chronic OVA treatment caused a significant increase in peribronchiolar and perivascular lung inflammation (Figure 3E) and mucin production (Figure 3A) but did not influence the differential cell counts in BAL (Figure 2A). In contrast to human asthma, the levels of Th2-type cytokines (IL-4 and IL-13) as well as IL-1β and the chemokine CCL5 (RANTES) were not elevated in OVA-treated mice, relative to alum-treated mice (Figure 2B) (Barnes, 2008). However, consistent with human asthma, IL-5 was significantly elevated after 6 weeks of OVA treatment. Albuterol treatment did not alter the BAL concentration of any of the measured cytokines, except for that of IL-13 which was reduced (Figure 2B).
Figure 2.
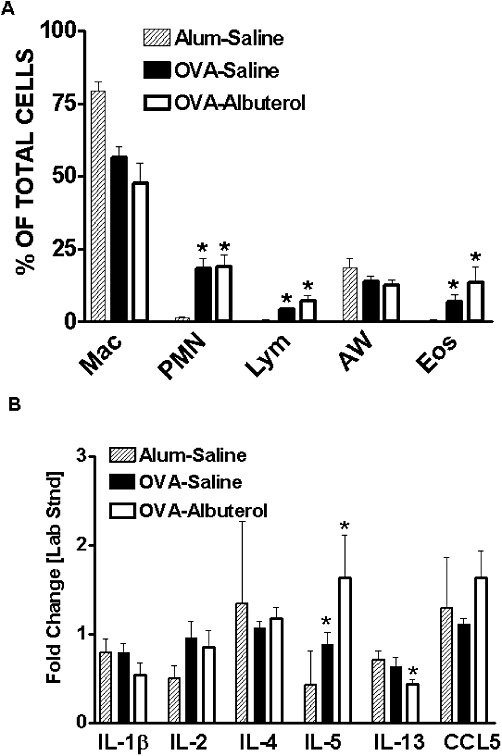
Assessment of lung inflammation. Relative to alum-treated mice, chronic OVA treatment increased (A) lung lavage neutrophils (PMNs), lymphocytes (Lym) and eosinophils (Eos), but not macrophages (Mac) or airway epithelial cells (AW). (B) Cytokine IL-5 was elevated by OVA treatment. Chronic administration of albuterol to OVA-treated mice had no significant effect on any of these measurements except that IL-13 was reduced. Values are mean ± SE from 3 to 13 mice per group. *P < 0.05 compared to alum–saline.
Figure 3.
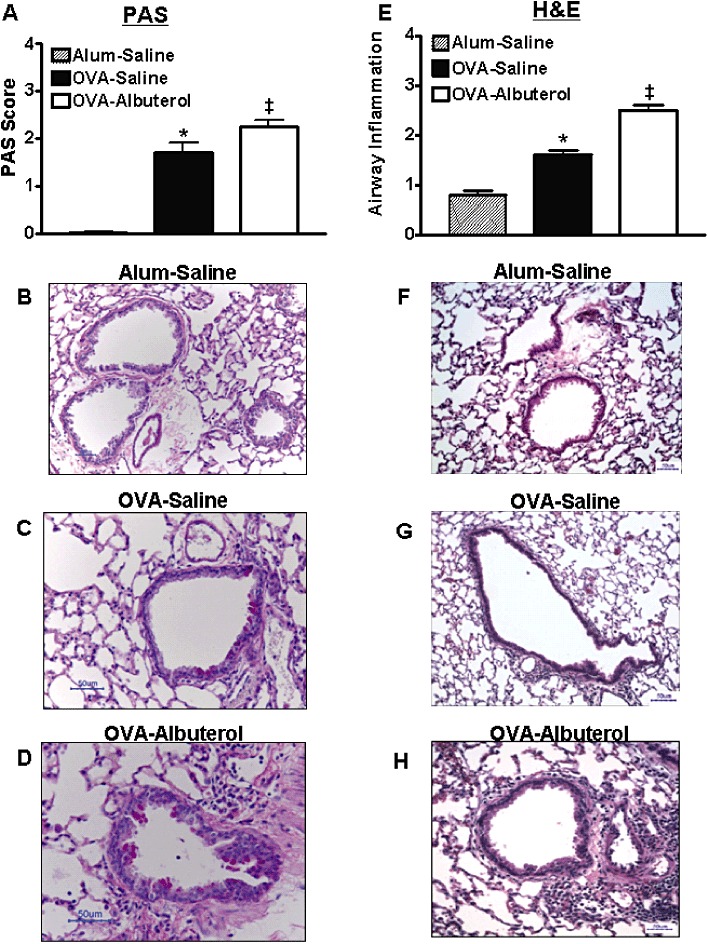
Assessment of airway mucous metaplasia and inflammation. Relative to alum–saline-treated mice, chronic OVA treatment increased(A–D) airway mucous metaplasia and (E–H) peribronchiolar and perivascular inflammation. The additional chronic administration of albuterol exacerbated both of these phenotypes. Values are mean ± SE from 8 to 10 mice per group. *P < 0.05 compared with alum–saline; ‡P < 0.05 compared with alum– and OVA–saline.
