

Graphical abstract

Keywords: α-GalCer, Dimer, Multivalency, CD1d, iNKT cells, Click chemistry
Abstract
A library of dimeric CD1d ligands, containing two α-galactosyl ceramide (α-GalCer) units linked by spacers of varying lengths has been synthesised. The key dimerisation reactions were carried out via copper-catalysed click reactions between a 6″-azido-6″-deoxy-α-galactosyl ceramide derivative and various diynes. Each α-GalCer dimer was tested for its ability to stimulate iNKT cells.
1. Introduction
A multimeric version of a monomeric ligand can achieve higher affinity and specificity for a target receptor.1, 2, 3 As a result, incorporating multiple copies of an active pharmacophore within a single molecule can increase biological activity by several orders of magnitude. These observations have opened up the development of so-called multivalent drug candidates as an active area of research.4, 5 There are now numerous examples of synthetic homodimers, which represent the simplest form of a multivalent compound, exhibiting significantly improved potency over their monomeric counterparts.6, 7, 8, 9, 10, 11 For example, dimeric derivative 2 of zanamivir (1), an influenza virus neuraminidase inhibitor, was found to be 100-fold more potent an inhibitor of influenza virus replication, both in vitro and in vivo, than its monomer analogue.6 In a second example, certain dimeric versions (including 4) of the DNA intercalator N-[(2-dimethylamino)ethyl]acridine (DACA, 3) displayed five times the cytotoxic potency of the parent monomer.12, 13 In other cases, the dimeric molecule exhibits different properties entirely from the corresponding monomer. For example, artemisinin (5) is an antimalarial agent, whilst its homodimeric analogues 6a–c exhibit potent anti-tumour activity (Fig. 1).14
Figure 1.

Selected examples of synthetic homodimers which possess increased, or different, biological activity compared with their natural monomeric counterparts.
As part of a wider research programme into the development of novel CD1d ligands,15, 16, 17, 18 we targeted a series of dimeric analogues of the prototypical invariant natural killer T (iNKT) cell agonist, α-galactosyl ceramide (α-GalCer, KRN7000 (7), Figure 2). α-GalCer19 7 is a synthetic glycolipid, which binds to the non-polymorphic MHC-class-I-like molecule, CD1d. The resulting glycolipid–CD1d complex is recognised by T cell receptors (TCRs) located on the surface of iNKT cells. Following recognition of the α-GalCer–CD1d complex, iNKT cells rapidly release a diverse array of both pro-inflammatory (Th1) and regulatory (Th2) cytokines and initiate a potent immune response. This method of activating the immune system is currently being explored as a mechanism for ‘boosting’ current vaccination strategies.20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32
Figure 2.
The prototypical iNKT cell agonist KRN7000 (α-GalCer) 7.
Localised clustering of CD1d molecules has been shown to play an important role in antigen presentation. Park et al. have shown that lipid rafts are essential for efficient TCR recognition of CD1d,33 whilst Im et al. have reported that α-GalCer 7 requires intracellular loading on to CD1d molecules, which leads to the organised transport of α-GalCer–CD1d complexes into cholesterol-rich lipid rafts.34 This localisation of CD1d molecules in membrane rafts serves to concentrate CD1d–lipid complexes in the plasma membrane.35 We hypothesised that bivalent ligands of the α-GalCer pharmacophore might serve to stabilise these lipid rafts by forming a cross-linked matrix within the raft structure. This, in turn, might result in more stable CD1d–glycolipid complexes as a result of a chelating effect.36, 37, 38 With an appropriate linker separating the two α-GalCer units, the dimer might be able to bind two CD1d molecules whilst still allowing space for presentation to iNKT TCRs.39, 40 The length of the linking unit in such ligands would be of key importance for optimal divalent binding; too long and it might provide too large a containment volume for the second pharmacophore and would result in the two α-GalCer moieties effectively behaving as unconnected entities; too short,41, 42 and steric hindrance might block the approach of a second CD1d molecule, although in this case, enhanced binding might still be observed as the result of a statistical re-binding effect.10 In the event that a dimer with a shorter linker is able to bind two CD1d molecules, the presence of a second α-GalCer–CD1d complex in close proximity might still serve to block the approach of the TCR.39, 40 In this way, a dimer could function as an antagonist and yet has signal transduction applications.11, 43 We now report the synthesis and initial biological results of a range of homodimers of α-GalCer, which vary in both the length and type of linker unit.
2. Design and synthesis
Our first consideration in the design of bivalent CD1d ligands was the selection of an appropriate position from which to append a linker to the α-GalCer molecule. The crystal structure of the CD1d–glycolipid–iNKT TCR complex39, 40 reveals that the 2-, 3- and 4-hydroxyl groups of the sugar head group are all involved in hydrogen bonding to either the CD1d molecule or iNKT TCR; modifying these positions leads to reduced, or even complete loss, of activity.44, 45, 46 In contrast, it has been shown that the iNKT TCR–glycolipid–CD1d interaction can tolerate derivatisation at C6 of the galactose unit and there are a number of biologically active CD1d agonists where the 6-hydroxyl group of the sugar has been replaced with another functional group.47, 48, 49, 50 These results are supported by the crystal structure of the CD1d–α-GalCer complex39 and a crystal structure of a TCR–α-GalCer–CD1d complex,40 which reveal that the 6-OH of the α-GalCer sugar head group is not directly involved in hydrogen bonding to either the CD1d molecule or the iNKT TCR. For these reasons, we selected the 6-position of the sugar head group as the most suitable site through which to link together our α-GalCer monomer units.
We have recently developed a facile route to 6″-azido-6″-deoxy-α-galactosyl ceramide 8,51 which we considered a potential advanced intermediate for building α-GalCer dimers. We have shown that the azido moiety of 8 can serve as a masked amine, allowing further elaboration to amides, carbamates and ureas; however more directly, the 6″-azido group is also primed for click chemistry. We therefore postulated that a series of 1,2,3-triazole derivatives would be readily available through a simple Huisgen reaction with an appropriate alkyne.52, 53, 54 We envisaged that α-GalCer dimers could be accessed in a single step via a ‘double click’ reaction between 2 equiv of azide 8 and an appropriate diyne (Scheme 1). This approach would also deliver symmetrical homodimers, which would simplify characterisation and analysis.
Scheme 1.

Proposed synthesis of α-GalCer homodimers.
Poly(ethylene glycol) spacers were chosen to link the α-GalCer units together. These units offer compatibility with biological environments55 as well as increased solubility of the amphiphilic glycolipid units in both aqueous and organic solvents.56, 57 We also chose to investigate alkylene linkers as a hydrophobic comparison. Alkyne-terminated versions of these two types of linkers are readily accessible and would allow the systematic variation of linker lengths.
In order to test the tolerance of the CD1d–glycolipid–iNKT TCR complex to these types of linker units, monomeric α-GalCer analogues 10b and 12 were first synthesised from azide 8 from alkynes 9b and 11, respectively (Scheme 2), and incubated with splenocytes from wildtype C57 BL/6 mice. The presence of IFNγ was then determined by ELISA as previously described by Reddy et al.58 Pleasingly, triazole 10b was found to stimulate iNKT cells in vitro at similar levels to α-GalCer 7, whilst triazole 12 stimulated iNKT cells, albeit to a lesser extent (Fig. 5, vide infra).
Scheme 2.

Synthesis of triazole-containing α-GalCer monomers.
Figure 5.

In vitro activation of iNKT cells by monomers 12 and 10a–e. Splenocytes from C57 BL/6 mice were cultured in the presence of various concentrations of lipids (12, panel A; 10a–e, panel B). The supernatants were analysed for the presence of IFNγ by ELISA following 48 h of culture.58
With the knowledge that the CD1d–glycolipid–iNKT TCR interaction tolerated this type of C6 derivatisation, our next consideration was the length of the spacer units to be employed in our dimer syntheses. Major histocompatibility complex (MHC) molecules, which present peptide fragments to T-cells, are structurally similar to CD1d molecules, and in a related study, Cebecauer et al. have studied MHC–peptide dimers.59 Dimers containing short linkers, 10–30 Å in length,† efficiently triggered intracellular calcium mobilisation and phosphorylation in cloned cytotoxic T lymphocytes, whereas dimers with longer linkers did not. The fact that MHC–peptide dimers containing linkers as short as 10 Å could still interact with the TCR raised the question of how the two MHC molecules align themselves. The authors describe a dimeric binding model in which two TCRs engage with their α3 domains in an anti-parallel manner, two MHC–peptide complexes facing each other. Models of adjacent MHC molecules revealed distances varying from 12 Å to 46 Å between the α3 domains.59 Applying similar orientations to CD1d models (Fig. 3, top), we proposed a range of α-GalCer dimers where the lengths of the spacer units separating the glycolipid pharmacophores could be systematically varied across a similar range. More specifically, we targeted α-GalCer dimers 13a–e and 14a–c, in which the fully extended linker lengths range from 9 Å (for 13a) to 38 Å (for 13e), as estimated from models using Pro-DRG server (Fig. 3, bottom).
Figure 3.
