

Abstract
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder characterized by accumulation of sulfatides in glial cells and neurons, the result of an inherited deficiency of arylsulfatase A (ARSA; EC 3.1.6.8) and myelin degeneration in the central and peripheral nervous systems. No effective treatment is currently available for the most frequent late infantile (LI) form of MLD, which results in rapid neurological degradation and early death after the onset of clinical manifestations. To potentially arrest or reverse disease progression, ARSA enzyme must be rapidly delivered to brain oligodendrocytes of patients with LI MLD. We previously showed that brain gene therapy with adeno-associated virus serotype 5 (AAV5) driving the expression of human ARSA cDNA under the control of the murine phosphoglycerate kinase (PGK) promoter alleviated most long-term disease manifestations in MLD mice. Herein, we evaluated the short-term effects of AAVrh.10 driving the expression of human ARSA cDNA under the control of the cytomegalovirus/β-actin hybrid (CAG/cu) promoter in 8-month-old MLD mice that already show marked sulfatide accumulation and brain pathology. Within 2 months, and in contrast to results with the AAV5-PGK-ARSA vector, a single intrastriatal injection of AAVrh.10cuARSA resulted in correction of brain sulfatide storage, accumulation of specific sulfatide species in oligodendrocytes, and associated brain pathology in the injected hemisphere. Better potency of the AAVrh.10cuARSA vector was mediated by higher neuronal and oligodendrocyte transduction, axonal transport of the AAVrh.10 vector and ARSA enzyme, as well as higher CAG/cu promoter driven expression of ARSA enzyme. These results strongly support the use of AAVrh.10cuARSA vector for intracerebral gene therapy in rapidly progressing early-onset forms of MLD.
Piguet and colleagues examine the efficacy of AAVrh.10-mediated overexpression of arylsulfatase A (ARSA) in a mouse model of metachromatic leukodystrophy (MLD). They show that a single intrastriatal injection of AAVrh.10cuARSA results in correction of brain sulfatide storage, accumulation of specific sulfatide species in oligodendrocytes, and associated brain pathology in the injected hemisphere. Better potency of AAVrh.10cuARSA vector was mediated by higher neuronal and oligodendrocyte transduction and axonal transport of AAVrh.10 vector and ARSA enzyme.
Introduction
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder (LSD) caused by an inherited deficiency of arylsulfatase A (ARSA; EC 3.1.6.8), or more rarely of its activator protein saposin B (SAP-B; OMIM 249900) (von Figura et al., 2001). ARSA enzyme catalyzes the first step in the degradation pathway of 3-O-sulfogalactosylceramides (sulfatides). Patients with MLD develop neurological symptoms that result from sulfatide accumulation in oligodendrocytes in the CNS and Schwann cells in the peripheral nerves. Sulfatides accumulate also in brain neurons, contributing significantly to additional pathology (von Figura et al., 2001; Wittke et al., 2004). The disease is usually classified, according to age of onset, as late infantile, juvenile, and adult forms. Patients with the late infantile (LI) form of MLD, the most frequent phenotype of the disease, develop between 12 and 24 months of age a devastating neurological degradation leading to a vegetative state or death within a few months or years. No effective treatment is currently available for patients with the LI form of MLD. Arresting their rapid neurological degradation is a challenge that requires rapid brain delivery of ARSA enzyme into oligodendrocytes, and likely also into neurons, before irreversible damage occurs.
Allogeneic hematopoietic stem cell transplantation has no effect in patients with LI MLD (Peters and Steward, 2003), even when the procedure is performed in presymptomatic neonates (Bredius et al., 2007). Autologous hematopoietic stem cell (HSC)-based gene therapy relying on ARSA overexpression in bone marrow-derived microglia was shown to result in better efficacy than wild-type hematopoietic stem cell transplantation in a mouse model of MLD (Biffi et al., 2004, 2006), and a phase I/II clinical trial of HSC gene therapy using lentiviral vector has been initiated (NCT01560182; http://clinicaltrials.gov). However, this gene therapy strategy will not change the slow rate at which lentivirus-transduced autologous HSC/or myeloid progenitors will penetrate into the brain and differentiate into macrophages/microglia overexpressing the ARSA enzyme. Autologous HSC gene therapy might therefore represent a potentially efficient therapeutic strategy only if it is performed at a presymptomatic stage. Enzyme replacement therapy is another option (Platt and Lachmann, 2009), but the lysosomal enzyme does not cross the blood–brain barrier efficiently, even if results obtained in vitro and in MLD mice suggest that ARSA is able to cross the blood–brain barrier to some extent (Matzner et al., 2009; Matthes et al., 2011). Enzyme replacement therapy using intrathecal delivery of recombinant ARSA enzyme could be an alternative, and a phase I/II clinical trial has just been initiated (NCT01510028; http://clinicaltrials.gov).
Although it will not correct the peripheral neuropathy that is often present in patients with LI MLD, intracerebral delivery of a viral vector encoding ARSA might potentially be more efficient than any other therapeutic strategy to arrest rapidly the neurodegenerative process in the brain of patients with LI MLD. One limitation of brain gene therapy for demyelinating diseases such as MLD is that the majority of gene therapy vectors, including adeno-associated virus (AAVs), transduce mostly neurons in vivo (Cearley and Wolfe, 2006; Sondhi et al., 2007; Taymans et al., 2007). This limitation can be overcome by the capacity of ARSA enzyme to be secreted by transduced neurons and taken up at a distance by untransduced oligodendrocytes. The efficacy of this cross-correction likely requires that a high number of neurons be transduced and that these cells express high levels of ARSA enzyme. Cross-correction has provided the rationale for several therapeutic strategies in LSDs (Parenti, 2009; Platt and Lachmann, 2009), and it has been shown that oligodendrocytes can endocytose ARSA produced by other cells (Sangalli et al., 1998; Martino et al., 2005; Consiglio et al., 2007). The ARSA enzyme, like other lysosomal enzymes, could also be transported along axons, a property that could enhance the bioavailability of the therapeutic enzyme within the brain (Passini et al., 2002; Luca et al., 2005; Chen et al., 2006). Also important, a significant level of transgene expression can be obtained in the brain 1 week after the intracerebral injection of AAV vector (Klein et al., 2008), which is mandatory for efficient therapeutic intervention in the LI form of MLD.
Using an adeno-associated vector of serotype 5 (AAV5) to express the human ARSA cDNA under the control of the mouse phosphoglycerate kinase (PGK) promoter, we previously demonstrated the long-term efficacy of the intracerebral injection of this vector in MLD mice (Sevin et al., 2006, 2007), and more recently the efficacy of this vector in normal nonhuman primates (Colle et al., 2010). A long-term decrease in sulfatide storage was observed in the brain of treated mice at the neuropathological and biochemical levels, but we did not evaluate how rapidly this beneficial effect of brain gene therapy occurred after injection of AAV5 vector and whether sulfatides that specifically accumulate in oligodendrocytes were corrected at the biochemical level. Both issues are crucial in determining the potential therapeutic effect of brain gene therapy in patients with the LI form of MLD.