Airway and lung remodelling
Subepithelial collagen deposition is a classic characteristic of airway remodelling in human asthma, as is thickening of airway smooth muscle (Vignola et al., 2003). To assess airway remodelling in chronically allergen-challenged mice, we stained lung sections with Masson-trichrome and morphometrically semi-quantitated the intensity of collagen staining per length of basement membrane in the airway subepithelial layer (Figure 4A–D). Chronic OVA treatment led to increased subepithelial collagen deposition, and this was not appreciably altered by chronic albuterol treatment. To assess morphological changes in airway smooth muscle, slides were semi-quantitated for volume % of α-SMA staining. We found no effect of chronic OVA, or OVA–albuterol treatment on the volume of smooth muscle around the airway (data not shown). However, we observed that lungs from mice chronically treated with OVA–albuterol displayed a significant increase in lung parenchymal α-SMA expression relative to alum–saline-treated mice (Figure 4E–H). As hyaluronan, a pro-fibrotic glycosaminoglycan, is associated with collagen regulation, we measured its levels in BAL. OVA treatment significantly increased BAL–hyaluronan relative to alum-treated control mice, and this was further significantly increased by the addition of albuterol treatment (Figure 5A–D). Some asthmatics display obstruction resulting from loss of elastic recoil and we therefore assessed elastin deposition in lung parenchyma (Figure 5E). Lung elastin deposition was not significantly altered by OVA or OVA-albuterol treatment. This mouse model displayed many of the airway remodelling features characteristic of human asthma.
Figure 4.
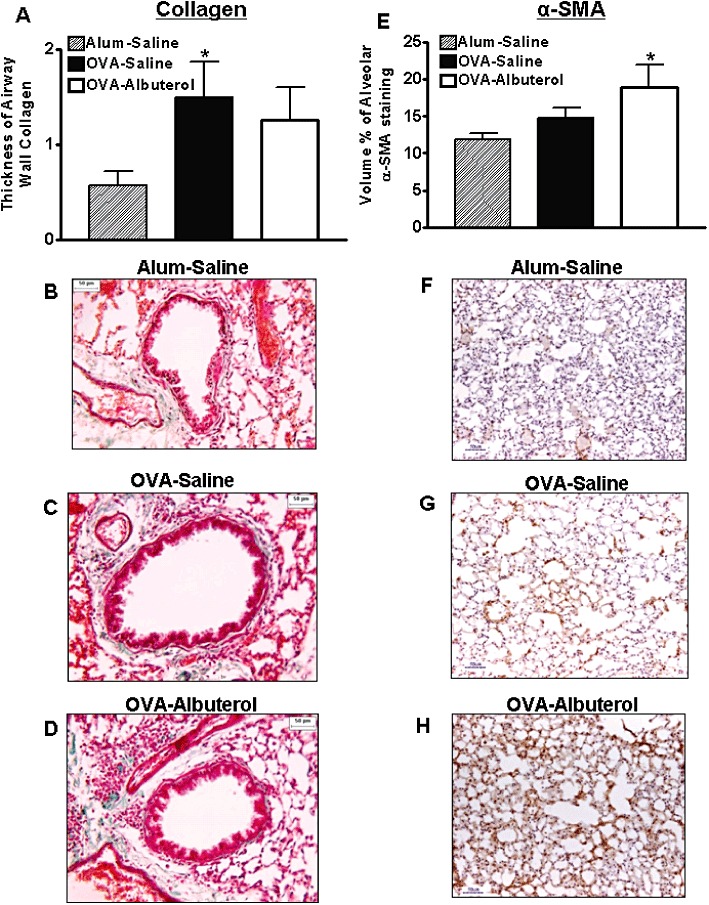
Assessment of airway remodelling. (A–D) Airway wall collagen thickness was significantly elevated in chronically OVA-treated mice, but not those that additionally received albuterol. (E–H) Only mice that chronically received combined OVA–albuterol treatment demonstrated a significant increase in alveolar α-SMA. Values are mean ± SE from 6 to 14 mice per group. *P < 0.05 compared with alum–saline.
Figure 5.
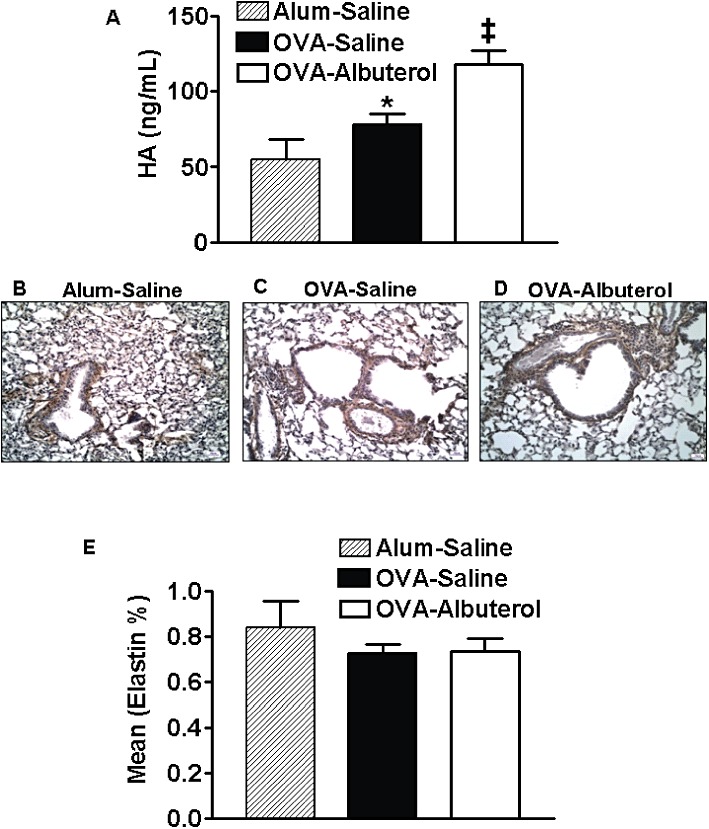
Assessment of hyaluronan and airway remodelling. (A) Hyaluronan (HA) levels in BAL were significantly elevated in chronically OVA-treated mice and this was significantly enhanced in OVA–albuterol-treated mice. (B–D) Representative micrographs of hyaluronan-stained lung slices are shown for all three treatment groups. (E) Neither OVA nor combined OVA–albuterol treatment was associated with significant changes in alveolar elastin. Values are mean ± SE from 6 to 10 mice per group. *P < 0.05 compared with alum–saline; ‡P < 0.05 compared with alum– and OVA–saline.