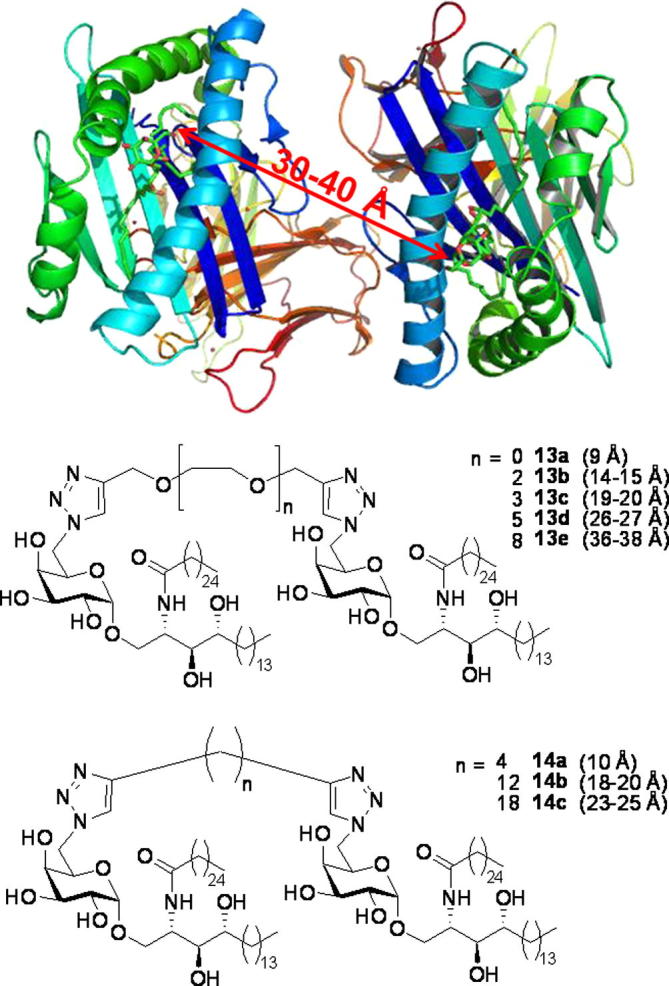
Distances between pharmacophores of target α-GalCer dimers 13a–e and 14a–c.
We first required our series of diynes. Dipropargyl ether 16a is available commercially, whilst bis-propargylated di-, tri-, penta- and octa(ethylene glycol)s 16b–e, respectively, were prepared in good yield by double deprotonation of the appropriate diol 15a–d with an excess of sodium hydride, followed by addition of an excess of propargyl bromide in the presence of tetrabutylammonium iodide (TBAI). For the alkylene diyne precursors, 1,7-octadiyne (18a) is commercially available, whilst 1,15-hexadecadiyne (18b) and 1,21-docosadiyne (18c) were synthesised in moderate yields by treating 1,12-dibromododecane (17a) and 1,18-dibromooctadecane (17b), respectively, with 2 equiv of lithium acetylide–ethylenediamine (EDA) complex (Scheme 3).
Scheme 3.

Synthesis of diynes 16b,c and 18b,c.
Diynes 16a–e and 18a–c were next employed in click reactions with azide 8. Thus, two equivalents of azide 8 and one equivalent of diyne 16a–e or 18a–c were heated in the presence of copper(II) sulfate and sodium ascorbate, to provide bis-triazole dimers 13a–e and 14a–c, respectively, in excellent yields (Scheme 4). These compounds were then tested for their ability to stimulate iNKT cells.
Scheme 4.

Synthesis of α-GalCer dimers 13a–e and 14a–c.
3. Biological results
All of the α-GalCer dimers were found to be active following incubation with splenocytes from wildtype C57 BL/6 mice (Fig. 4);58 PEG-linked dimers 13a–e stimulated iNKT cells in vitro at similar levels to α-GalCer 7 at high concentrations (>10 nM), although there was no obvious sensitivity to linker length. At lower concentrations (<1 nM), dimers 13a and 13b, both of which possess short linkers between the α-GalCer residues, were less active than dimers 13c–e, which contain longer linkers. Alkylene-linked dimers 14a–c also stimulated iNKT cells in vitro, but to a lesser extent than α-GalCer 7. No detectable IFNγ was observed when compounds 13a–e and 14a–c were incubated with splenocytes from CD1d−/− (iNKT cell-deficient) mice (data not shown), indicating that the observed iNKT cell stimulation by our α-GalCer dimers is CD1d-dependent.
Figure 4.

In vitro activation of iNKT cells by dimers 14a–c and 13a–e. Splenocytes from C57 BL/6 mice were cultured in the presence of various concentrations of lipids (14a–c, panel A; 13a–e, panel B). The supernatants were analysed for the presence of IFNγ by ELISA following 48 h of culture.58
In order to test the ability of the iNKT TCR to recognise the CD1d–glycolipid complex, in vitro binding experiments were performed (Fig. 6).60 Dimers 13c–e were comparable to α-GalCer 7 at high concentrations (>10 nM), whilst dimers 13a,b again proved to be considerably weaker than α-GalCer 7. At lower concentrations (<5 nM), dimers 13c–e, which contain the longer linker units, displayed greater iNKT-cell recognition than α-GalCer 7. Dimer 13d, which contains a penta(ethylene glycol) linker (∼26–27 Å fully extended length), proved optimal. Alkylene-linked dimers 14a–c showed weaker CD1d binding than α-GalCer 7 (Fig. 6) and we tentatively postulate that the lower activity of this series of dimers might be a result of the hydrophobic alkylene linker coiling to minimise contact with the aqueous medium. This coiling will serve to decrease the effective length of the linker and may account for the observed activity of these dimers more closely resembling that observed for the ethylene glycol dimers containing shorter linkers (i.e., 13a,b).
Figure 6.

iNKT cell recognition of 14a–c and 13a–e. The recognition of 14a–c (panel A) and 13a–e (panel B) by human iNKT TCR tetramer was assessed by flow cytometry following co-incubation of fluorescent human iNKT cell TCR and hCD1d C1R cells loaded with the indicated lipids.60 MFI = Median Fluorescent Intensity.
In order to investigate whether or not the trends observed with dimers 13a–e were a consequence of the presence of the second pharmacophore or due to the presence of the linker unit alone, we targeted a series of monomers 10a–e each containing a PEG tail length which corresponded to the same linker lengths separating the α-GalCer residues in dimers 13a–e. Monomers 10a–e were readily accessed via click reactions between azide 8 and alkynes 9a–e. Alkyne 9a was available commercially, whilst alkynes 9b–e were readily synthesised from alcohols 19a–d (Scheme 5).
Scheme 5.

Synthesis of monomers 10a–e.
Each of the monomers 10a–e was tested for its ability to stimulate iNKT cells (Fig. 5). α-GalCer 7 served as a reference compound, enabling us to compare the data for monomers 10a–e with the data for the corresponding dimers 13a–e. At high concentrations (>10 nM), the level of iNKT-cell stimulation for all of the monomers 10a–e was not significantly different to that observed for the corresponding dimers (relative to α-GalCer 7 reference). Surprisingly, at lower concentrations (<1 nM), monomer 10d proved to be a more potent activator of iNKT cells than its dimeric equivalent (13d), relative to α-GalCer 7, whilst 10a, the control for 13a, was again inactive at low concentrations in vitro. The fact that dimers 13a–e do not show any enhanced biological activity over their corresponding monomers 10a–e suggests that a bivalent effect is not taking place in vitro. A comparison of the data for monomer 12, containing an octyl chain, and the alkylene-linked dimers 14a–c also provides little evidence for a bivalent effect.
The effect of lipid rafts and clustering effects on iNKT-cell activation might be expected to be more apparent in vivo,61 and the in vivo results were indeed more interesting. PEG-linked dimers 13c–e, which had proven to be the most active from the in vitro studies, were injected intravenously into C57 BL/6 WT mice. Blood serum was taken at 18 h and the presence of IFNγ determined by ELISA (Fig. 7).62 iNKT-cell stimulation was found to increase with linker length, up to the point where the dimer with the longest octa(ethylene glycol) linker, 13e, had almost the same activity as α-GalCer 7. As before, CD1d−/− (iNKT cell-deficient) mice did not have any detectable IFNγ (data not shown), indicating that these results are again CD1d-dependent. These in vivo results raise the question of how this trend of iNKT-cell stimulation versus linker length would extrapolate with even longer linkers.
Figure 7.

Wildtype C57 BL/6 (n = 3/group) mice were injected intravenously with 1 μg of analogue or vehicle. IFNγ levels in blood serum were determined by ELISA at 18 h post injection.62
In summary, we have synthesised two series of biologically active dimeric α-GalCer analogues (13a–e and 14a–c). Initial in vitro experiments showed that dimers based on poly(ethylene glycol) linkers (13a–e) stimulated iNKT cells to a similar extent as α-GalCer 7 for longer linker lengths (13c–e), whilst those with shorter lengths (13a,b) were less-effective CD1d agonists. We considered the possibility that this trend might be due to the linking poly(ethylene glycol) unit itself rather than as a result of a bivalent effect and therefore synthesised and tested a series of monomers (10a–e) containing a poly(ethylene glycol) tail length corresponding to each dimer linker length. These monomers showed a similar trend to the corresponding dimers, with 10a,b being the least active. Surprisingly, monomer 10d was more active than its corresponding dimer, 13d. Dimers based on alkylene linkers proved to be less active than α-GalCer 7 in vitro, and showed no obvious sensitivity to linker length. Overall, the in vitro results show little evidence for a bivalent effect. Preliminary in vivo experiments show an increase in activity with increasing linker length, with the octa(ethylene glycol)-linked dimer 13e being the most potent activator of iNKT cells, having a similar activity to α-GalCer 7. Future work will involve producing dimers and multimers with longer linker lengths and further probing the multivalent effects using different techniques.
4. Experimental
4.1. General methods
Infra-red spectra were recorded neat as thin films between sodium chloride discs, or as potassium bromide discs. The intensity of each band is described as s (strong), m (medium) or w (weak) and with the prefix v (very) and suffix br (broad) where appropriate. The poor solubility of all α-GalCer dimers at rt prevented us from obtaining reliable optical rotation data. 1H NMR and 13C NMR chemical shifts are reported as δ values (ppm) referenced to the following solvent signals: CHCl3, δH 7.26; CDCl3, δC 77.0; MeOH, δH 3.31; CD3OD, δC 49.0. The term ‘stack’ is used to describe a region where resonances arising from non-equivalent nuclei are coincident, and multiplet, m, to describe a resonance arising from a single nucleus (or equivalent nuclei) but where coupling constants cannot be readily assigned. Mass spectra were recorded on a LCT spectrometer utilising electrospray ionisation with a methanol mobile phase, or electron impact ionisation, and are reported as (m/z (%)). HRMS were recorded on a LCT spectrometer using a lock mass incorporated into the mobile phase. Melting points were determined using open capillaries and are uncorrected.