Among new serotypes of AAV, AAVrh.10, isolated from nonhuman primates, was shown to spread more markedly away from intracerebral injection sites than AAV2, AAV1, AAV5, AAV7, and AAV8 (Cearley and Wolfe, 2006, 2007; Sondhi et al., 2007; Klein et al., 2008). Better potency of AAVrh.10 compared with AAV5 was shown in a mouse model of Batten disease, another brain LSD due to the deficiency of tripeptidyl-peptidase-I (TPPI) enzyme. In comparison with AAV5 vector, intracerebral injection of AAVrh.10 resulted in higher TPPI expression, significant reduction of CNS pathology, and clinical improvement. In those experiments, the expression of TPPI enzyme was under the control of the strong cytomegalovirus/β-actin hybrid (CAG/cu) promoter (Sondhi et al., 2007, 2008).
Therefore, as an AAVrh.10 vector could potentially be more efficient than an AAV5 vector, we evaluated, at the neuropathological and biochemical levels, the short-term efficiency of a single intrastriatal injection of AAVrh.10cuARSA vector in the brain of 8-month-old MLD mice that have already accumulated marked amounts of sulfatides. Herein, we demonstrate that a single intrastriatal injection of AAVrh.10cuARSA vector results in more robust and diffuse expression of ARSA enzyme than the injection of AAV5-PGK-ARSA vector. Importantly, the intracerebral injection of AAVrh.10cuARSA vector allowed us to correct, within 2 months, the sulfatide accumulation in oligodendrocytes and the brain pathology, whereas the injection of AAV5-PGK-ARSA vector was much less efficient.
Materials and Methods
Adeno-associated viral vector construction and production
AAVrh.10cuARSA vector
Human ARSA (hARSA) cDNA was subcloned in a pAAV2-CAG plasmid (Sondhi et al., 2007) to produce pAAV2-CAG-hARSA that included the left-hand viral inverted terminal repeat (ITR) from AAV2; the cytomegalovirus/β-actin hybrid promoter (CAG/cu), consisting of the enhancer from the cytomegalovirus immediate-early gene, the promoter, splice donor, and intron from the chicken β-actin gene, and the splice acceptor from rabbit β-globin; the human ARSA cDNA with an optimized Kozak consensus translation initiation signal before the start codon; the poly(A) site from rabbit β-globin; and the right-hand ITR of AAV2. AAVrh.10cuARSA vector was produced by transient transfection in the 293 human embryonic kidney packaging line and purified using iodixanol gradients (Sondhi et al., 2007). The final titer was 2.5×1013 vector genomes (VG)/ml.
AAVrh.10cuGFP vector
The green fluorescent protein (GFP) cDNA sequence was subcloned in the same pAAV2-CAG plasmid (Sondhi et al., 2007) to produce AAVrh.10cuGFP, with a final titer of 2.3×1013 VG/ml.
AAV5-PGK-ARSA vector
The construction and production of AAV5-PGK-ARSA vector (1.15×1012 VG/ml) has been described elsewhere (Sevin et al., 2006). Expression of the human ARSA cDNA is under the control of the mouse phosphoglycerate kinase (PGK) promoter.
Animal model
ARSA-deficient mice (MLD mice) were bred from homozygous founders (Hess et al., 1996) and were housed in a pathogen-free animal facility. Animal experiments were approved by the Institutional Animal Care and Use Committee of the Institut National de la Santé et de la Recherche Médicale (INSERM, Paris, France), and were carried out in compliance with guidelines and recommendations concerning ethical issues and animal welfare as provided by the 2010/63/EU directive covering the use of animals for scientific purposes.
Injection of AAV vectors into the striatum and ventral tegmental area
Eight-month-old MLD mice were anesthetized by intraperitoneal injection of ketamine and xylazine (0.1 and 0.05 mg, respectively, per gram body weight) and positioned on a stereotactic frame (David Kopf Instruments, Tujunga, CA). Animals were injected unilaterally with 2 μl (for injection into the right striatum) or 1 μl (for injection into the right ventral tegmental area, VTA) of viral vector, using a 30-gauge blunt micropipette attached to a 10-μl Hamilton syringe (Hamilton, Reno, NV) at a rate of 0.2 μl/min. The stereotactic coordinates were as follows: anteroposterior (AP),+0.5 mm; mediolateral (ML), −2.2 mm; dorsoventral (DV), −3.35 mm from the bregma for injection into the striatum; and AP, −3.1 mm; ML, −0.5 mm; DV, −4.5 mm from the bregma for injection into the VTA.
To evaluate the tropism and axonal transport of AAVrh.10 vector, 2.3×109 VG of AAVrh.10cuGFP or AAVrh.10cuARSA vector was injected into the right VTA (n=5 mice for each vector) or into the right striatum (n=3 mice for each vector). To compare the efficiency of AAV5-PGK-ARSA and AAVrh.10cuARSA vectors in MLD mice, the right striatum of animals was injected with 2.3×109 VG of each vector (n=11 or 12 mice per vector).
In addition, the right striatum of 16-month-old MLD mice (n=4) was injected with AAVrh.10cuARSA vector (2.3×109 VG) to evaluate the sulfatide storage correction at a more advanced stage of their disease.
Tissue processing
Mice were killed 2 months after injection (10 or 18 months of age). Deeply anesthetized animals were transcardially infused with phosphate-buffered saline (PBS). For mice injected via the right striatum with AAV5-PGK-ARSA or AAVrh.10cuARSA vector, the brain and spinal cord were removed and processed as follows: (1) for protein and molecular (DNA and RNA) analysis (n=5 mice per group), brain samples were placed on a mouse coronal brain matrix (World Precision Instruments, Stevenage, UK) and eight 1-mm-thick left (uninjected hemisphere) and right (injected hemisphere) coronal brain sections were cut and stored at −80°C. For DNA and RNA extraction from the same samples, tissue samples were crushed in liquid nitrogen and divided into two equal parts, one for each analysis; (2) for lipid analysis (n=3 mice per group), each cerebral hemisphere (injected and noninjected) was divided into two parts: a rostral sample (containing the injection site in the right hemisphere) and a caudal sample, which were stored at −20°C; and (3) for histological studies (n=3 mice per group), brain tissues were postfixed in 4% paraformaldehyde (PFA), cryoprotected in 30% sucrose, embedded in Tissue-Tek OCT compound (Sakura Finetek USA, Torrance, CA) at −55°C, and cut into 10-μm sagittal sections in a cryostat (Leica, Langham, TX). Brain samples of untreated (UT) and wild-type (WT) mice were processed identically. Mice injected with AAVrh.10cuGFP vector were processed only for histological studies.