Lung β-adrenoceptor expression
Since β-agonist effectiveness depends, in part, on β2-adrenoceptor expression, we measured lung β-adrenoceptor density by radioligand binding. As shown in Figure 6, lung expression of β-adrenoceptors was significantly diminished in association with chronic OVA treatment but was not further altered by the addition of chronic albuterol treatment. Importantly, the KD values remained unchanged between treatment groups (alum–saline, 75.7 ± 7.0 pM; OVA–saline, 84.4 ± 8.9 pM; OVA–albuterol, 63.7 ± 3.3 pM), showing that neither OVA nor albuterol treatment influenced β-adrenoceptor density.
Figure 6.
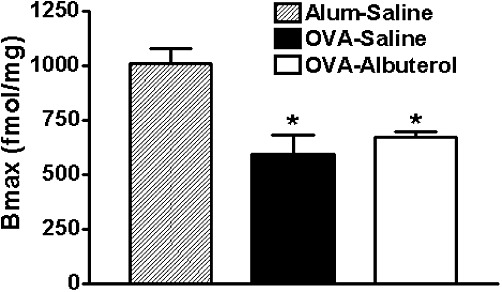
Assessment of β-adrenoceptor expression. Radioligand binding experiments showed that relative to alum–saline-treated mice, chronically OVA-treated mice demonstrate a decrease in lung β-adrenoceptor expression, that is not significantly altered by additional chronic oropharyngeal administration of albuterol. Values are mean ± SE from four mice per group. *P < 0.05 compared with alum–saline.
Discussion
β-Agonists are very important therapeutic agents for the treatment of asthma. However, when used chronically, their effectiveness wanes, and they may actually promote disease progression. This report provides two important advances to the study of chronic β-agonist effects on β2-adrenoceptor function and regulation in asthma. Initially, this study provides the first unequivocal evidence that, as in humans, murine bronchorelaxation is mediated by the β2-adrenoceptors. This finding establishes the mouse as a suitable species with which to model loss of bronchoprotection and β2-adrenoceptor desensitization. Secondly, this is the first report that provides a detailed description of the pro-inflammatory effects of chronic β-agonist treatment in the context of allergic inflammatory airway disease and associates these changes with functional loss of bronchoprotection. This finding is important because it provides evidence that chronic exposure to β-agonists can exacerbate allergic inflammation, implicates β2-adrenoceptor dysfunction as an underlying mechanism of inflammation and provides an animal model with which to further explore this association.
Mice have been widely used as a model to study allergic inflammatory airway disease; however, their suitability as a model for the study of β-adrenoceptor-mediated bronchodilation and desensitization has been questioned. Prior to this report, the distribution, density and function of β-adrenoceptor subtypes in murine airways distal to the trachea were unknown. Moreover, based on work from Henry and colleagues, it appeared that, in contrast to the functional dominance of β2-adrenoceptors clearly demonstrated in humans, mice might mediate bronchorelaxation via β1-adrenoceptors. They showed that the density and function of β1-adrenoceptors predominated in murine tracheal smooth muscle; however, no one had mapped the smooth muscle distribution or density of β-adrenoceptor subtypes in murine bronchioles, where the greatest resistance to airflow occurs in healthy and asthmatic humans. Although we did not directly localize β-adrenoceptor subtypes in mouse airways, the observation that albuterol provides no significant bronchoprotection in β2-adrenoceptor-KO mice demonstrated that the β2-adrenoceptor subtype was the predominant mediator of albuterol-induced bronchorelaxation in mice.
It is likely, but not certain, that the functional dominance of murine bronchiolar β2-adrenoceptors is coincident with elevated receptor density. The idea that the density of β2-adrenoceptors may increase along the tracheobronchial tree in mice is supported by a similar pattern demonstrated in human airways (Johnson, 1998). Additionally, other species, such as guinea pig and dog demonstrate a predominant β2-adrenoceptor density and function (Tamaoki et al., 1993; Kompa et al., 1995) respectively, in bronchial smooth muscle.
Our study cannot completely rule out a role for β1-adrenoceptors in mediating bronchorelaxation; however, our finding adds credibility to those murine studies where only the relatively weak selectivity of albuterol for β2-, over β1-adrenoceptors was used as evidence of β2-adrenoceptor-mediated bronchorelaxation (Finney et al., 2000; Callaerts-Vegh et al., 2004).