4.2. Chemicals
All reagents were obtained from commercial sources and used without further purification unless specified otherwise. Tetrahydrofuran was freshly distilled under nitrogen from sodium benzophenone ketyl. All solutions are aqueous and saturated unless specified otherwise.
4.3. General procedures
4.3.1. General procedure for bis-propargylation of diols 15a–d and mono-propargylation of alcohols 19a–d (synthesis of diynes 16b–e and alkynes 9b–e)
NaH (60% w/w in mineral oil, 3 equiv (for bis-propargylation) or 1.5 equiv (for mono-propargylation)) was added to a solution of appropriate diol 15ba–d or alcohol 19a–d (1 equiv) in anhydrous THF (0.2 M) at 0 °C under an N2 atmosphere. A catalytic amount (spatula tip) of Bu4NI (0.05 equiv) and propargyl bromide (3 equiv (for bis-propargylation) or 1.5 equiv (for mono-propargylation)) was added sequentially. The mixture was stirred at rt overnight, before being concentrated under reduced pressure. Purification of the residue by flash column chromatography afforded diynes 16b–e and alkynes 9b–e as pale yellow oils.
4.3.2. General procedure for lithium acetylide addition to dibromoalkanes 17a and 17b (synthesis of diynes 18b and 18c)
Lithium acetylide–ethylenediamine complex (2 equiv) was added to a solution of dibromoalkane 17a or 17b (1 equiv) in DMSO (0.2 M) at 0 °C. The mixture was allowed to warm to rt and stirred overnight. H2O (5 × volume) and hexane (5 × volume) were added and the phases were separated. The aqueous phase was extracted with hexane (5 × volume). The combined organic extracts were washed with brine (5 × volume), dried (MgSO4), filtered and concentrated under reduced pressure. Purification of the residue by flash column chromatography (hexane) afforded diyne 18b or 18c both as white solids.
4.3.3. General procedure for click chemistry (synthesis of dimers 13a–e and 14a–c and monomers 10a–e and 12)
CuSO4 solution (5 μL of 0.5 M solution, 2.5 μmol) and sodium ascorbate solution (18 μL of a 1.0 M solution, 18 μmol) were added to a solution of azide 8 (20 mg, 0.023 mmol) and diyne 14a–e or 16a–c (0.011 mmol) or alkyne 9a–e or 11 (0.023 mmol) in tBuOH/H2O (1 mL, 1:1) at rt. The reaction mixture was heated for 10 h at 50 °C and then diluted with CHCl3 (10 mL), and washed with brine (3 mL). The phases were separated and the aqueous layer was extracted with CHCl3 (2 × 5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification of the residue by flash column chromatography afforded dimers 13a–e and 14a–c and monomers 10a–e and 12.
4.4. Characterisation
4.4.1. Bis-O-propargyl-di(ethylene glycol) (16b)
Diyne 16b was prepared from di(ethylene glycol) 15a (1.00 g, 9.43 mmol) and propargyl bromide (2.54 mL of a 80% w/w solution in toluene, 29.5 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (25% EtOAc in hexane) afforded diyne 16b as a yellow oil (1.38 g, 81%): Rf = 0.28 (25% EtOAc in hexane); IR (film) ν 3252m ( C–H), 2866m, 2114w (C C), 1443m, 1349m, 1288w, 1136s, 1094s, 1033m, 918m, 877w, 843m; 1H NMR (300 MHz, CDCl3) δ 2.42 (t, 2H, J 2.4 Hz), 3.65–3.73 (stack, 8H, OCH2CH2O), 4.19 (d, 4H, J 2.4 Hz); 13C NMR: (75 MHz, CDCl3) δ 58.1 (CH2), 68.9 (CH2), 70.2 (CH2), 74.4 (CH), 79.4 (quat. C); m/z (TOF ES+) 205.1 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 205.0847. C10H14NaO3 requires 205.0841.
4.4.2. Bis-O-propargyl-tri(ethylene glycol) (16c)
Diyne 16c was prepared from tri(ethylene glycol) 15b (1.42 g, 9.47 mmol) and propargyl bromide (2.54 mL of a 80% w/w solution in toluene, 29.5 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (40% EtOAc in hexane) afforded diyne 16c as a yellow oil (1.52 g, 71%): Rf = 0.31 (40% EtOAc in hexane); IR (film) ν 3251m ( C–H), 2868m, 2114w (C C), 1443m, 1349m, 1248m, 1093s, 1031s, 917s, 877s, 842s, 731s; 1H NMR (300 MHz, CDCl3) δ 2.43 (t, 2H, J 2.4 Hz), 3.65–3.78 (12H, stack), 4.21 (d, 4H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.2 (CH2,), 68.9 (CH2), 70.2 (CH2), 70.4 (CH2), 74.4 (CH), 79.5 (quat. C); m/z (TOF ES+) 249.1 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 249.1094. C12H18NaO4 requires 249.1103.
4.4.3. Bis-O-propargyl-penta(ethylene glycol) (16d)
Diyne 16d was prepared from penta(ethylene glycol) 15c (2.25 g, 9.45 mmol) and propargyl bromide (2.54 mL of 80% w/w solution in toluene, 29.5 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (60% EtOAc in hexane) afforded diyne 16d as a yellow oil (2.17 g, 73%): Rf = 0.26 (60% EtOAc in hexane); IR (film) ν 3247m ( C–H), 2867s, 2113w (C C), 1458m, 1349m, 1289m, 1248m, 1093s, 1032s, 948m, 919m, 842m, 669s; 1H NMR (300 MHz, CDCl3) δ 2.43 (t, 2H, J 2.4 Hz), 3.64–3.73 (stack, 20H), 4.20 (d, 4H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.3 (CH2), 69.1 (CH2), 70.3 (CH2), 70.5 (CH2), 74.5 (CH), 79.6 (quat. C), some overlapping ethylene glycol resonances; m/z (TOF ES+) 337.2 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 337.1615. C16H26NaO6 requires 337.1627.
4.4.4. Bis-O-propargyl-octa(ethylene glycol) (16e)
Diyne 16e was prepared from octa(ethylene glycol) 15d (1.00 g, 2.70 mmol) and propargyl bromide (0.73 mL of a 80% w/w solution in toluene, 8.20 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (6% MeOH in CHCl3) afforded diyne 16e as a yellow oil (1.03 g, 85%): Rf = 0.29 (6% MeOH in CHCl3); IR (film) ν 3247m ( C–H), 2919m, 2864m, 2112w (C C), 1956w, 1458m, 1349m, 1291m, 1248m, 1093s, 1032m, 946m, 840m, 703m, 688m, 675m, 660m; 1H NMR (300 MHz, CDCl3) δ 2.43 (t, 2H, J 2.4 Hz), 3.64–3.73 (stack, 32H), 4.20 (d, 4H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.2 (CH2), 68.9 (CH2), 70.1 (CH2), 70.3 (CH2), 74.4 (CH), 79.5 (quat. C), some overlapping ethylene glycol resonances; m/z (TOF ES+) 469.2 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 469.2406. C22H38NaO9 requires 469.2414.
4.4.5. Hexadeca-1,15-diyne (18b)
Lithium acetylide–ethylenediamine complex (693 mg, 7.50 mmol) and dibromide 17a (1.15 g, 3.51 mmol) were reacted according to Section 4.3.2. After stirring overnight, work-up and purification of the residue by flash column chromatography (hexane) afforded diyne 18b as a white solid (62%): mp 43–45 °C, lit.63 43–44 °C; Rf = 0.65 (hexane); IR (KBr) ν 3286m ( C–H), 2917m, 2849m, 2115w (C C), 1472m, 1462m, 1419w, 1339w, 1280w, 732m, 720m, 666s; 1H NMR (300 MHz, CDCl3) δ 1.13–1.44 (stack, 16H), 1.46–1.57 (m, 4H), 1.93 (t, 2H, J 2.6 Hz), 2.17 (td, 4H, J 6.9, 2.6 Hz); 13C NMR (75 MHz, CDCl3) δ [18.4, 28.5, 28.8, 29.1, 29.5, 29.6 (CH2, overlapping alkyl chain resonances)], 68.0 (CH, C CH), 84.8 (quat. C, C CH); m/z (TOF ES+) 241.2 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 241.1941. C16H26Na requires 241.1932.
4.4.6. Docosa-1,21-diyne (18c)
Lithium acetylide–ethylenediamine complex (36 mg, 0.39 mmol) and dibromide 17b (75 mg, 0.18 mmol) were reacted according to Section 4.3.2. After stirring overnight, work-up and purification of the residue by flash column chromatography (hexane) afforded diyne 18c as a white solid (58%): mp 64–65 °C; Rf = 0.73 (hexane); IR (KBr) ν 3906m ( C–H), 2917m, 2849m, 2115w (C C), 1472m, 1462m, 1419w, 1340w, 1280w, 733m, 720m, 665s; 1H NMR (300 MHz, CDCl3) δ 1.17–1.45 (stack, 28H), 1.46–1.57 (m, 4H), 1.93 (t, 2H, J 2.6 Hz), 2.18 (td, 4H, J 7.1, 2.6 Hz); 13C NMR (75 MHz, CDCl3) δ [18.4, 28.5, 28.8, 29.1, 29.5, 29.7 (CH2, overlapping alkyl chain resonances)], 68.0 (CH), 84.8 (quat. C); m/z (TOF ES+) 325.3 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 325.2867. C22H38Na requires 325.2871.