Quantitative PCR and real-time RT-PCR
AAV5-PGK-ARSA and AAVrh.10cuARSA vector genome copy numbers were measured by quantitative PCR in the brain, cerebellum, brainstem, and spinal cord, using an ABI PRISM 7900 sequence detection system (Life Technologies, Carlsbad, CA) and a SYBR green PCR core reagents kit (Life Technologies) as described (Sevin et al., 2006). Briefly, vector sequences and mouse genomic albumin (internal control) sequences were simultaneously amplified, and each sample was expressed in terms of its albumin content. The results (vector genome copy number per cell, VGC) were expressed as n-fold differences in the transgene sequence copy number relative to the albumin gene copy number (number of viral genome copies for 2N genome). Samples were considered eligible for the study if the albumin sequence Ct value was <26, and were scored vector-negative if the transgene sequence Ct value was >35. The thermal cycling conditions comprised an initial denaturing step at 95°C for 10 min, followed by 50 cycles at 95°C for 15 sec and at 65°C for 1 min. Each sample was analyzed in duplicate. The nucleotide sequences of primers are available on request.
Real-time RT-PCR was performed as described. Quantification of murine TATA box-binding protein (TBP) mRNA was performed as an internal control and each sample was normalized on the basis of its TBP mRNA content.
Results, expressed as N-fold differences in ARSA gene expression relative to the TBP gene, termed NARSA, were determined by the formula: 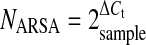 , where the ΔCt value of the sample is determined by subtracting the Ct value of the target gene from the Ct value of the TBP gene.
, where the ΔCt value of the sample is determined by subtracting the Ct value of the target gene from the Ct value of the TBP gene.
NARSA values of the samples were subsequently normalized to a “basal mRNA level,” that is, normalized to the smallest amount of ARSA mRNA quantifiable (target gene Ct value=35, and then scored as the smallest NARSA value, i.e., Ntarget value=1). Samples with low levels of ARSA mRNA, not quantifiable by means of the real-time quantitative RT-PCR assay (Ct<35), were considered “nonexpressing” (NARSA value=0).
The nucleotide sequences of primers for murine TBP and human ARSA are available on request. To avoid amplification of contaminating genomic DNA, one of the two primers was placed at the junction between two exons. The thermal cycling conditions comprised an initial denaturation step at 95°C for 10 min and 50 cycles at 95°C for 15 sec and 65°C for 1 min.
ARSA mRNA expression levels were normalized to the vector genome copy number for each vector in order to compare ARSA expression under the CAG/cu and PGK promoters.
ARSA expression and activity
Samples were homogenized in 0.3 ml (brain) or 0.5 ml (spinal cord, brainstem and cerebellum) of lysis buffer (100 mM Trizma base, 150 mM NaCl, 0.3% Triton; pH 7) and incubated for 30 min on ice. Samples were sonicated on ice (1 min 30 sec in pulse mode 5 sec/5 sec; amplitude, 25 Hz) and centrifuged. The supernatant was collected for the determination of (1) protein content (bicinchoninic acid [BCA] protein assay kit; Pierce Biotechnology/Thermo Fisher Scientific, Rockville, IL); (2) the concentration of recombinant ARSA, using an indirect sandwich ELISA as described (Matzner et al., 2000). Assays were performed in triplicate and results are expressed as nanograms of hARSA per milligram of protein (mean±SEM of five mice for each group); and (3) ARSA activity, using the artificial p-nitrocatechol sulfate (pNCS) substrate assay (Sigma-Aldrich, Lyon, France) (Bass et al., 1970; Sevin et al., 2006). Assays were performed in triplicate and results are expressed as nanomoles of 4-nitrocatechol (4NC) per hour per milligram of protein (mean±SEM of five mice for each group).
Galactose-6-sulfatase activity
Assessment of galactose-6-sulfatase activity was performed as described (Tylki-Szymanska et al., 1998).
Analysis of brain lipids
Total lipids were extracted in chloroform–methanol–water (14:28:5, v/v/v) and subjected to alkaline saponification as described (Sevin et al., 2006). Sulfatide (Sulf) content and galactosylceramide (GalCer) content were identified by thin-layer chromatography (TLC) (Fujita et al., 1996; Sevin et al., 2006). Detection was performed with orcinol–sulfuric acid and quantification of sulfatides and GalCer was performed with a CAMAG (Muttenz, Switzerland) chromatogram immersion device III and densitometry at 580 nm with a CAMAG TLC scanner model II. Three deposits of sulfatide (0.8, 1.6, and 2.4 μg; each in duplicate) or GalCer (1.2, 1.8, and 2 μg; each in duplicate) standards were run with brain samples on the same TLC plate, to realize a standard curve. Assays were performed in triplicate and results are expressed as micrograms of sulfatides or GalCer per milligram of wet tissue (mean±SEM of three mice for each group). The Sulf/GalCer ratio was calculated and expressed as the mean±SEM.
Determination of sulfatide isoforms, using tandem mass spectrometry
Electrospray (ESI)-tandem mass spectrometry (MS/MS) was carried out on an API 3200 MS/MS triple quadrupole mass spectrometer (AB SCIEX, Toronto, ON, Canada) equipped with a TurboIonSpray source heated at 300°C. Two binary pumps, a vacuum degasser, and a high-performance autosampler controlled by an Instant Pilot, used for solvent delivery and automated sample introduction, were from the Agilent 1200 series (Agilent Technologies, Massy, France). Nitrogen, produced by an NM30L nitrogen generator (Peak Scientific, Inchinnan, Renfrewshire, UK), was used for desolvation and collision gas. All results were acquired with Analyst version 1.5 software (AB SCIEX).
MS/MS was performed in negative ionization mode. Parameters used were as follows (in arbitrary units): curtain (CUR) gas, 20; collision gas, 6; ion source gas 1 (GS1), 20; ion source gas 2 (GS2), 55; Q1 and Q3 resolution, unit; ion spray voltage (IS), −4500 V; entrance potential (EP), −9.5 V. Declustering potential (DP) and collision energy (CE) were optimized at −140 V and −94 eV, respectively.
Determination of sulfatide isoforms was performed by scanning m/z 97 precursor (30 cumulated scans of 1 sec from m/z 750 to 950) by infusing 1:9 dilutions in chloroform–methanol (2:1) of brain sample lipid extracts at 10 μl/min. This allowed the identification of 23 sulfatide species ranging from C16 to C26, with the following m/z mass of the [M-H]– ion in multiple reaction-monitoring (MRM) mode: C16:0 (m/z=778.6), C16:0-OH (m/z=794.6), C18:0 (m/z=806.6), C18:0-D3 (m/z=809.6), C18:1-OH (m/z=820.6), C18:0-OH (m/z=822.6), C20:0 (m/z=834.6), C20:1-OH (m/z=848.6), C20:0-OH (m/z=850.6), C22:1 (m/z=860.6), C22:0 (m/z=862.6), C23:0-OH or C22:1-OH (m/z=876.6), C22:0-OH (m/z=878.6), C24:1 (m/z=888.6), C24:0 (m/z=890.6), C23:0 (m/z=OH 892.6), C25:1 (m/z=902.6), C25:0-OH or C24:1-OH (m/z=904.6), C24:0-OH (m/z=906.6), C26:1 (m/z=916.6), C26:0 (m/z=918.6), C25:0-OH (m/z=920.6), C26:1-OH (m/z=932.6) and C26:0-OH (m/z=934.6).