Tolerance or subsensitivity to the bronchoprotective effect of β-agonists is a reproducible event in humans (Jackson and Lipworth, 2004) and is demonstrated in our mouse model. Chronic β-agonist-induced loss of bronchoprotection is clinically relevant for at least three reasons. First, although there are reports to the contrary (Wilding et al., 1997; Korosec et al., 1999), studies have shown that loss of bronchoprotection associated with repeated use of long-acting β-agonists (LABAs) can result in cross-tolerance to short-acting β-agonists (SABAs) (Newnham et al., 1994; 1995; Grove and Lipworth, 1995), thereby reducing the effectiveness of rescue bronchodilators. Secondly, the inability of β-agonists to inhibit bronchoconstriction mediated by naturally clinically important stimuli (such as allergens) may have significant deleterious consequences during an asthma exacerbation (Cockcroft et al., 1993; Jokic et al., 2001). Finally, the β2-adrenoceptor desensitization mechanism underlying loss of bronchoprotection has been implicated in exacerbating asthma pathogenesis and worsening of asthma control (see Deshpande and Penn, 2006) presumably via pro-inflammatory effects [reviewed in (Cazzola et al., 2011)]. It is theorized that, in the setting of asthma, chronic β-agonist treatment drives β2-adrenoceptor signalling through a non-canonical, β-arrestin-dependent, adverse signalling pathway that exacerbates airway inflammation and mucin phenotypes (Dickey et al., 2010; Walker et al., 2011).
It comes as no surprise that studies describing the effect of β-agonist therapy on asthmatic airway inflammation have provided mixed results. Whereas some clinical studies report anti-inflammatory effects of β-agonists (see Remington and Digiovine, 2005), others suggest inflammation is unaffected or increased by inhaled β-agonist therapy (reviewed in Loza et al., 2008). More recent reviews conclude that the body of evidence from human-based studies is weighted towards β-agonists being pro-inflammatory (see Taylor, 2009; Cazzola et al., 2011). This conclusion is compellingly supported by work from the Bond laboratory that has shown, through pharmacological and genetic methods, that β2-adrenoceptor signalling is required for full development of the asthma phenotype in mice (Callaerts-Vegh et al., 2004; Nguyen et al., 2008; 2009). Although Bond's work focused on loss of β2-adrenoceptor function, and our present work focuses on enhanced β2-adrenoceptor function, taken together, these studies support the notion that chronic therapeutic use of β-agonists could accelerate asthma severity.
Efforts to reveal the mechanisms underlying β2-adrenoceptor desensitization and associated deleterious events have been hampered by clinical research limitations including the relative dearth of β-agonist naïve patients, the supposition that only a subpopulation of asthmatics will experience β-agonist induced adverse events (Taylor, 2009) and restrictions on the amount and type of tissue that can be collected from humans. Advances in our understanding have also been slowed by animal models that fail to combine the optimal dose, route and duration of β-agonist treatment with sufficient duration of allergen exposure, and which lack an in-depth assessment of the asthma phenotype. In particular, airway hyperresponsiveness to the bronchoconstrictor MCh is typically reported for allergen-treated mice (Lundblad et al., 2011) rather than an assessment of bronchoprotection. Despite being influenced by β-adrenoceptor expression (McGraw et al., 2003), MCh (or other bronchoconstrictor) responsiveness is not a direct reflection of β-adrenoceptor desensitization. Only one animal study to date has assessed the effect of chronic β-agonist treatment on bronchoprotection in the context of allergen sensitization and challenge; however, only a cursory description of the inflammatory phenotype was included in that study (Callaerts-Vegh et al., 2004). Consistent with our results, they showed that chronic albuterol treatment led to loss of bronchoprotective effect, and because they administered albuterol via osmotic pump, we further conclude that the route of β-agonist delivery may not affect the process of β2-adrenoceptor desensitization.
The observation in humans and animals that repeated β2-adrenoceptor activation leads to functional receptor desensitization is not surprising since receptor desensitization is a normal homeostatic process that presumably serves to protect cells from excessive stimulation (Penn and Benovic, 2008). One obvious mechanism by which chronic β-agonist treatment might lead to β2-adrenoceptor tolerance is through receptor down-regulation. However, our results, which are consistent with those of others (Callaerts-Vegh et al., 2004), showed that OVA-sensitization and -challenge significantly down-regulated lung β-adrenoceptor expression, but that repeated albuterol treatment did not further reduce receptor expression. Thus, β2-adrenoceptor subsensitivity in the present study was likely to have derived from receptor modification or altered signalling independent of whole cell receptor loss, and this conclusion is in keeping with in vitro studies on human cells that point to changes in β-adrenoceptor signalling elements and regulatory events as the explanation for agonist-induced loss of β2-adrenoceptors function (see Deshpande and Penn, 2006). One caveat to our interpretation of the binding data is that our measurements were made using whole lung membranes and a non-receptor subtype specific radioligand (ICYP), so we cannot rule out the possibility that a change in receptor expression in airway smooth muscle is being masked by an equal and opposite change in another lung tissue or that a decline in β2-adrenoceptor expression is hidden by a concomitant increase in β1-adrenoceptors expression.
The chronic OVA treatment that we employed in this study successfully reproduced in mice many of the remodelling features characteristic of human asthma including mucous metaplasia (see Sugiura et al., 2007), increased basement membrane collagen deposition (Vignola et al., 2003) and the appearance of myofibroblasts in the lung parenchyma (Bergeron et al., 2005). However, the loss of bronchoprotection observed in our model cannot be explained by changes in airway remodelling.