4.4.7. Bis-1,2,3-triazole 13a (PEG-0 link)
Azide 8 (20 mg, 0.023 mmol) and diyne 16a (1.0 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded bis-1,2,3-triazole 13a as a colourless paste (15 mg, 72%): Rf = 0.22 (15% MeOH in CHCl3); IR (KBr) ν 3353br s (O–H), 2917s, 2850s, 1638m (C O), 1544w, 1468m, 1343w, 1230w, 1148m, 1066s, 1035s, 784w, 720s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.85 (app. t, 12H, J 6.6 Hz), 1.14–1.36 (stack, 136H), 1.42–1.63 (stack, 8H), 2.14 (app. t, 4H, J 6.7 Hz), 3.36–3.40 (m, 2H), 3.45–3.52 (stack, 6H), 3.71 (dd, 2H, J 9.9, 3.0 Hz), 3.75–3.83 (stack, 4H), 4.03–4.12 (m, 2H), 4.16 (app. t, 2H, J 5.7 Hz), 4.53–4.62 (stack, 8H), 4.86 (d, 2H, J 3.6 Hz), 7.89 (br s, 2H), exchangeable protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 13.5 (CH3), [20.6, 22.2, 25.4, 28.79. 28.85, 28.92, 29.1, 29.2, 31.4, 31.9, 35.9 (CH2, overlapping alkyl chain resonances)], 49.6 (CH), 50.5 (CH2), 62.6 (CH2), 66.6 (CH), 68.1 (CH), 68.9 (2 × CH, resonance overlap), 69.4 (CH2), 71.4 (CH), 74.0 (CH), 99.1 (CH), 124.5 (CH), 144.9 (quat. C), 174.0 (quat. C); m/z (TOF ES−) 1859.3 ([M−H]−, 100%), 1691.0 (43), 1521.7 (63).
4.4.8. Bis-1,2,3-triazole 13b (PEG-2 link)
Azide 8 (20 mg, 0.023 mmol) and diyne 16b (2.0 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded bis-1,2,3-triazole 13b as a colourless paste (18 mg, 84%): Rf = 0.23 (15% MeOH in CHCl3); IR (KBr) ν 3351m br (O–H), 2917s, 2850s, 1631m (C O), 1547w, 1467m, 1349w, 1230w, 1141m, 1064s, 1034s, 785w, 720s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 12H, J 6.6), 1.14–1.32 (stack, 136H), 1.46–1.61 (stack, 8H), 2.14 (app. t, 4H, J 6.7 Hz), 3.32–3.40 (m, 2H), 3.41–3.50 (stack, 6H), 3.61–3.73 (stack, 10H), 3.75–3.83 (stack, 4H), 4.04–4.12 (m, 2H), 4.18 (dt, 2H, J 3.3, 3.0 Hz), 4.52–4.59 (stack, 4H), 4.60–4.66 (stack, 4H), 4.85 (d, 2H, J 3.6 Hz), 7.84 (br d, 2H, J 3.0 Hz), exchangeable protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 13.5 (CH3), [20.6, 22.2, 25.5, 28.9, 29.0, 29.1, 29.2, 29.3, 29.3, 29.4, 29.8, 31.5, 32.2, 33.8, 36.1 (CH2, overlapping alkyl chain resonances)], 49.7 (CH), 50.4 (CH2), 63.8 (CH2), 66.9 (CH2), 68.3 (CH), 68.9 (CH), 69.0 (CH), 69.2 (CH2), 69.5 (CH), 70.0 (CH2), 71.6 (CH), 74.2 (CH), 99.2 (CH), 124.4 (CH), 144.8 (quat. C), 174.0 (quat. C); m/z (TOF ES+) 1947.9 ([M+H]+, 80%), 1107.9 (42), 1025.9 (100).
4.4.9. Bis-1,2,3-triazole 13c (PEG-3 link)
Azide 8 (20 mg, 0.023 mmol) and diyne 16c (2.5 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded bis-1,2,3-triazole 13c as a colourless paste (19 mg, 87%): Rf = 0.23 (15% MeOH in CHCl3); IR (KBr) ν 3368m br (O–H), 2918s, 2850s, 1634m (C O), 1552w, 1467s, 1349w, 1231w, 1147m, 1071s, 1037s, 785w, 719m; 1H NMR (400 MHz, CDCl3/CD3OD, 2:1) δ 0.85 (app. t, 12H, J 6.6 Hz), 1.15–1.33 (stack, 136H), 1.45–1.62 (stack, 8H), 2.14 (app. t, 4H, J 7.2 Hz), 3.36–3.43 (m, 2H), 3.43–4.52 (stack, 6H), 4.53–3.60 (m, 2H), 3.60–3.73 (stack, 14H), 3.76–3.81 (m, 2H), 4.06–4.11 (m, 2H), 4.18 (app. t, 2H, J 6.0 Hz), 4.55–4.59 (stack, 4H), 4.60–4.64 (stack, 4H), 4.85 (d, 2H, J 3.6 Hz), 7.84 (br s, 2H), exchangeable protons not observed; 13C NMR (100 MHz, CDCl3/CD3OD, 2:1) δ 13.3 (CH3), [22.1, 25.4, 28.7, 28.82, 28.88, 28.93, 29.1, 29.3, 31.4, 31.9, 35.9 (CH2, overlapping alkyl chain resonances)], 49.6 (CH), 50.4 (CH2), 63.6 (CH2), 66.6 (CH2), 68.2 (CH), 68.9 (CH), 69.0 (CH), 69.1 (CH2), 69.4 (CH), 69.8 (CH2), 69.9 (CH2), 71.4 (CH), 74.0 (CH), 99.1 (CH), 124.3 (CH), 144.3 (quat. C), 173.9 (quat. C); m/z (TOF ES+) 1992.0 ([M+H]+, 30%), 1070.0 (100).
4.4.10. Bis-1,2,3-triazole 13d (PEG-5 link)
Azide 8 (20 mg, 0.023 mmol) and diyne 16d (3.5 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (20% MeOH in CHCl3) afforded bis-1,2,3-triazole 13d as a colourless paste (18 mg, 79%): Rf = 0.23 (20% MeOH in CHCl3); IR (KBr) ν 3369m br (O–H), 2918s, 2850s, 1634m (C O), 1552w, 1467s, 1349w, 1231w, 1147m, 1070s, 1035s, 785w, 719m; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.85 (app. t, 12H, J 6.8 Hz), 1.14–1.39 (stack, 136H), 1.47–1.65 (stack, 8H), 2.14 (app. t, 4H, J 7.5 Hz), 3.36–3.43 (m, 2H), 3.44–3.53 (stack, 6H), 3.60–3.67 (stack, 20H), 3.71 (dd, 2H, J 9.9, 3.3 Hz), 3.76–3.83 (stack, 4H,), 4.03–4.13 (m, 2H), 4.17 (app. t, 2H, J 6.5 Hz), 4.53–4.63 (stack, 8H), 4.86 (d, 2H, J 3.6 Hz), 7.42 (d, 2H, J 8.7 Hz), 7.84 (s, 2H), alcoholic protons not observed; 13C NMR (100 MHz, CDCl3/CD3OD, 2:1) δ 13.2 (CH3), [22.1, 25.3, 28.78, 28.83, 28.9, 29.1, 29.09, 29.13, 31.4, 31.8, 35.8 (CH2, overlapping alkyl chain resonances)], 49.6 (CH), 50.4 (CH2), 63.5 (CH2), 66.6 (CH2), 68.1 (CH), 68.9 (CH), 69.0 (CH), 69.4 (CH), 69.8 (CH2, overlapping ethylene glycol resonances), 71.4 (CH), 73.9 (CH), 99.1 (CH), 124.3 (CH), 144.1 (quat. C), 173.9 (quat. C); m/z (TOF ES+) 2079.2 ([M+Na]+, 100%), 694.5 (70, [ceramide]+).
4.4.11. Bis-1,2,3-triazole 13e (PEG-8 link)
Azide 8 (20 mg, 0.023 mmol) and diyne 16e (4.9 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (20% MeOH in CHCl3) afforded bis-1,2,3-triazole 13e as a colourless paste (21 mg, 86%): Rf = 0.25 (20% MeOH in CHCl3); IR (KBr) ν 3340m br (O–H), 2917s, 2850s, 1632m (C O), 1549w, 1467m, 1349m, 1300w, 1232w, 1138m, 1082s, 1039s, 948m, 786m, 719s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.85 (app. t, 12H, J 6.9 Hz), 1.14–1.49 (stack, 136H), 1.43–1.67 (stack, 8H), 2.14 (app. t, 4H, J 7.2 Hz), 3.36–3.52 (stack, 8H), 3.60–3.68 (stack, 32H), 3.68–3.75 (m, 2H), 3.76–3.83 (stack, 4H), 4.01–4.09 (m, 2H), 4.17 (app. t, 2H, J 6.2 Hz), 4.52–4.63 (stack, 8H), 4.85 (d, 2H, J 2.4 Hz), 7.51 (d, 2H, J 8.1 Hz), 7.83 (s, 2H), alcoholic protons not observed; 13C NMR (100 MHz, CDCl3/CD3OD, 2:1) δ 13.3 (CH3), [22.1, 25.4, 28.8, 28.89, 28.94, 29.06, 29.13, 29.2, 29.3, 31.4, 31.9, 35.9 (CH2, overlapping alkyl chain resonances)], 49.7 (CH), 50.5 (CH2), 63.6 (CH2), 66.6 (CH2), 68.2 (CH), 68.9 (CH), 69.0 (CH), 69.4 (CH), [69.6, 69.7 (CH2, overlapping ethylene glycol resonances)] 71.4 (CH), 73.9 (CH), 99.1 (CH), 124.3 (CH), 144.8 (quat. C), 173.9 (quat. C); m/z (TOF ES+) 2211.6 ([M+H]+, 45%), 694.6 (35, [ceramide]+), 450.2 (100).