For quantification, 600 pmol of C18:0-D3 (m/z=809.6) sulfatide internal standard (N-octadecanoyl-D3-sulfatide; Matreya, Pleasant Gap, PA) diluted in chloroform–methanol (2:1) was mixed 1:9 with diluted brain lipid extract. Fifty microliters of this mixture was injected into the mobile phase (chloroform–methanol [2:1]) at 100 μl/min for MS/MS analysis.
Quantification was performed in MRM mode. Each isoform concentration was calculated according to the internal standard concentration. Total sulfatides was the sum of all isoform results.
Histopathology
Ten-micrometer frozen coronal sections were stained with hematoxylin–eosin for standard histological analysis. To evaluate sulfatide storage, frozen sections were postfixed in 4% PFA, stained with Alcian blue (A5268; Sigma-Aldrich) (0.05% in 0.025 M sodium acetate buffer, pH 5.7, containing 0.3 M MgCl2 and 1% PFA), rinsed in the same buffer without dye, and mounted.
ARSA immunohistochemistry was performed as described previously (Sevin et al., 2006), using a rabbit polyclonal anti-hARSA antibody (from V. Gieselmann, Bonn, Germany). GFP fluorescence was directly observed after fixation of the slices with 4% PFA or using a goat anti-GFP antibody (BA-0702, diluted 1:200; Abcys, Paris, France). Colocalization of ARSA and GFP with specific cellular markers was performed with the following primary antibodies: mouse anti-NeuN (neuronal nuclei) (MAB377, diluted 1:200; Chemicon International, Temecula, CA) for neurons; and mouse anti-CNPase (2′,3′-cyclic nucleotide-3′-phosphodiesterase) (clone 11-5B, diluted 1:100; Sigma-Aldrich) for oligodendrocytes.
Stereological cell counts
Stereological counts were performed by two independent investigators, using Histolab image analyzer software (Microvision Instruments, Paris, France).
For mice injected via the right striatum with AAVrh.10cuGFP or AAVrh.10cuARSA vector, GFP-positive or ARSA-positive cells were assessed in the VTA, a distant area connected to the injection site (n=3 mice per vector, n=6 sections per mouse). For each section, 300 cells (stained with 4′,6-diamidino-2-phenylindole [DAPI]) were counted in three random areas of 0.2 mm2 at 20-fold magnification. The percentage of GFP-positive or ARSA-positive cells was assessed among these cells and expressed as the mean±SEM.
For mice injected via the right striatum with AAVrh.10cuGFP or AAVrh.10cuARSA, the percentages of CNPase-positive oligodendrocytes expressing ARSA or GFP were evaluated in three sections of the white matter in the right internal capsule. For each section, 300 cells expressing CNPase were counted in 3 random areas of 0.2 mm2 at 20-fold magnification. The percentage of GFP-positive and ARSA-positive cells was assessed among these cells.
Statistical analysis
Statistical analysis was carried out with GraphPad Prism software (GraphPad Software, La Jolla, CA). Statistical analyses were performed by two-sample t test or analysis of variance (ANOVA) followed by post-hoc Tukey HSD (honestly significant difference) test.
Results
Vector genome copies in CNS of MLD mice injected with AAV.rh10 and AAV5 vectors
A single injection of AAVrh.10cuARSA or AAV5-PGK-ARSA vector (total dose, 2.3×109 VG) was performed in the right striatum of 8-month-old MLD mice (n=11 or 12 mice per vector). Mice were killed 2 months postinjection. For the same number of injected viral genomes, the mean number of vector genome copies per cell (VGC) in the whole injected hemisphere was 3.6-fold higher in mice after injection of AAVrh.10cuARSA vector in comparison with AAV5-PGK-ARSA vector (67.5±14.4 vs. 18.4±4.8 VGC; p=0.002) (Fig. 1A). As expected, maximal VGC values were observed at the injection site (S3–S4) for both vectors but maximal VGC values were higher with the AAVrh.10 vector (Fig. 1B). Within 2.0 mm of the intrastriatal injection site (S2 or S5; see inset in Fig. 1B), the mean VGC was also significantly higher after the injection of AAVrh.10 vector (p=0.01 for S2 and p=0.03 for S5) (Fig. 1B). In more distant areas from the intrastriatal injection site, the mean VGC value was still higher after injection of AAVrh.10 vector, but the difference was not statistically significant. Viral genomes of AAVrh.10 and AAV5 vectors were also detected, but at lower values (0.01 to 0.04 VGC), in the cerebellum, brainstem, and spinal cord as well as in the noninjected hemisphere, without significant difference between the two vectors (data not shown).
FIG. 1.

AAVrh.10 (solid columns) and AAV5 (shaded columns) vector genome copy number per 2n genome (VGC) in whole brain (A) and in serial coronal brain sections of the injected hemisphere (B) in 10-month-old MLD mice, 2 months after injection of AAVrh.10cuARSA or AAV5-PGK-ARSA vector into the right striatum. S1 to S8 refer to 1-mm coronal brain sections. The gray asterisk (B, inset) indicates the injection site in the striatum. All results are expressed as mean±SEM of five mice. *p<0.05; **p<0.01.
In comparison with AAV5-PGK-ARSA vector, intracerebral injection of AAVrh.10cuARSA vector results in more robust expression of ARSA enzyme in the brain of MLD mice
For the same number of injected vector genomes in the right striatum of MLD mice, expression of human ARSA assessed by ELISA in the whole ipsilateral injected hemisphere was 3.9-fold higher after injection of AAVrh.10cuARSA vector, in comparison with AAV5-PGK-ARSA vector (335.5±86.8 vs. 85.6±16.3 ng/mg protein; p<0.0001; Fig. 2A). ARSA expression was significantly higher up to 2 mm from the injection site in the rostral part of the injected hemisphere (S2, 24-fold increase; p<0.01) and up to 5 mm from the injection site in the caudal part of the injected hemisphere (S8, 6-fold increase; p<0.05) in AAVrh.10-treated mice (Fig. 2B). Significantly higher amounts of ARSA protein were also detected in the cerebellum (11.5±3.11 vs. 1.6±1.1 ng/mg protein; p=0.04), brainstem (11.1±5.8 vs. 0.96±0.31; p=0.01), spinal cord (8.4±4.3 vs. 2±0.5; p=0.005), and noninjected hemisphere (28.8±5.6 vs. 13.9±1.98 ng/mg protein; p=0.009) after intrastriatal injection of AAVrh.10cuARSA vector (Fig. 2B and C).
FIG. 2.