Despite the fact that subepithelial collagen deposition (Kuhn et al., 2000; McParland et al., 2003) and lung parenchymal elastin content can affect airway mechanics, neither was significantly changed by β-agonist treatment in this study. Similarly, although myofibroblasts can develop mechanical tension in small airways (Sugiura et al., 2007), β-agonist treatment did not significantly alter α-SMA staining. Thus, myofibroblast activity does not contribute to β2-adrenoceptor tolerance in our model. Interestingly, myofibroblasts can release the tissue repair and remodelling regulatory molecule, hyaluronan, which is an extracellular matrix glycosaminoglycan. Hyaluronan becomes fragmented in the context of lung inflammation and, in so doing, becomes pro-inflammatory and pro-fibrotic (Jiang et al., 2007). The mechanism by which chronic β-agonist treatment significantly elevated BAL–hyaluronan accumulation in OVA-treated mice is unknown but is consistent with the idea that such treatment is pro-inflammatory. In contrast to models of lung fibrosis, where hyaluronan is positively associated with collagen accumulation, our results demonstrate no enhanced increase in airway wall collagen in OVA–albuterol-treated mice. However, in pulmonary fibrosis, the pro-fibrotic effect of hyaluronan is manifest outside airways (Jiang et al., 2007), a location not assessed in our study.
Consistent with other chronic allergen models, we demonstrated airway epithelial mucous metaplasia in chronically OVA-treated mice. Interestingly, the addition of chronic albuterol treatment resulted in a modest, but significant, increase in this remodelling phenotype, which is consistent with previous reports (Kamachi et al., 2001; Nguyen et al., 2009). It appears that combined β-agonist and allergen treatment needs to be of sufficient duration to elicit the enhanced airway mucous phenotype, as others who used acute models showed no effect of albuterol treatment on mucin content in the airway epithelium (Nguyen et al., 2008) and no effect of albuterol on airway epithelial density of PAS-positive cells (Riesenfeld et al., 2010). However, based on their associated lung mechanics and microCT-imaging results, Riesenfeld et al., concluded that a physiologically significant change in epithelial mucous cell density was indeed associated with prolonged β-agonist administration. If β-agonist administration can promote epithelial cell mucous metaplasia in humans, then this might provide one explanation for the β-agonist associated worsening of asthma control observed in some human asthmatics.
Conclusion
The animal model described here provides an advanced tool that can be used to facilitate our mechanistic understanding of the temporal effects of β-agonism on β-adrenoceptor function in asthma. Additionally, the chronic effects of other β-adrenoceptor ligands (i.e. ultra-long acting β-agonists) or β-adrenoceptor modulating drugs (i.e. corticosteroids) on β2-adrenoceptor desensitization and severity of asthma phenotype could be assessed using this model. Finally, the outcomes measured in this model, which extend beyond standard pulmonary mechanics and conventional reductionist endpoints, may aid the discovery of new β-adrenoceptor ligands for the treatment of asthma.
Acknowledgments
The authors are grateful to Raymond B Penn for critical review of the manuscript. The authors gratefully receive support from the National Institutes of Health (grants HL084123, HL093103 to JKLW; AI052201 to PWN; HL60532 and ES06766 to ARB), the Durham Veterans Administration (JKLW) and an Established Investigator Award from the American Asthma Foundation (MES).
Glossary
- α-SMA
α-smooth muscle actin
- APTI
airway pressure time index
- BAL
bronchoalveolar lavage
- bHABP
biotinylated hyaluronan-binding protein
- Bmax
maximal binding
- H&E
haematoxylin and eosin
- ICYP
iodocyanopindolol
- LABA
long-acting β-agonist
- MCh
methacholine
- OVA
ovalbumin
- PAS
periodic acid Schiff
- PEEP
positive end expiratory pressure
- SABA
short-acting β-agonist
Conflicts of interest
None.