4.4.12. Bis-1,2,3-triazole 14a (–(CH2)4− link)
Azide 8 (20 mg, 0.023 mmol) and diyne 18a (1.2 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded bis-1,2,3-triazole 14a as a colourless paste (16 mg, 81%): Rf = 0.24 (15% MeOH in CHCl3); IR (KBr) ν 3359m br (O–H), 2921s, 2849s, 1636m (C O), 1555m, 1468s, 1351w, 1222w, 1150m, 1057s, 904w, 787m, 724s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 12H, J 6.7 Hz), 1.14–1.38 (stack, 136H), 1.45–1.72 (stack, 12H), 2.14 (app. t, 4H, J 7.5 Hz), 2.63–2.74 (m, 4H, J 7.2 Hz), 3.38 (dd, 2H, J 10.6, 4.17 Hz), 3.43–3.58 (stack, 6H), 3.66–3.72 (m, 2H), 3.73–3.82 (stack, 4H), 4.07–4.20 (stack, 4H), 4.48–4.57 (stack, 4H), 4.85 (d, 2H, J 3.6 Hz), 7.41 (d, 2H, J 8.8 Hz), 7.56 (s, 2H), alcoholic protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 13.4 (CH3), [22.2, 24.5, 25.3, 25.4, 25.5, 28.2, 28.9, 28.96, 29.02, 29.2, 29.3, 29.4, 31.5, 32.2, 36.0 (CH2, overlapping alkyl chain resonances)], 49.7 (CH), 50.2 (CH2), 66.8 (CH2), 68.2 (CH), 68.8 (CH), 69.0 (CH), 69.5 (CH), 71.5 (CH), 74.3 (CH), 99.2 (CH), 122.6 (CH), 147.5 (quat. C), 174.0 (quat. C); m/z (TOF ES+) 1894.5 ([M+Na]+, 100%).
4.4.13. Bis-1,2,3-triazole 14b (–(CH2)12− link)
Azide 8 (20 mg, 0.023 mmol) and diyne 18b (2.1 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded bis-1,2,3-triazole 14b as a colourless paste (19 mg, 88%): Rf = 0.26 (15% MeOH in CHCl3); IR (KBr) ν 3351m br (O–H), 2917s, 2850s, 1635m (C O), 1550m, 1467s, 1344w, 1221w, 1150m, 1058s, 906w, 784m, 720m; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.85 (app. t, 12H, J 6.6 Hz), 1.15–1.49 (stack, 152H), 1.47–1.68 (stack, 12H), 2.14 (app. t, 4H, J 7.5 Hz), 2.65 (app. t, 4H, J 7.2 Hz), 3.36–3.43 (m, 2H), 3.44–3.55 (stack, 6H), 3.67–3.74 (m, 2H), 3.76–3.83 (stack, 4H), 4.07–4.21 (stack, 4H), 4.47–4.60 (stack, 4H), 4.86 (d, 2H, J 3.3 Hz), 7.56 (s, 2H), exchangeable protons not observed; 13C NMR (100 MHz, CDCl3/CD3OD, 2:1) δ 13.1 (CH3), [22.0, 24.8, 25.2, 25.3, 28.7, 28.76, 28.83, 29.0, 29.1, 29.2, 31.3, 35.8 (CH2, overlapping alkyl chain resonances)], 49.5 (CH), 50.2 (CH2), 66.4 (CH2), 68.1 (CH), 68.9 (CH), 69.0 (CH), 69.4 (CH), 71.3 (CH), 74.0 (CH), 99.0 (CH), 122.3 (CH), 147.6 (quat. C), 174.0 (quat. C); m/z (TOF ES−) 1983.1 ([M−H]− 100%).
4.4.14. Bis-1,2,3-triazole 14c (–(CH2)18− link)
Azide 8 (20 mg, 0.023 mmol) and diyne 18c (3.3 mg, 0.011 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded bis-1,2,3-triazole 14c as a colourless paste (20 mg, 87%): Rf = 0.27 (15% MeOH in CHCl3); IR (film) ν 3386m br (O–H), 2918s, 2851s, 1637m (C O), 1537w, 1467s, 1438w, 1345w, 1204m, 1151s, 1065s, 1036s, 801m, 720s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.85 (app. t, 12H, J 6.6 Hz), 1.14–1.49 (stack, 164H), 1.47–1.68 (stack, 12H), 2.14 (app. t, 4H, J 7.7 Hz), 2.65 (app. t, 4H, J 7.5 Hz), 3.38 (dd, 2H, J 10.2, 4.2 Hz), 3.43–3.55 (stack, 6H), 3.69 (dd, 2H, J 10.2, 3.0 Hz), 3.76–3.82 (stack, 4H), 4.07–4.20 (stack, 4H), 4.45–4.60 (stack, 4H), 4.87 (d, 2H, J 3.6 Hz), 7.54 (br s, 2H), exchangeable protons not observed; 13C NMR (100 MHz, CDCl3/CD3OD, 2:1) δ 13.2 (CH3), [22.1, 24.8, 25.28, 25.32, 28.77, 28.82, 28.87, 28.92, 29.00, 29.09, 29.13, 29.3, 31.4, 32.0, 35.8 (CH2, overlapping alkyl resonances)], 49.6 (CH), 50.2 (CH2), 66.4 (CH2), 68.1 (CH), 68.9 (CH), 69.0 (CH), 69.4 (CH), 71.3 (CH), 74.1 (CH), 99.1 (CH), 122.2 (CH), 147.7 (quat. C), 173.8 (quat. C); m/z (TOF ES+) 2067.7 ([M+Na]+, 30%), 1885.3 (30), 694.6 (100, [ceramide]+).
4.4.15. O-Propargyl-O′-methyl-di(ethylene glycol) (9b)
Alkyne 9b was prepared from di(ethylene glycol) monomethyl ether (19a) (0.50 g, 4.16 mmol) and propargyl bromide (0.53 mL of a 80% w/w solution in toluene, 6.24 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (25% EtOAc in hexane) afforded alkyne 9b as a yellow oil (0.57 g, 87%): Rf = 0.26 (25% EtOAc in hexane); IR (film) ν 3251m ( C–H), 2866m, 2113w (C C), 1442m, 1350m, 1295w, 1288w, 1136s, 1094s, 1033m, 918m, 877w, 843m, 734m; 1H NMR (300 MHz, CDCl3) δ 2.41 (t, 1H, J 2.4 Hz), 3.35 (s, 3H), 3.50–3.58 (m, 2H), 3.59–3.72 (stack, 6H), 4.17 (d, 2H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.3 (CH), 58.9 (CH3), 69.0 (CH2), 70.3 (CH2), 70.5 (CH2), 71.8 (CH2), 74.4 (CH), 79.6 (quat. C); m/z (TOF ES+) 181.1 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 181.0837. C10H18NaO3 requires 181.0841.
4.4.16. O-Propargyl-O′-methyl-triethylene glycol (9c)
Alkyne 9c was prepared from tri(ethylene glycol) monomethyl ether (19b) (250 mg, 1.52 mmol) and propargyl bromide (190 μL of a 80% w/w solution in toluene, 2.28 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (40% EtOAc in hexane) afforded alkyne 9c as a yellow oil (279 mg, 91%): Rf = 0.30 (40% EtOAc in hexane); IR (film) ν 3248m ( C–H), 2865m, 2114w (C C), 1442m, 1348m, 1246m, 1092s, 1031s, 918s, 879s, 842s, 756m, 731s; 1H NMR (300 MHz, CDCl3) δ 2.41 (t, 1H, J 2.4 Hz), 3.35 (s, 3H), 3.50–3.59 (m, 2H), 3.59–3.74 (stack, 10H), 4.17 (d, 2H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.3 (CH), 58.9 (CH3), 69.0 (CH2), 70.3 (CH2), 70.4 (CH2), 70.5 (CH2, overlapping ethylene glycol resonances), 71.8 (CH2), 74.4 (CH), 79.6 (quat. C); m/z (TOF ES+) 225.1 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 225.1110. C10H18NaO4 requires 225.1103.
4.4.17. O-Propargyl-O′-methyl-penta(ethylene glycol) (9d)
Alkyne 9d was prepared from penta(ethylene glycol) monomethyl ether (19c) (250 mg, 0.99 mmol) and propargyl bromide (126 μL of a 80% w/w solution in toluene, 1.49 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (60% EtOAc in hexane) afforded alkyne 9d as a yellow oil (244 mg, 85%): Rf = 0.24 (60% EtOAc in hexane); IR (film) ν 3253m ( C–H), 2869m, 2113w (C C), 1447m, 1346m, 1247m, 1093s, 1029s, 917s, 879s, 842s, 748m, 731s; 1H NMR (300 MHz, CDCl3) δ 2.42 (t, 1H, J 2.4 Hz), 3.36 (s, 3H), 3.50–3.58 (m, 2H), 3.60–3.73 (stack, 18H), 4.18 (d, 2H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.3 (CH), 59.0 (CH3), 69.0 (CH2), 70.3 (CH2), 70.45 (CH2), 70.51 (CH2, overlapping ethylene glycol resonances), 71.9 (CH2), 74.4 (CH), 79.6 (quat. C); m/z (TOF ES+) 313.3 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 313.1624. C14H26NaO6 requires 313.1627.