(A–C) Arylsulfatase A (ARSA) expression (ng/mg protein) assessed by ELISA in the injected hemisphere (A), serial 1 mm-coronal brain sections of the injected hemisphere (B), and in the noninjected hemisphere (C) of 10-month-old MLD mice, 2 months after injection of AAVrh.10cuARSA or AAV5-PGK-ARSA vector into the right striatum. (D) ARSA activity in the whole injected hemisphere of the same AAVrh.10-or AAV5-treated mice, assessed by the degradation of an artificial substrate of the enzyme (pNCS, p-nitrocatechol sulfate). (E) ARSA mRNA levels driven by the CAG/cu (CAG) or PGK promoter. Results are expressed as means±SEM of three or four samples (arbitrary unit). 4NC, 4-nitrocatechol; Bs, brainstem, Cb, cerebellum; CeSC, cervical spinal cord; S2, S4, S6, and S8 refer to 1-mm coronal brain sections; UT, untreated; WT, wild type. Results are expressed as means±SEM of four or five mice. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Immunohistochemistry with anti-hARSA antibody showed that recombinant ARSA enzyme was more readily detected in the injected hemisphere of AAVrh.10-treated mice than in the injected hemisphere of AAV5-treated mice (Fig. 3A–D). For both vectors, ARSA immunostaining was detected in the striatum (Fig. 3E), cortex (Fig. 3F), and hippocampus (Fig. 3G). However, AAVrh.10-treated, but not AAV5-treated, mice displayed ARSA-positive cells in numerous brain areas that are or are not synaptically connected to the injection site in the striatum, particularly the posterior part of the forebrain, cerebellum, and brainstem (Fig. 3A, B, and H–J).
FIG. 3.

(A and B) Cartography of ARSA expression in the brain of 10-month-old MLD mice, 2 months after a single injection of AAVrh.10cuARSA (A) or AAV5-PGK-ARSA (B) vector into the right striatum. (C and D) Representative immunostaining of human ARSA enzyme in a sagittal brain section of AAVrh.10-treated (C) and AAV5-treated mice (D). (E–J) After a single injection of AAVrh.10cuARSA vector into the right striatum, ARSA-positive cells were observed in several brain regions including the striatum (E), cortex (F), hippocampus (G), thalamus (H), ventral tegmental area (I), and interposed nucleus (J). cc, corpus callosum; ctx, cortex; hpf, hippocampus formation; hy, hypothalamus; ip, interposed nucleus; mgp, medial globus pallidus; pcg, pontine central nucleus; pg, pontine nucleus; sc, superior colliculus; sn, substantia nigra; STR, striatum; th, thalamus; vta, ventral tegmental area. Scale bars: 50 μm.
Intracerebral injection of AAVrh.10cuARSA vector also results in higher ARSA activity in brain of MLD mice
ARSA activity was measured in the brain of 10-month-old MLD mice, 2 months after a single intrastriatal injection of AAV5-PGK-ARSA or AAVrh.10cuARSA vector, using the degradation of pNCS, an artificial substrate for ARSA but also for other arylsulfatases. With this enzymatic assay, the measurement of pNCS degradation was not significantly different in the brains of wild-type and untreated MLD mice, the pNCS substrate being hydrolyzed by other sulfatases (Matzner et al., 2000; Sevin et al., 2006) (Fig. 2D). Whereas a single intrastriatal injection of AAV5-PGK-ARSA vector did not significantly increase ARSA activity in the injected hemisphere of MLD mice, AAVrh.10-treated mice displayed a more than 60% increase in ARSA activity, in comparison with untreated MLD mice (686.4±61.5 vs. 425.7±20.1 nmol of 4NC/hr/mg protein; p=0.002) (Fig. 2D).
A difference in the strength of the promoters (CAG/cu for AAVrh.10 vs. PGK for the AAV5 vector) might be a factor that contributed to higher expression of ARSA in AAVrh.10-treated mice. We therefore quantified ARSA mRNA levels in different brain sections of AAVrh.10- and AAV5-treated mice by quantitative RT-PCR. After adjusting mRNA levels for the total number of vector genome copies per cell (VGC) measured in the same sample, the CAG/cu promoter was shown to drive transcription of 40-fold higher mRNA levels for human ARSA than did the PGK promoter (Fig. 2E). This increase translated into a 4-fold increase in ARSA protein expression based on ELISA (Fig. 2A).
ARSA is expressed in oligodendrocytes after intracerebral injection of AAVrh.10cuARSA vector in MLD mice
In mice injected with AAV5-PGK-ARSA vector, most ARSA-positive cells were neurons and we failed to detect any oligodendrocyte expressing the recombinant enzyme, as already described (Sevin et al., 2006, 2007). After injection of AAVrh.10cuARSA vector, most ARSA-expressing cells (>90%) were neurons, as also observed after the intracerebral injection of AAVrh.10cuGFP vector (data not shown). However, in myelinated areas such as the corpus callosum or internal capsules, the number of cells having the morphology of oligodendrocytes and expressing ARSA was 2- to 3-fold higher after the injection of AAVrh.10cuARSA vector compared with the number of cells having the morphology of oligodendrocytes and expressing GFP after the injection of AAVrh.10cuGFP vector (Fig. 4A and B). In the right internal capsule, double immunostaining with anti-CNPase and anti-ARSA or anti-GFP antibodies showed that 21.4±1.1% of oligodendrocytes expressed the ARSA enzyme after injection of AAVrh.10-ARSA vector into the striatum (Fig. 4A), whereas 9.1±0.7% expressed GFP after injection of AAVrh.10cuGFP vector into the same brain structure (p<0.0001) (Fig. 4B). This indicates that the AAVrh.10 vector, unlike the AAV5 vector, is able to transduce oligodendrocytes, but also that a significant amount of ARSA was recaptured by oligodendrocytes that were not transduced by the AAVrh.10 vector, suggesting in vivo cross-correction of these cells.
FIG. 4.
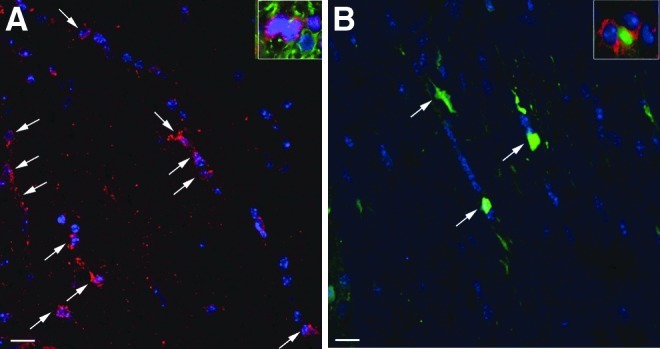
Immunostaining with anti-ARSA (A) and anti-GFP (B) antibodies in the right internal capsule after a single injection of AAVrh.10cuARSA or AAVrh.10cuGFP vector into the right striatum. Cells having the morphology of oligodendrocytes and expressing ARSA (arrows in A) were 2-fold more abundant than cells having the morphology of oligodendrocytes and expressing GFP (arrows in B), suggesting a cross-correction of oligodendrocytes after injection of the AAVrh.10cuARSA vector. Nuclei are stained blue. Scale bars: 25 μm. Insets: Double immunostaining with CNPase (a marker for oligodendrocytes) and (A) ARSA or (B) GFP.