References
- Abisheganaden J, Boushey H. Long-acting inhaled beta 2-agonists and the loss of ‘bronchoprotective’ efficacy. Am J Med. 1998;104:494–497. doi: 10.1016/s0002-9343(98)00092-8. [DOI] [PubMed] [Google Scholar]
- Baker JG. The selectivity of β-adrenoceptor antagonists at the human β1, β2 and β3 adrenoceptors. Br J Pharmacol. 2005;144:317–322. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman E, Nelson H, Bousquet J, Kral K, Sutton L, Ortega H, et al. Meta-analysis: effects of adding salmeterol to inhaled corticosteroids on serious asthma-related events. Ann Intern Med. 2008;149:33–42. doi: 10.7326/0003-4819-149-1-200807010-00229. [DOI] [PubMed] [Google Scholar]
- Bergeron C, Hauber HP, Gotfried M, Newman K, Dhanda R, Servi RJ, et al. Evidence of remodeling in peripheral airways of patients with mild to moderate asthma: effect of hydrofluoroalkane-flunisolide. J Allergy Clin Immunol. 2005;116:983–989. doi: 10.1016/j.jaci.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Bhagat R, Kalra S, Swystun V, Cockcroft D. Rapid onset of tolerance to the bronchoprotective effect of salmeterol. Chest. 1995;108:1235–1239. doi: 10.1378/chest.108.5.1235. [DOI] [PubMed] [Google Scholar]
- Bond RA. Is paradoxical pharmacology a strategy worth pursuing? Trends Pharmacol Sci. 2001;22:273–276. doi: 10.1016/s0165-6147(00)01711-9. [DOI] [PubMed] [Google Scholar]
- Callaerts-Vegh Z, Evans KLJ, Dudekula N, Cuba D, Knoll BJ, Callaerts PFK, et al. Effects of acute and chronic administration of β-adrenoceptor ligands on airway function in a murine model of asthma. Proc Natl Acad Sci U S A. 2004;101:4948–4953. doi: 10.1073/pnas.0400452101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Calzetta L, Matera MG. β2-adrenoceptor agonists: current and future direction. Br J Pharmacol. 2011;163:4–17. doi: 10.1111/j.1476-5381.2011.01216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung D, Timmers M, Zwinderman A, Bel E, Dijkman J, Sterk P. Long-term effects of a long-acting beta 2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma [see comments] N Engl J Med. 1992;327:1198–1203. doi: 10.1056/NEJM199210223271703. [DOI] [PubMed] [Google Scholar]
- Cockcroft DW, McParland CP, Britto SA, Swystun VA, Rutherford BC. Regular inhaled salbutamol and airway responsiveness to allergen. Lancet. 1993;342:833–837. doi: 10.1016/0140-6736(93)92695-p. [DOI] [PubMed] [Google Scholar]
- Dennis SM, Sharp SJ, Vickers MR, Frost CD, Crompton GK, Barnes PJ, et al. Regular inhaled salbutamol and asthma control: the TRUST randomised trial. Lancet. 2000;355:1675–1679. doi: 10.1016/s0140-6736(00)02238-8. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Penn RB. Targeting G protein-coupled receptor signaling in asthma. Cell Signal. 2006;18:2105–2120. doi: 10.1016/j.cellsig.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, Walker JKL. β-arrestins specifically constrain β2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J. 2008;22:2134–2141. doi: 10.1096/fj.07-102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey BF, Walker JK, Hanania NA, Bond RA. β-adrenoceptor inverse agonists in asthma. Curr Opin Pharmacol. 2010;10:254–259. doi: 10.1016/j.coph.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazen JM, Israel E, Boushey HA, Chinchilli VM, Fahy JV, Fish JE, et al. Comparison of regularly scheduled with as-needed use of albuterol in mild asthma. NEJM. 1996;335:841–847. doi: 10.1056/NEJM199609193351202. [DOI] [PubMed] [Google Scholar]
- Drotar DE, Davis EE, Cockcroft DW. Tolerance to the bronchoprotective effect of salmeterol 12 hours after starting twice daily treatment. Ann Allergy Asthma Immunol. 1998;80:31–34. doi: 10.1016/S1081-1206(10)62935-3. [DOI] [PubMed] [Google Scholar]
- Finney P, Donnelly L, Belvisi M, Chuang T, Birrell M, Harris A, et al. Chronic systemic administration of salmeterol to rats promotes pulmonary beta[2]-adrenoceptor desensitization and down-regulation of G(s alpha) Br J Pharmacol. 2001;132:1261–1270. doi: 10.1038/sj.bjp.0703946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney PA, Belvisi MG, Donnelly LE, Chuang T-T, Mak JCW, Scorer C, et al. Albuterol-induced downregulation of Gsα accounts for pulmonary β2-adrenoceptor desensitization in vivo. J Clin Invest. 2000;106:125–135. doi: 10.1172/JCI8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove A, Lipworth BJ. Bronchodilator subsensitivity to salbutamol after twice daily salmeterol in asthmatic patients. Lancet. 1995;346:201–206. doi: 10.1016/s0140-6736(95)91265-7. [DOI] [PubMed] [Google Scholar]
- Henry PJ, Goldie RG. Beta 1-adrenoceptors mediate smooth muscle relaxation in mouse isolated trachea. Br J Pharmacol. 1990;99:131–135. doi: 10.1111/j.1476-5381.1990.tb14666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry PJ, Rigby PJ, Goldie RG. Distribution of beta 1- and beta 2-adrenoceptors in mouse trachea and lung: a quantitative autoradiographic study. Br J Pharmacol. 1990;99:136–144. doi: 10.1111/j.1476-5381.1990.tb14667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth JW, Theriot BS, Li Z, Lawson BL, Sunday M, Schwartz DA, et al. Both hematopoietic-derived and non-hematopoietic-derived β-arrestin-2 regulates murine allergic airway disease. Am J Respir Cell Mol Biol. 2010;43:269–275. doi: 10.1165/rcmb.2009-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CM, Lipworth B. Benefit-risk assessment of long-acting β2-agonists in asthma. Drug Safety. 2004;27:243–270. doi: 10.2165/00002018-200427040-00003. [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- Johnson M. The β-adrenoceptor. Am J Respir Crit Care Med. 1998;158:S146–S153. doi: 10.1164/ajrccm.158.supplement_2.13tac110. [DOI] [PubMed] [Google Scholar]