4.4.18. O-Propargyl-O′-methyl-octa(ethylene glycol) (9e)
Alkyne 9e was prepared from octa(ethylene glycol) monomethyl ether (19d) (100 mg, 0.26 mmol) and propargyl bromide (32 μL of a 80% w/w solution in toluene, 0.39 mmol) according to Section 4.3.1. After stirring overnight, removal of the solvent and purification of the residue by flash column chromatography (5% MeOH in CHCl3) afforded alkyne 9e as a yellow oil (244 mg, 85%): Rf = 0.23 (5% MeOH in CHCl3); IR (film) ν 3248m ( C–H), 2916m, 2864m, 2114w (C C), 1956w, 1457m, 1346m, 1292m, 1246m, 1094s, 1030m, 946m, 839m, 764m, 703m; 1H NMR (300 MHz, CDCl3) δ 2.41 (t, 1H, J 2.4 Hz), 3.32 (s, 3H), 3.46–3.54 (m, 2H), 3.57–3.70 (stack, 30H), 4.15 (d, 2H, J 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ 58.2 (CH), 58.8 (CH3), 68.9 (CH2), 70.2 (CH2), 70.4 (CH2, some overlapping ethylene glycol resonances), 71.8 (CH2), 74.4 (CH), 79.5 (quat. C); m/z (TOF ES+) 445.2 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 445.2421. C20H38NaO9 requires 445.2414.
4.4.19. 1,2,3-triazole 10a (PEG-0 tail)
Azide 8 (20 mg, 0.023 mmol) and alkyne 9a (1.6 mg, 0.023 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded 1,2,3-triazole 10a as a colourless paste (20 mg, 92%): Rf = 0.29 (15% MeOH in CHCl3); IR (KBr) ν 3352br s (O–H), 2914s, 2851s, 1638m (C O), 1543w, 1467m, 1343w, 1229w, 1150m, 1067s, 1034s, 784w, 720s, 717w, 668m; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 6H, J 6.6 Hz), 1.14–1.38 (stack, 68H), 1.43–1.67 (stack, 4H), 2.13 (app. t, 2H, J 7.5 Hz), 3.33–3.40 (stack including [3.37 (s, 3H)], 4H), 3.42–3.52 (stack, 3H), 3.69 (dd, 1H, J 9.8, 3.3 Hz), 3.74–3.83 (stack, 2H), 4.03–4.13 (m, 1H), 4.13–4.20 (m, 1H), 4.52 (s, 2H), 4.53–4.59 (m, 2H), 4.85 (d, 1H, J 3.7 Hz), 7.28 (d, 1H, J 8.79 Hz), 7.78 (s, 1H), alcoholic protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 14.2 (CH3), [23.0, 26.2, 29.7, 30.0, 32.3, 33.0, 36.8 (CH2, overlapping alkyl resonances)], 50.5 (CH), 51.3 (CH2), 58.4 (CH3), 65.8 (CH2), 67.5 (CH2), 69.0 (CH), 69.7 (CH), 69.8 (CH), 70.3 (CH), 72.3 (CH), 75.1 (CH), 100.0 (CH), 125.0 (CH), 144.8 (quat. C), 174.7 (quat. C); m/z (TOF ES+) 975.8 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 975.7711. C54H104N4NaO9 requires 975.7701.
4.4.20. 1,2,3-triazole 10b (PEG-2 tail)
Azide 8 (20 mg, 0.023 mmol) and alkyne 9b (4.2 mg, 0.023 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded 1,2,3-triazole 10b as a colourless paste (22 mg, 94%): Rf = 0.27 (15% MeOH in CHCl3); IR (KBr) ν 3347m br (O–H), 2917s, 2849s, 1632m (C O), 1544w, 1469m, 1349w, 1347w, 1230w, 1141m, 1063s, 1036s, 785w, 732m, 720s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 6H, J 6.6 Hz), 1.13–1.38 (stack, 68H), 1.46–1.63 (stack, 4H), 2.13 (app. t, 2H, J 7.4 Hz), 3.33–3.39 (stack including [3.35 (s, 3H)]), 4H, 3.42–3.51 (stack, 3H), 3.51–3.55 (stack, 2H), 3.58–3.64 (stack, 6H), 3.69 (dd, 1H, J 9.6, 3.2 Hz), 3.76–3.80 (stack, 2H), 4.09 (dd, 1H, J 8.4, 4.8 Hz), 4.15–4.20 (m, 1H), 4.52–4.58 (m, 2H), 4.62 (s, 2H), 4.85 (d, 1H, J 4.0 Hz), 7.80 (s, 1H), exchangeable protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 14.2 (CH3), [23.0, 26.2, 29.7, 30.0, 32.3, 33.0, 36.8 (CH2, overlapping alkyl resonances)], 50.5 (CH), 51.3 (CH2), 59.1 (CH3), 64.5 (CH2), 67.5 (CH2), 69.0 (CH), 69.7 (CH), 69.8 (CH), 70.0 (CH2), 70.3 (CH), 70.67 (CH2), 70.73 (CH2), 72.2 (CH2), 72.3 (CH), 75.0 (CH), 100.0 (CH), 125.1 (CH), 145.0 (quat. C), 174.7 (quat. C); m/z (TOF ES+) 1064.0 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 1063.8217. C58H112N4NaO11 requires 1063.8225.
4.4.21. 1,2,3-Triazole 10c (PEG-3 tail)
Azide 8 (20 mg, 0.023 mmol) and alkyne 9c (4.6 mg, 0.023 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded 1,2,3-triazole 10c as a colourless paste (23 mg, 91%): Rf = 0.27 (15% MeOH in CHCl3); IR (KBr) ν 3368m br (O–H), 2916s, 2849s, 1630m (C O), 1551w, 1467s, 1349w, 1232w, 1147m, 1071s, 1062w, 1035s, 786w, 720m; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.86 (app. t, 6H, J 6.6 Hz), 1.12–1.39 (stack, 68H), 1.44–1.67 (stack, 4H), 2.13 (app. t, 2H, J 7.5 Hz), 3.33–3.41 (stack including [3.36 (s, 3H)]), 4H, 3.42–3.50 (stack, 3H), 3.50–3.56 (stack, 2H), 3.58–3.64 (stack, 10H), 3.69 (dd, 1H, J 9.6, 3.3 Hz), 3.75–3.81 (stack, 2H), 4.04–4.12 (m, 1H), 4.17 (app. t, 1H, J 6.9 Hz), 4.53–4.59 (m, 2H), 4.62 (s, 2H), 4.85 (d, 1H, J 3.9 Hz), 7.31 (d, 1H, J 9.0 Hz), 7.80 (s, 1H), alcoholic protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 14.2 (CH3), [23.0, 26.2, 29.7, 29.8, 30.1, 32.3, 33.0, 36.8 (CH2, some overlapping alkyl resonances)], 50.5 (CH), 51.3 (CH2), 59.1 (CH3), 64.6 (CH2), 67.5 (CH2), 69.0 (CH), 69.7 (CH), 69.8 (CH), 70.0 (CH2), 70.3 (CH), [70.67, 70.74, 70.8 (CH2, some overlapping ethylene glycol resonances)], 72.2 (CH2), 72.3 (CH), 75.0 (CH), 100.0 (CH), 125.1 (CH), 145.0 (quat. C), 174.7 (quat. C); m/z (TOF ES+) 1107.9 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 1107.8484. C60H116N4NaO12 requires 1107.8487.
4.4.22. 1,2,3-Triazole 10d (PEG-5 tail)
Azide 8 (20 mg, 0.023 mmol) and alkyne 9d (6.7 mg, 0.023 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (20% MeOH in CHCl3) afforded 1,2,3-triazole 10d as a colourless paste (20 mg, 87%): Rf = 0.28 (20% MeOH in CHCl3); IR (KBr) ν 3368m br (O–H), 2917s, 2848s, 1635m (C O), 1550w, 1467s, 1348w, 1231w, 1145m, 1070s, 1035s, 1001w, 785w, 719m, 685m; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 6H, J 6.5 Hz), 1.13–1.39 (stack, 68H), 1.44–1.68 (stack, 4H), 2.13 (app. t, 2H, J 7.4 Hz), 3.33–3.41 (stack including [3.35 (s, 3H)], 4H), 3.42–3.50 (stack, 3H), 3.50–3.55 (stack, 2H), 3.58–3.65 (stack, 18H), 3.69 (dd, 1H, J 9.6, 3.2 Hz), 3.74–3.82 (stack, 2H), 4.04–4.12 (m, 1H), 4.17 (app. t, 1H, J 6.7 Hz), 4.52–4.58 (m, 2H), 4.62 (s, 2H), 4.85 (d, 1H, J 3.6 Hz), 7.80 (s, 1H), exchangeable protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 14.3 (CH3), [23.0, 26.2, 29.7, 29.8, 30.1, 32.3, 32.9, 36.8 (CH2, some overlapping alkyl resonances)], 50.5 (CH), 51.3 (CH2), 59.1 (CH3), 64.6 (CH2), 67.5 (CH2), 69.0 (CH), 69.7 (CH), 69.8 (CH), 70.0 (CH2), 70.3 (CH), [70.66, 70.79 (CH2, some overlapping ethylene glycol resonances)], 72.2 (CH2), 72.3 (CH), 75.0 (CH), 100.0 (CH), 125.1 (CH), 145.0 (quat. C), 174.7 (quat. C); m/z (TOF ES+) 1196.6 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 1195.9045. C64H124N4NaO14 requires 1195.9045.