Mechanisms of ARSA diffusion after intracerebral injection of AAVrh.10 vector in MLD mice
We used GFP as a reporter gene to evaluate the axonal transport of AAVrh.10 vector after unilateral injection into the striatum, which receives and sends multiple connections from adjacent or distant parts of the brain. After a single injection of AAVrh.10cuGFP vector into the right striatum, GFP-positive cells were detected in the cellular bodies of neurons in distant areas of the ipsilateral hemisphere that send efferences to the striatum (i.e., thalamus, cortex, VTA, superior colliculus) (Fig. 5A), suggesting retrograde transport of the AAVrh.10 vector.
FIG. 5.

Schematic representation of GFP and ARSA expression in the ipsilateral hemisphere after a single injection of AAVrh.10cuGFP vector (A and B) or AAVrh.10cuARSA vector (C and D) into the right striatum (A and C) or the right VTA (B and D) of MLD mice. Solid lines represent reciprocal projections, dashed lines anterograde (efferent) projections, and dotted lines retrograde (afferent) projections. Insets for (A–D) show representative GFP or ARSA expression in selected brain areas. cc, corpus callosum; ctx, cortex; hpf, hippocampal formation; hy, hypothalamus; int, internal capsules; ip, interpositus nucleus; my, medulla; ml, medial lemniscus; soc, superior olivary complex; ob, olfactory bulbs; ot, olfactory tubercle; pag, periaqueductal gray; pcg, pontine central nucleus; pg, pontine nucleus; purkinje, Purkinje cells; sc, superior colliculus; STR/str, striatum; st term, stria terminalis; th, thalamus; vta, ventral tegmental area. Scale bars: 50 μm.
GFP staining was also observed in cellular bodies of neurons of the right medial (internal) globus pallidus, which receives inputs from the ipsilateral striatum, possibly reflecting anterograde transport of AAVrh.10 vector (Fig. 5A). However, the medial globus pallidus is relatively close to the injection site in the striatum, and therefore the expression of GFP in this structure could also reflect diffusion of the AAVrh.10 vector into the extracellular space and then transduction of neurons in the medial globus pallidus. GFP staining was not observed in the right substantia nigra, another structure anterogradely connected to the right striatum.
Similar observations concerning the axonal transport of AAVrh.10cuGFP vector were made after a single injection of this vector into the right VTA. Expression of GFP was observed in neuronal bodies from ipsilateral brain regions that are synaptically connected to the right VTA via reciprocal (thalamus, stria terminalis, periaqueductal gray), anterograde (striatum, interpositus nucleus in the cerebellum), or retrograde (superior colliculus) connections (Fig. 5B).
We then compared the distribution of ARSA and GFP expression after a single injection of the same number of AAVrh.10cuARSA and AAVrh.10cuGFP vector genomes (2.3×109 VG) into the right striatum or the right VTA of 8-month-old MLD mice to evaluate the potential of ARSA to undergo axonal transport.
After injection of AAVrh.10cuARSA or AAVrh.10cuGFP vector into the right striatum (Fig. 5A and C), ARSA-expressing cells were more abundant than GFP-expressing cells in distant areas that are synaptically connected to the striatum such as the cortex, thalamus, ventral tegmental area, and hippocampal formation. For example, in the ventral tegmental area that sends efferences to the ipsilateral striatum, the percentage of cells expressing ARSA was 2.3-fold higher than the percentage of cells expressing GFP (23.2±1.4 vs. 10.3±1.5; p<0.0001). This suggests that ARSA may have been secreted from transduced neurons in the striatum, endocytosed by connected axons, and transported along the axons to cellular bodies of neurons in the VTA.
However, ARSA-expressing cells, but not GFP-expressing cells, were also detected in distant areas that are not synaptically connected with the striatum, that is, the interpositus nucleus and pontine central gray nucleus (Fig. 5A and C). The same observations were made after the injection of AAVrh.10cuARSA or AAVrh.10cuGFP vector into the right VTA (Fig. 5B and D).
Thus, the expression of ARSA observed after a single intrastriatal injection of AAVrh.10cuARSA vector was mediated through two other mechanisms: (1) the secretion of ARSA by transduced neurons, mostly in the vicinity of the injection site, followed by the recapture of ARSA enzyme by distant nontransduced neurons and oligodendrocytes; and (2) the axonal transport of AAVrh.10 vector and ARSA enzyme.
In comparison with AAV5-PGK-ARSA vector, intracerebral injection of AAVrh.10cuARSA vector results in faster correction of sulfatide accumulation in oligodendrocytes of MLD mice
Sulfatide storage is already detectable at 3 months of age in MLD mice and increases progressively with age (Wittke et al., 2004; Sevin et al., 2007). Selective histochemical staining of sulfatides with Alcian blue showed that 10-month-old MLD mice display massive sulfatide storage in macrophages, neurons, and oligodendrocytes. Two months after a single injection of AAV5-PGK-ARSA vector into the right striatum, sulfatide storage assessed by Alcian blue staining was decreased but still clearly present in the ipsilateral hemisphere of MLD mice injected at 8 months of age, whereas it was almost undetectable in MLD mice injected with the AAVrh.10cuARSA vector at the same age (Fig. 6A).
FIG. 6.

(A) Correction of sulfatide storage as assessed by Alcian blue staining in the corpus callosum of wild-type (WT), untreated (UT), and AAVrh.10- and AAV5-treated MLD mice at 10 months of age, 2 months after treatment. In UT MLD mice, storage of sulfatides is particularly evident in macrophages and oligodendrocytes. Sulfatide accumulation is decreased but still present in AAV5-treated mice, whereas it is no longer detectable in AAVrh.10-treated mice. Insets: Higher magnification showing sulfatide storage in oligodendrocytes of UT mice that is corrected in AAVrh.10-treated mice, but not in AAV5-treated mice. (B) Immunostaining of astrocytes (glial fibrillary acidic protein [GFAP], red) in the anterior part of the corpus callosum, showing marked astrogliosis in UT mice that is prevented in AAVrh.10-treated mice, but to a much lesser extent in AAV5-treated mice at 10 months of age (2 months after treatment). (C) Immunostaining of microglia (Iba1, red) in the frontal cortex. Ten-month-old UT MLD mice display increased numbers of amoeboid microglial cells (yellow arrows), which correspond to activated microglia. At 10 months of age (2 months after treatment) these cells are not present in AAVrh.10-treated mice, which display only ramified microglia (blue arrows) corresponding to resting nonactivated microglia, as in WT mice, whereas they are still present in AAV5-treated mice. Scale bars: 100 μm.
The Sulf/GalCer ratio is a sensitive indicator of ARSA activity on sulfatide load. Fluctuations in the Sulf/GalCer ratio are minor during normal aging in mice, whereas these fluctuations are important in MLD mice, as the ratio increases by 2.5-fold at 10 months of age (Sevin et al., 2007) (Fig. 7A). A single injection of AAV5-PGK-ARSA vector into the right striatum of 8-month-old MLD mice did not lead to a decrease in the Sulf/GalCer ratio in the ipsilateral hemisphere 2 months later (Fig. 7A). In contrast, injection of AAVrh.10cuARSA vector led to a decrease in the Sulf/GalCer ratio by an average of 36% (0.49±0.01 vs. 0.77±0.04 in untreated MLD mice; p=0.012) in the rostral part of the brain and by an average of 24% (0.56±0.04 vs. 0.72±0.02; p=0.01) in the caudal part of the brain (Fig. 7A).