- Jokic R, Swystun VA, Davis BE, Cockcroft DW. Regular inhaled salbutamol. Chest. 2001;119:370–375. doi: 10.1378/chest.119.2.370. [DOI] [PubMed] [Google Scholar]
- Kamachi A, Munakata M, Nasuhara Y, Nishimura M, Ohtsuka Y, Amishima M, et al. Enhancement of goblet cell hyperplasia and airway hyperresponsiveness by salbutamol in a rat model of atopic asthma. Thorax. 2001;56:19–24. doi: 10.1136/thorax.56.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompa AR, Molenaar P, Summers RJ. Beta-adrenoceptor regulation and functional responses in the guinea-pig following chronic administration of the long-acting beta 2-adrenoceptor agonist formoterol. Naunyn Schmiedebergs Arch Pharmacol. 1995;351:576–588. doi: 10.1007/BF00170156. [DOI] [PubMed] [Google Scholar]
- Korosec M, Novak R, Myers E, Skowronski M, McFadden E. Salmeterol does not compromise the bronchodilator response to albuterol during acute episodes of asthma. Am J Med. 1999;107:209–213. doi: 10.1016/s0002-9343(99)00222-3. [DOI] [PubMed] [Google Scholar]
- Kuhn C, Homer RJ, Zhu Z, Ward N, Elias JA. Morphometry explains variation in airway responsiveness in transgenic mice overexpressing interleukin-6 and interleukin-11 in the lung. Chest. 2000;117(5 Suppl. 1):260S–262S. [PubMed] [Google Scholar]
- Levitt R, Mitzner W. Expression of airway hyperreactivity to acetylcholine as a simple autosomal recessive trait in mice. FASEB J. 1988;2:2605–2608. doi: 10.1096/fasebj.2.10.3384240. [DOI] [PubMed] [Google Scholar]
- Lipworth B, Tan S, Devlin M, Aiken T, Baker R, Hendrick D. Effects of treatment with formoterol on bronchoprotection against methacholine. Am J Med. 1998;104:431–438. doi: 10.1016/s0002-9343(98)00086-2. [DOI] [PubMed] [Google Scholar]
- Loza MJ, Foster S, Peters SP, Penn RB. Interactive effects of steroids and beta-agonists on accumulation of type 2 T cells. J Allergy Clin Immunol. 2008;121:750–755. doi: 10.1016/j.jaci.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Lundblad L, Rinaldi L, Poynter M, Riesenfeld E, Wu M, Aimi S, et al. Detrimental effects of albuterol on airway responsiveness requires airway inflammation and is independent of beta-receptor affinity in murine models of asthma. Respir Res. 2011;12:27–38. doi: 10.1186/1465-9921-12-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw DW, Forbes SL, Mak JCW, Witte DP, Carrigan PE, Leikauf GD, et al. Transgenic overexpression of beta 2-adrenergic receptors in airway epithelial cells decreases bronchoconstriction. Am J Physiol Lung Cell Mol Physiol. 2000;279:L379–L389. doi: 10.1152/ajplung.2000.279.2.L379. [DOI] [PubMed] [Google Scholar]
- McGraw DW, Almoosa KF, Paul RJ, Kobilka BK, Liggett SB. Antithetic regulation by β-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway β-agonist paradox. J Clin Invest. 2003;112:619–626. doi: 10.1172/JCI18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McParland B, Macklem P, Pare P. Airway wall remodeling: friend or foe? J Appl Physiol. 2003;95:426–434. doi: 10.1152/japplphysiol.00159.2003. [DOI] [PubMed] [Google Scholar]
- Newnham DM, McDevitt DG, Lipworth BJ. Bronchodilator subsensitivity after chronic dosing with eformoterol in patients with asthma. Am J Med. 1994;97:29–37. doi: 10.1016/0002-9343(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Newnham DM, Grove A, McDevitt DG, Lipworth BJ. Subsensitivity of bronchodilator and systemic beta 2 adrenoceptor responses after regular twice daily treatment with eformoterol dry powder in asthmatic patients. Thorax. 1995;50:497–504. doi: 10.1136/thx.50.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Omoluabi O, Parra S, Frieske JM, Clement C, Ammar-Aouchiche Z, et al. Chronic exposure to beta-blockers attenuates inflammation and mucin content in a murine asthma model. Am J Respir Cell Mol Biol. 2008;38:256–262. doi: 10.1165/rcmb.2007-0279RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ, et al. β2-Adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc Natl Acad Sci U S A. 2009;106:2435–2440. doi: 10.1073/pnas.0810902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Tamaoki J, Isono K, Aoshiba K, Nagai A. β-adrenergic receptor-mediated growth of human airway epithelial cell lines. Eur Respir J. 2002;20:353–358. doi: 10.1183/09031936.02.01352001. [DOI] [PubMed] [Google Scholar]
- Packer M. Is activation of the sympathetic nervous system beneficial or detrimental to the patient with chronic heart failure? Lessons learned from clinical trials with beta-adrenergic agonists and antagonists. J Cardiovasc Pharmacol. 1989;14(Suppl. 5):S38–S43. [PubMed] [Google Scholar]