4.4.23. 1,2,3-Triazole 10e (PEG-8 tail)
Azide 8 (20 mg, 0.023 mmol) and alkyne 9e (9.7 mg, 0.023 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (20% MeOH in CHCl3) afforded 1,2,3-triazole 10e as a colourless paste (29 mg, 97%): Rf = 0.27 (20% MeOH in CHCl3); IR (Kr) ν 3336m br (O–H), 2919s, 2849s, 1630m (C O), 1549w, 1467m, 1350m, 1301w, 1231w, 1138m, 1082s, 1039s, 1012w, 948m, 786m, 719s; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 6H, J 6.6 Hz), 1.12–1.39 (stack, 68H), 1.45–1.69 (stack, 4H), 2.13 (app. t, 2H, J 7.4 Hz), 3.33–3.41 (stack including [3.35 (s, 3H)], 4H), 3.41–3.50 (stack, 3H), 3.50–3.56 (stack, 2H), 3.58–3.65 (stack, 30H), 3.69 (dd, 1H, J 10.2, 3.0 Hz), 3.74–3.81 (stack, 2H), 4.05–4.13 (m, 1H), 4.18 (app. t, 1H, J 6.6 Hz), 4.52–4.58 (m, 2H), 4.62 (s, 2H), 4.85 (d, 1H, J 3.6 Hz), 7.28 (d, 1H, J 8.7 Hz), 7.80 (s, 1H), alcoholic protons not observed; 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 14.2 (CH3), [23.0, 26.2, 29.7, 30.0, 32.3, 33.0, 36.8 (CH2, some overlapping alkyl resonances)], 50.4 (CH), 51.2 (CH2), 59.1 (CH3), 64.5 (CH2), 67.5 (CH2), 69.0 (CH), 69.7 (CH), 69.8 (CH), 70.0 (CH2), 70.3 (CH), 70.8 (CH2, some overlapping ethylene glycol resonances), 72.2 (CH2), 72.3 (CH), 75.0 (CH), 100.0 (CH), 125.1 (CH), 145.0 (quat. C), 174.7 (quat. C); m/z (TOF ES+) 1328.8 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 1327.9788. C70H136N4NaO17 requires 1327.9798.
4.4.24. 1,2,3-Triazole 12 (octyl tail)
Azide 8 (20 mg, 0.023 mmol) and 1-decyne (11) (3.3 mg, 0.023 mmol) were reacted according to Section 4.3.3. After 10 h, work-up and purification of the residue by flash column chromatography (15% MeOH in CHCl3) afforded 1,2,3-triazole 12 as a colourless paste (22 mg, 95%): Rf = 0.30 (15% MeOH in CHCl3); IR (film) ν 3385m br (O–H), 2916s, 2852s, 1635m (C O), 1532w, 1468s, 1438w, 1345w, 1204m, 1151s, 1065s, 1036s, 801m, 732m, 725m; 1H NMR (300 MHz, CDCl3/CD3OD, 2:1) δ 0.84 (app. t, 9H, J 6.6 Hz), 1.12–1.43 (stack, 80H), 1.47–1.71 (stack, 6H), 2.13 (app. t, 2H, J 7.7 Hz), 2.64 (app. t, 2H, J 7.8 Hz), 3.37 (dd, 1H, J 10.5, 4.5 Hz), 3.42–3.55 (stack, 3H), 3.68 (dd, 1H, J 10.2, 3.0 Hz), 3.75–3.84 (stack, 2H), 4.07–4.21 (stack, 2H), 4.51–4.57 (m, 2H), 4.86 (d, 1H, J 3.6 Hz), 7.29 (d, 1H, J 8.7 Hz), 7.52 (s, 1H); 13C NMR (75 MHz, CDCl3/CD3OD, 2:1) δ 14.3 (CH3), [23.0, 25.8, 26.3, 29.7, 29.8, 30.1, 32.3, 33.0, 36.8 (CH2, some overlapping alkyl resonances)], 50.4 (CH), 51.1 (CH2), 67.4 (CH2), 69.1 (CH), 69.8 (CH), 69.9 (CH), 70.3 (CH), 72.3 (CH), 75.1 (CH), 100.0 (CH), 123.17 (CH), 148.6 (quat. C), 174.7 (quat. C); m/z (TOF ES+) 1043.9 ([M+Na]+, 100%); HRMS m/z (TOF ES+) 1043.8697. C60H116N4NaO8 requires 1043.8691.
4.5. Materials and methods for biology
4.5.1. Mice and reagents
C57BL/6 and CD1d−/− (iNKT cell-deficient) mice were used. Animal experiments were carried out under the authority of a UK Home Office Project License. Compounds were solubilised in 150 mM NaCl and 0.5% Tween 20 (vehicle).
4.5.2. In vitro and in vivo activation of iNKT cells
For in vitro activation of iNKT cells, 5 × 105 splenoctyes from C57BL/6 and CD1d−/− (iNKT cell-deficient) mice were pulsed with various concentrations of α-GalCer 7, 14a–c, 13a–e, 12, 10a–e or vehicle for 48 h. Supernatants were removed and the presence of IFNγ determined by ELISA.29 For in vivo activation of iNKT cells, C57 BL/6 WT or CD1d−/− mice were injected intravenously (iv) with 1 μg lipids and blood serum taken at 18 h and the presence of IFNγ determined by ELISA.33
4.5.3. Soluble human NKT TCR binding assay
Soluble iNKT TCR tetramers were prepared according to the protocol described by McCarthy et al.60 C1R-hCD1d cells were pulsed with lipids or vehicle at various concentrations overnight and following washes incubated with fluorescently labelled iNKT TCR tetramer. iNKT TCR–CD1d–lipid complexes were detected by flow cytometry on a FACScalibur device using CellQuest software.
Acknowledgment
G.S.B. acknowledges support in the form of a Personal Research Chair from Mr. James Bardrick, Royal Society Wolfson Research Merit Award, as a former Lister Institute-Jenner Research Fellow; The Wellcome Trust (084923/B/08/Z) for funding (to P.J.J.). V.C. acknowledges support from Cancer Research UK (C399/A2291), the UK Medical Research Council, The Wellcome Trust (084923/Z/08/Z) for funding (J.-P.J. and H.G.), the Cancer Research Institute and the Ludwig Institute for Cancer Research. NMR spectrometers used in this research were funded in part through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands and part-funded by the European Regional Development Fund.
Footnotes
Quoted lengths refer to the linker in its fully extended conformation.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.carres.2012.02.034.
Supplementary data
Spectral data.
References
- 1.Borman S. Chem. Eng. News. 2000;78:48–53. [Google Scholar]
- 2.Kiessling L.L., Pohl N.L. Chem. Biol. 1996;3:71–77. doi: 10.1016/s1074-5521(96)90280-x. [DOI] [PubMed] [Google Scholar]
- 3.Dam T.K., Brewer C.F. Biochemistry. 2008;47:8470–8476. doi: 10.1021/bi801208b. [DOI] [PubMed] [Google Scholar]
- 4.Kiessling L.L., Strong L.E., Gestwicki J.E. Annu. Rep. Med. Chem. 2000;35:21–330. [Google Scholar]
- 5.Jung H., Robison A.D., Cremer P.S. J. Struct. Biol. 2009;168:90–94. doi: 10.1016/j.jsb.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacDonald S.J.F., Watson K.G., Cameron R., Chalmers D.K., Demaine D.A., Fenton R.J., Gower D., Hamblin J.N., Hamilton S., Hart G.J., Inglis G.G.A., Jin B., Jones H.T., McConnell D.B., Mason A.M., Nguyen V., Owens I.J., Parry N., Reece P.A., Shanahan S.E., Smith D., Wu W.-Y., Tucker S.P. Antimicrob. Agents Chemother. 2004;48:4542–4549. doi: 10.1128/AAC.48.12.4542-4549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadden M.K., Blagg B.S.J. Anticancer Agents Med. Chem. 2008;8:807–816. doi: 10.2174/187152008785914743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arav-Boger R., He R., Chiou C.J., Liu J., Woodward L., Rosenthal A., Jones-Brando L., Forman M., Posner G. PLoS One. 2010;5:e10370. doi: 10.1371/journal.pone.0010370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riley A.M., Morris S.A., Nerou E.P., Correa V., Potter B.V.L., Taylor C.W. J. Biol. Chem. 2002;277:40290–40295. doi: 10.1074/jbc.M206925200. [DOI] [PubMed] [Google Scholar]
- 10.Keissling L.L., Gestwicki J.E., Strong L.E. Angew. Chem., Int. Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spencer D.M., Wandless T.J., Schreiber S.L., Crabtree G.R. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 12.Spicer J.A., Finlay G.J., Baguley B.C., Velea L., Graves D.E., Denny W.A. Anti-Cancer Drug Des. 1999;14:37–45. [PubMed] [Google Scholar]
- 13.Gamage S.A., Spicer J.A., Atwell G.J., Findlay G.J., Baguley B.C., Denny W.A. J. Med. Chem. 1999;42:2383–2393. doi: 10.1021/jm980687m. [DOI] [PubMed] [Google Scholar]
- 14.Paik I.-H., Xie S., Shapiro T.A., Labonte T., Narducci Sarjeant A.A., Baege A.C., Posner G.H. J. Med. Chem. 2006;49:2731–2734. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 15.Veerapen N., Brigl M., Garg S., Cerundolo V., Cox L.R., Brenner M.B., Besra G.S. Bioorg. Med. Chem. Lett. 2009;19:4288–4291. doi: 10.1016/j.bmcl.2009.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Diaz Y.R., Wojno J., Cox L.R., Besra G.S. Tetrahedron: Asymmetry. 2009;20:747–753. [Google Scholar]
- 17.Veerapen N., Leadbetter E.A., Brenner M.B., Cox L.R., Besra G.S. Bioconjugate Chem. 2010;21:741–747. doi: 10.1021/bc9005255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jervis P.J., Veerapen N., Bricard G., Cox L.R., Porcelli S.A., Besra G.S. Bioorg. Med. Chem. Lett. 2010;20:3475–3478. doi: 10.1016/j.bmcl.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawano T., Cui J., Koezuka Y., Motoki K., Ueno H., Nakagawa R., Sato H., Kondo E., Koseki H., Taniguchi M. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 20.Hermans I.F., Silk J.D., Gileadi U., Salio M., Mathew B., Ritter G., Schmidt R., Harris A.L., Old L., Cerundolo V. J. Immunol. 2003;171:5140–5147. doi: 10.4049/jimmunol.171.10.5140. [DOI] [PubMed] [Google Scholar]