FIG. 7.

Ratio of sulfatide content to galactosylceramide content (Sulf/GalCer ratio) in the brain of 10-month-old (A) and 18-month old (B) MLD mice, 2 months after a single intrastriatal injection of AAVrh.10cuARSA or AAV5-PGK-ARSA vector. (C) Sulfatide species identified by their fatty acid moiety in the brains of normal (WT), untreated (UT), and AAV5- or AAVrh.10-treated MLD mice at 10 months of age (2 months after treatment). Results are expressed as means±SEM (three mice per group). *p<0.05, **p<0.01, ***p<0.001.
AAVrh.10cuARSA vector was also injected into the right striatum of 16-month-old MLD mice, when the Sulf/GalCer ratio is even higher, and killed 2 months after injection. The Sulf/GalCer ratio decreased on average by 33% in treated MLD mice (0.57±0.01 vs. 0.85±0.03 in untreated animals; p=0.0002) (Fig. 7B).
Sulfatides comprise several molecular species that are highly cell-specific: long-chain fatty acid species (C18–C20) predominate in neurons and astrocytes whereas very long-chain fatty acid species (C24–C26) are more abundant in oligodendrocytes (Isaac et al., 2006). Two months after AAV5-PGK-ARSA vector injection into the right striatum of 8-month-old MLD mice, levels of C18:0 and C24:1 sulfatide species were reduced by only 16% (p=0.02) and 6% (p=not significant) in the injected hemisphere and in comparison with untreated mice. In contrast, 2 months after a single injection of AAVrh.10cuARSA vector into the right striatum of 8-month-old MLD mice, levels of these sulfatide species decreased by 63% (p=0.005) and 31% (p=0.01) (Fig. 7C). Thus, the AAVrh.10cuARSA vector was more efficient than the AAV5-PGK-ARSA vector, rapidly reducing the C18 sulfatide species that accumulate mostly in neurons and astrocytes, and also nearly completely correcting the accumulation of C24 sulfatide species that are present mostly in oligodendrocytes.
Similar results were obtained in MLD mice treated at 16 months and killed 2 months after injection. Within 2 months, the injection of AAVrh.10cuARSA vector was also able to reduce the levels of C18:0 sulfatide species by 62% (p=0.003) and the levels of C24:1 sulfatide species by 40% (p=0.018). As for MLD mice treated at 8 months of age, the accumulation of C24:1 sulfatide species was almost completely corrected within 2 months (data not shown).
In comparison with AAV5-PGK-ARSA vector, brain pathology of MLD mice is more rapidly corrected after intracerebral injection of AAVrh.10cuARSA vector
Whereas 10-month-old MLD mice do not show evidence of cerebral demyelination, they display extensive astrogliosis in the white matter of the corpus callosum and cerebellum (Hess et al., 1996). Astrogliosis was markedly reduced in the ipsilateral hemisphere, 2 months after a single injection of AAVrh.10cuARSA vector into the right striatum of 8-month-old MLD mice, but not after injection of AAV5-PGK-ARSA vector (Fig. 6B).
Ten-month-old MLD mice also display an increased number of Iba1-positive cells in the white or gray matter, which show mostly a morphological aspect of activated microglia (round or amoeboid shaped cells) (Fig. 6C). A marked reduction of activated microglia was observed in the ipsilateral hemisphere, 2 months after a single injection of AAVrh.10cuARSA vector into the right striatum of 8-month-old MLD mice. Most microglial cells had a ramified morphology of resting microglia, similar to wild-type mice. In contrast, a single injection of AAV5-PGK-ARSA vector resulted in only a mild decrease in the number of activated microglial cells (Fig. 6C).
Overexpression of ARSA does not impair activity of galactose-6-sulfatase
The activity of all sulfatase enzymes (A to E) requires an essential posttranslational modification, that is, the formylglycination of a cysteine residue at the catalytic site of sulfatase enzymes, which is catalyzed by the formylglycine-generating enzyme (FGE). To assess whether ARSA overexpression could potentially affect the posttranslational modification of other sulfatases, we measured the brain activity of galactose-6-sulfatase, another sulfatase, in untreated MLD mice and in MLD mice injected with the AAVrh.10cuARSA or AAV5-PGK-ARSA vector at 8 months of age. Galactose-6-sulfatase activity was identical in the brains of normal mice, untreated MLD mice, AAV5-treated MLD mice, and AAVrh.10-treated MLD mice at 10 months of age (data not shown).
Discussion
Several neurological diseases of the CNS have been attributed to oligodendrocyte or astrocyte rather than neuronal dysfunction. This is the case of metachromatic leukodystrophy (MLD), in which deficiency of lysosomal ARSA enzyme results in sulfatide accumulation that affects oligodendrocytes, resulting in myelin loss in the CNS (von Figura et al., 2001). However, in MLD, neurons are also damaged by the sulfatide storage (von Figura et al., 2001; Wittke et al., 2004). Of note, various molecular species accumulate in oligodendrocytes and neurons from MLD mice (Isaac et al., 2006). Given the rapid and devastating progression of cerebral disease in patients with the LI form of MLD, the challenge for any gene therapy strategy is to deliver rapidly the ARSA enzyme to both oligodendrocytes and neurons in the whole brain. Our results demonstrate clearly that the intracerebral injection of AAVrh.10cuARSA vector into MLD mice corrects more rapidly and more efficiently the specific sulfatide species that accumulate in oligodendrocytes and neurons than does the AAV5-PGK-ARSA vector we previously used (Sevin et al., 2006, 2007). Accordingly, the sulfatide load in the whole injected hemisphere evaluated at the neuropathological level and at the biochemical level, as well as the associated brain pathology (microgliosis, astrogliosis), were also more rapidly corrected.
MLD mice were injected at 8 months of age, a time by which they have already developed marked sulfatide accumulation in the brain as well as moderate balance and motor coordination deficits (Sevin et al., 2006). This stage corresponds approximately to the clinical stage at which patients with symptomatic LI MLD are diagnosed. Our study in MLD mice was designed to evaluate the short-term biochemical and neuropathological effects of the AAVrh.10cuARSA vector, precluding evaluation of its potential benefit on motor deficits. Indeed, as shown previously (Sevin et al., 2006), motor deficits developed by the MLD mouse remain moderate at 8 months of age and do not aggravate significantly until 15 months of age (see Fig. 9A in Sevin et al., 2006). Hence, other experiments with a large number of treated and untreated MLD mice are warranted to evaluate the long-term benefits of intracerebral injection of AAVrh.10cuARSA vector on MLD mouse motor functions.