- Pearce N, Beasley R, Crane J, Burgess C, Jackson R. End of the New Zealand asthma mortality epidemic. Lancet. 1995;345:41–44. doi: 10.1016/s0140-6736(95)91159-6. [DOI] [PubMed] [Google Scholar]
- Penn RB, Benovic JL. Regulation of heterotrimeric G protein signaling in airway smooth muscle. Proc Am Thorac Soc. 2008;5:47–57. doi: 10.1513/pats.200705-054VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrofski JA, Koch WJ. The β-adrenergic receptor kinase in heart failure. J Mol Cell Cardiol. 2003;35:1167–1174. doi: 10.1016/s0022-2828(03)00243-8. [DOI] [PubMed] [Google Scholar]
- Proskocil BJ, Fryer AD. β2-agonist and anticholinergic drugs in the treatment of lung disease. Proc Am Thorac Soc. 2005;2:305–310. doi: 10.1513/pats.200504-038SR. [DOI] [PubMed] [Google Scholar]
- Remington TL, Digiovine B. Long-acting beta-agonists: anti-inflammatory properties and synergy with corticosteroids in asthma. Curr Opin Pulm Med. 2005;11:74–78. doi: 10.1097/01.mcp.0000146784.56834.ff. [DOI] [PubMed] [Google Scholar]
- Riesenfeld E, Sullivan M, Thompson-Figueroa J, Haverkamp H, Lundblad L, Bates J, et al. Inhaled salmeterol and/or fluticasone alters structure/function in a murine model of allergic airways disease. Respir Res. 2010;11:22–32. doi: 10.1186/1465-9921-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both β1- and β2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- Salpeter S, Buckley N, Ormiston T, Salpeter E. Meta-analysis: effect of long-acting {beta}-agonists on severe asthma exacerbations and asthma-related deaths. Ann Intern Med. 2006;144:904–912. doi: 10.7326/0003-4819-144-12-200606200-00126. [DOI] [PubMed] [Google Scholar]
- Schuessler TF, Bates JHT. A computer-controlled research ventilator for small animals: design and evaluation. IEEE Trans Biomed Eng. 1995;42:860–866. doi: 10.1109/10.412653. [DOI] [PubMed] [Google Scholar]
- Sears MR. Adverse effects of [beta]-agonists. J Allergy Clin Immun. 2002;110(6 Part2):S322–S328. doi: 10.1067/mai.2002.129966. [DOI] [PubMed] [Google Scholar]
- Spitzer WO, Suissa S, Ernst P, Horwitz RI, Habbick B, Cockcroft D, et al. The use of β-agonists and the risk of death and near death from asthma. NEJM. 1992;326:501–506. doi: 10.1056/NEJM199202203260801. [DOI] [PubMed] [Google Scholar]
- Stolley PD, Schinnar R. Association between asthma mortality and isoproterenol aerosols: a review. Prev Med. 1978;7:519–538. doi: 10.1016/0091-7435(78)90265-7. [DOI] [PubMed] [Google Scholar]
- Sugiura H, Liu X, Duan F, Kawasaki S, Togo S, Kamio K, et al. Cultured lung fibroblasts from ovalbumin-challenged ‘asthmatic’ mice differ functionally from normal. Am J Respir Cell Mol Biol. 2007;37:424–430. doi: 10.1165/rcmb.2007-0089OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaoki J, Yamauchi F, Chiyotani A, Yamawaki I, Takeuchi S, Konno K. Atypical beta-adrenoceptor- (beta 3-adrenoceptor) mediated relaxation of canine isolated bronchial smooth muscle. J Appl Physiol. 1993;74:297–302. doi: 10.1152/jappl.1993.74.1.297. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Tagaya E, Kawatani K, Nakata J, Endo Y, Nagai A. Airway mucosal thickening and bronchial hyperresponsiveness induced by inhaled β2-agonist in mice. Chest. 2004;126:205–212. doi: 10.1378/chest.126.1.205. [DOI] [PubMed] [Google Scholar]
- Taylor DR. The {beta}-agonist saga and its clinical relevance: on and on it goes. Am J Respir Crit Care Med. 2009;179:976–978. doi: 10.1164/rccm.200901-0055CC. [DOI] [PubMed] [Google Scholar]
- Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Puré E, et al. Resolution of lung inflammation by CD44. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- Veness-Meehan KA, Pierce RA, Moats-Staats BM, Stiles AD. Retinoic acid attenuates O2-induced inhibition of lung septation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L971–L980. doi: 10.1152/ajplung.00266.2001. [DOI] [PubMed] [Google Scholar]
- Vignola AM, Mirabella F, Costanzo G, Di Giorgi R, Gjomarkaj M, Bellia V, et al. Airway remodeling in asthma. Chest. 2003;123(3 Suppl.):417S–422S. doi: 10.1378/chest.123.3_suppl.417s. [DOI] [PubMed] [Google Scholar]
- Walker JKL, Peppel K, Lefkowitz RJ, Caron MG, Fisher JT. Altered airway and cardiac responses in mice lacking G protein-coupled receptor kinase 3. Am J Physiol Regul Integr Comp Physiol. 1999;276:R1214–R1221. doi: 10.1152/ajpregu.1999.276.4.R1214. [DOI] [PubMed] [Google Scholar]
- Walker JKL, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, et al. β-Arrestin-2 regulates the development of allergic asthma. J Clin Invest. 2003;112:566–574. doi: 10.1172/JCI17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JKL, Penn RB, Hanania NA, Dickey BF, Bond RA. New perspectives regarding β2-adrenoceptor ligands in the treatment of asthma. Br J Pharmacol. 2011;163:18–28. doi: 10.1111/j.1476-5381.2010.01178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding P, Clark M, Coon JT, Lewis S, Rushton L, Bennett J, et al. Effect of long term treatment with salmeterol on asthma control: a double blind, randomised crossover study. BMJ. 1997;314:1441–1446. doi: 10.1136/bmj.314.7092.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]