- 21.Fujii S., Shimizu K., Smith C., Bonifaz L., Steinman R.M. J. Exp. Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carnaud C., Lee D., Donnars O., Park S.-H., Beavis A., Koesuka Y., Bendelac A. J. Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- 23.Cerundolo V., Silk J.D., Masri S.H., Salio M. Nat. Rev. Immunol. 2009;9:28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- 24.Fujii S., Liu K., Smith C., Bonito A.J., Steinman R.M. J. Exp. Med. 2004;199:1607–1618. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silk J.D., Hermans I.F., Gileadi U., Chong T.W., Shepherd D., Salio M., Mathew B., Schmidt R.R., Lunt S.J., Williams K.J., Stratford I.J., Harris A.L., Cerundolo V. J. Clin. Invest. 2004;114:1800–1811. doi: 10.1172/JCI22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barral P., Eckl-Dorna J., Harwood N.E., De Santo C., Salio M., Illarionov P., Besra G.S., Cerundolo V., Batista F.D. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8345–8350. doi: 10.1073/pnas.0802968105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujio M., Wu D., Garcia-Navarro R., Ho D.D., Tsuji M., Wong C.-H. J. Am. Chem. Soc. 2006;128:9022–9023. doi: 10.1021/ja062740z. [DOI] [PubMed] [Google Scholar]
- 28.Schmieg J., Yang G., Franck R.W., Tsuji M. J. Exp. Med. 2003;198:1631–1641. doi: 10.1084/jem.20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leadbetter E.A., Brigl M., Illarionov P., Cohen N., Luteran M.C., Pillai S., Besra G.S., Brenner M.B. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8339–8344. doi: 10.1073/pnas.0801375105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Aseguinolaza G., Van Kaer L., Bergmann C.C., Wilson J.M., Schmieg J., Kronenberg M., Nakayama T., Taniguchi M., Koezuka Y., Tsuji M. J. Exp. Med. 2002;195:617–624. doi: 10.1084/jem.20011889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyamoto K., Miyake S., Yamamura T. Nature. 2001;413:531–534. doi: 10.1038/35097097. [DOI] [PubMed] [Google Scholar]
- 32.Li X., Fujio M., Imamura M., Wu D., Vasan S., Wong C.H., Ho D.D., Tsuji M. Proc. Natl. Acad. Sci. U.S.A. 2010;107:13010–13015. doi: 10.1073/pnas.1006662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park Y.-K., Lee J.-W., Ko Y.-G., Hong S., Park S.-H. Biochem. Biophys. Res. Commun. 2005;327:1143–1154. doi: 10.1016/j.bbrc.2004.12.121. [DOI] [PubMed] [Google Scholar]
- 34.Im J.S., Arora P., Bricard G., Molano A., Venkataswamy M.M., Baine I., Jerud E.S., Goldberg M.F., Baena A., Yu K.O.A., Ndonye R.M., Howell A.R., Yuan W., Cresswell P., Chang Y.-T., Illarionov P.A., Besra G.S., Porcelli S.A. Immunity. 2009;30:888–898. doi: 10.1016/j.immuni.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu K.O., Im J.S., Molano A., Dutronc Y., Illarionov P.A., Forestier C., Fujiwara N., Arias I., Miyake S., Yamamura T., Chang Y.T., Besra G.S., Porcelli S.A. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3383–3388. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarzenbach G. Helv. Chim. Acta. 1952;35:2344–2359. [Google Scholar]
- 37.Chung C.-S. Inorg. Chem. 1979;18:1321–1324. [Google Scholar]
- 38.Breslow R., Belvedere S., Gershell L., Leung D. Pure Appl. Chem. 2000;72:333–342. [Google Scholar]
- 39.Koch M., Stronge V.S., Shepherd D., Gadola S.D., Mathew B., Ritter G., Fersht A.R., Besra G.S., Schmidt R.R., Jones E.Y., Cerundolo V. Nat. Immunol. 2005;6:819–826. doi: 10.1038/ni1225. [DOI] [PubMed] [Google Scholar]
- 40.Borg N.A., Wun K.S., Kjer-Nielsen L., Wilce M.C.J., Pellicci D.G., Koh R., Besra G.S., Bharadwaj M., Godfrey D.I., McCluskey J., Rossjohn J. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- 41.Dieudé M., Striegl H., Tyznik A.J., Wang J., Behar S.M., Piccirillo C.A., Levine J.S., Zajonc D.M., Rauch J. J. Immunol. 2011;186:4771–4781. doi: 10.4049/jimmunol.1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dvir H., Wang J., Ly N., Dascher C.C., Zajonc D.M. J. Immunol. 2010;184:2504–2511. doi: 10.4049/jimmunol.0903509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klemm J.D., Schreiber S.L., Crabtree G.R. Annu. Rev. Immunol. 1998;16:569–592. doi: 10.1146/annurev.immunol.16.1.569. [DOI] [PubMed] [Google Scholar]
- 44.Wun K.S., Cameron G., Patel O., Pang S.S., Pellicci D.G., Sullivan L.C., Keshipeddy S., Young M.H., Uldrich A.P., Thankur M.S., Richardson S.K., Howell A.R., Illarionov P.A., Brooks A.G., Besra G.S., McCluskey J., Gapin L., Porcelli S.A., Godfrey D.I., Rossjohn J. Immunity. 2011;34:327–339. doi: 10.1016/j.immuni.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wun K.S., Borg N.A., Kjer-Nielson L., Beddoe T., Koh R., Richardson S.K., Thakur M., Howell A.R., Scott-Browne J.P., Gapin L., Godfrey D.I., McCluskey J., Rossjohn J. J. Exp. Med. 2008;205:939–949. doi: 10.1084/jem.20072141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajan R., Mathew T., Buffa R., Bornancin F., Cavallari M., Nussbaumer P., De Libero G., Vasella A. Helv. Chim. Acta. 2009;92:918–927. [Google Scholar]
- 47.Trappeniers M., Van Beneden K., Decruy T., Hillaert U., Linclau B., Elewaut D., Van Calenbergh S. J. Am. Chem. Soc. 2008;130:16468–16469. doi: 10.1021/ja8064182. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y., Goff R.D., Zhou D., Mattner J., Sullivan B.A., Khurana A., Cantu C., III, Ravkov E.V., Ibegbu C.C., Altman J.D., Teyton L., Bendalac A., Savage P.B. J. Immunol. Methods. 2006;312:34–39. doi: 10.1016/j.jim.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Xia C., Zhang W., Zhang Y., Woodward R.L., Wang J., Wang P.G. Tetrahedron. 2009;65:6390–6395. [Google Scholar]
- 50.Zhou X.-T., Forestier C., Goff R.D., Li C., Teyton L., Bendalac A., Savage P.B. Org. Lett. 2002;4:1267–1270. doi: 10.1021/ol025565+. [DOI] [PubMed] [Google Scholar]
- 51.Jervis P.J., Cox L.R., Besra G.S. J. Org. Chem. 2011;76:320–323. doi: 10.1021/jo102064p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. Angew. Chem., Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 53.Kolb H.C., Finn M.G., Sharpless K.B. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 54.Moses J.E., Moorhouse A.D. Chem. Soc. Rev. 2007;36:1249–1262. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 55.Natarajan A., Du W., Xiong C.-Y., DeNardo G.L., DeNardo S.J., Gervay-Hague J. Chem. Commun. 2007:695–697. doi: 10.1039/b611636a. Poly(ethylene glycol) are commonly used linkers, see for example: [DOI] [PubMed] [Google Scholar]
- 56.Veronese F.M., Pasut G. Drug Discovery Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 57.Zalipsky S., Harris J.M. ACS Symp. Ser. 1997;680:1–13. doi: 10.1021/bk-1997-0680.ch001. Chapter. [DOI] [Google Scholar]
- 58.Reddy B.G., Silk J.D., Salio M., Balamurugan R., Shepherd D., Ritter G., Cerundolo V., Schmidt R.R. ChemMedChem. 2009;4:171–175. doi: 10.1002/cmdc.200800354. [DOI] [PubMed] [Google Scholar]
- 59.Cebecauer M., Guillaume P., Mark S., Michielin O., Boucheron N., Bezard M., Meyers B.H., Segura J.-M., Vogel H., Leuscher I.F. J. Biol. Chem. 2005;280:23820–23828. doi: 10.1074/jbc.M500654200. [DOI] [PubMed] [Google Scholar]
- 60.McCarthy C., Shepherd D., Fleire S., Stronge V.S., Koch M., Illarionov P.A., Bossi G., Salio M., Denkberg G., Reddington F., Tarlton A., Reddy B.G., Schmidt R.R., Reiter Y., Griffiths G.M., van der Merwe P.A., Besra G.S., Jones E.Y., Batista F.D., Cerundolo V. J. Exp. Med. 2007;204:1131–1144. doi: 10.1084/jem.20062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lingwood D., Simons K. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 62.Silk J.D., Salio M., Reddy B.G., Shepherd D., Gileadi U., Brown J., Masri S.H., Polzella P., Ritter G., Besra G.S., Jones E.Y., Schmidt R.R., Cerundolo V. J. Immunol. 2008;180:6452–6456. doi: 10.4049/jimmunol.180.10.6452. [DOI] [PubMed] [Google Scholar]
- 63.Moretto A.F., Zhang H.C., Maryanoff B.E. J. Am. Chem. Soc. 2001;123:3157–3158. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectral data.