On the basis of viral genome copy quantification, our data demonstrate that, in comparison with AAV5 vector, the intracerebral injection of AAVrh.10 vector leads to higher numbers of vector genome copies not only at injection sites but also at a distance from the injection sites, in agreement with previous observations (Cearley and Wolfe, 2006, 2007; Sondhi et al., 2007, 2008). Although we did not use a labeled AAVrh.10 vector, our data strongly suggest better diffusion of the AAVrh.10 vector from intracerebral injection sites in comparison with AAV5. This was likely mediated through better diffusion of AAVrh.10 viral particles into the extracellular space but may also have occurred through retrograde axonal transport. Several AAV serotypes have been shown to be transported along neuronal projections, either from the cell body to the synapse (anterograde) or from the axon to the cell body (retrograde). In rodents, and based on different methodologies (CY3 capsid markers, quantitative PCR, and/or mRNA in situ hybridization), retrograde transport was described for AAV1, AAV2, AAV8, AAV9, and AAVrh.10 vectors, even with sometimes contradictory results (Kaspar et al., 2002, 2003; Cearley and Wolfe, 2006, 2007; Cearley et al., 2008; Hollis et al., 2008; Foust et al., 2009). Anterograde transport of AAV vector is more challenging to demonstrate because it requires showing axonal transport of the vector from the cellular body of the neuron to the synapse and then the transsynaptic passage of the vector. In mice, anterograde transport was suggested for AAV1, AAV2, AAV5, AAV8, and AAV9 (Cearley and Wolfe, 2006, 2007; Cearley et al., 2008; Foust et al., 2008). Anterograde transport of AAV was also suggested in nonhuman primates after the injection of AAV1, AAV5, and AAV8 vector encoding GFP into the caudate nucleus or putamen (Dodiya et al., 2010) or the injection of AAV2 vector encoding GFP into the thalamus (Kells et al., 2009). Our results with AAVrh.10 vector are similar to those reported with AAV9 after injection into the ventral tegmental area or striatum (Cearley and Wolfe, 2007). They suggest that AAVrh.10 vector is transported mostly in axons in a retrograde manner.
Our results indicate also that the ARSA enzyme can be transported along neuronal projections, like other lysosomal enzymes, potentially increasing enzymatic correction at a distance from the injection site (Passini et al., 2002; Hennig et al., 2003; Luca et al., 2005; Cearley and Wolfe, 2006, 2007). Intrastriatal injection of AAVrh.10cuARSA or AAVrh.10cuGFP vector led to significantly higher expression of ARSA than GFP in the VTA, which is connected to the striatum.
AAVrh.10 vector transduces mostly neurons but also oligodendrocytes, although to a much less extent. After injection of AAVrh.10cuGFP vector into the striatum, 9% of oligodendrocytes localized in the white matter of internal capsules were transduced. When using AAVrh.10cuARSA vector, the percentage of oligodendrocytes expressing ARSA increased to 21% owing to additional ARSA enzyme recapture by these cells. Such a percentage of oligodendrocyte correction likely explains the beneficial effect that was observed at the biochemical level in MLD mice. Several other AAV vectors have been shown to transduce nonneuronal cells in vivo to some extent (Wang et al., 2003; Cearley et al., 2008; Foust et al., 2009; Hadaczek et al., 2009; Lawlor et al., 2009; Airavaara et al., 2010), but these results have not yet been translated into any mouse model of CNS disease affecting primarily oligodendrocytes, as is the case for MLD. The use of an oligodendrocyte-specific promoter might reveal higher transduction of oligodendrocytes (Chen et al., 1999), but the drawback for MLD is that neurons that degenerate from sulfatide accumulation would not be corrected.
The beneficial effect of AAVrh.10 vector was also mediated by the use of the CAG/cu promoter, which allows high expression of ARSA in transduced cells. Overexpression of ARSA enzyme could potentially exceed the capacity of formylglycine-generating enzyme (encoded by the sulfatase-modifying factor-1 [SUMF1] gene) to fully activate the ARSA enzyme, and also other sulfatases. Our results indicate that the overexpression of ARSA did not affect the activity of galactose-6-sulfatase, another sulfatase that requires posttranslational modification by formylglycine-generating enzyme. Using AAV1 vector in combination with the CAG/cu promoter, Kurai and colleagues showed that ARSA activity was enhanced by the cointroduction of SUMF1 cDNA in the AAV1 vector (Kurai et al., 2007). Using the AAV5 vector and a PGK promoter, we did not observe any beneficial effect of SUMF1 coexpression on ARSA activity (our unpublished results). It is, however, necessary to verify whether brain injection of AAVrh10.cu.ARSA vector coexpressing SUMF1 could potentially increase higher level ARSA activity in treated MLD mice.
All together, the intracerebral injection of AAVrh.10cu.ARSA vector resulted in an unprecedented level of oligodendrocyte correction that may have implications for a brain gene therapy approach to another lysosomal disorder of myelin, globoid cell leukodystrophy. Those results provide strong support for implementing AAVrh.10 brain gene therapy in MLD patients with rapidly progressive forms of the disease. Tolerance and efficacy studies are currently in progress in nonhuman primates before translation to human patients, in particular to evaluate a potential immune reaction against the ARSA transgene.
Acknowledgments
The authors thank Laurence Dubreil for technical assistance in the immunohistochemical studies and the production of photomicrographs, Philippe Moullier and the vector core facility of the University Hospital of Nantes for AAV5 vector, and Irène Dang for technical assistance. This work was supported by grants from INSERM, the European Leukodystrophy Association (2009-010C5A), the Association Française contre les Myopathies, the European FP7 Program (HEALTH-F2-2010-241622), the Fondation Jérôme Lejeune, and the National Institutes of Health (U01 NS047458).
Author Disclosure Statement
No competing financial interests exist.
References
- Airavaara M. Chiocco M.J. Howard D.B., et al. Widespread cortical expression of MANF by AAV serotype 7: Localization and protection against ischemic brain injury. Exp. Neurol. 2010;225:104–113. doi: 10.1016/j.expneurol.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass N.H. Witmer E.J. Dreifuss F.E. A pedigree study of metachromatic leukodystrophy: Biochemical identification of the carrier state. Neurology. 1970;20:52–62. doi: 10.1212/wnl.20.1.52. [DOI] [PubMed] [Google Scholar]
- Biffi A. De Palma M. Quattrini A., et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest. 2004;113:1118–1129. doi: 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A. Capotondo A. Fasano S., et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Invest. 2006;116:3070–3082. doi: 10.1172/JCI28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredius R.G. Laan L.A. Lankester A.C., et al. Early marrow transplantation in a pre-symptomatic neonate with late infantile metachromatic leukodystrophy does not halt disease progression. Bone Marrow Transplant. 2007;39:309–310. doi: 10.1038/sj.bmt.1705581. [DOI] [PubMed] [Google Scholar]
- Cearley C.N. Wolfe J.H. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol. Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Cearley C.N. Wolfe J.H. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J. Neurosci. 2007;27:9928–9940. doi: 10.1523/JNEUROSCI.2185-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cearley C.N. Vandenberghe L.H. Parente M.K., et al. Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol. Ther. 2008;16:1710–1718. doi: 10.1038/mt.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F. Vitry S. Hocquemiller M., et al. α-l-Iduronidase transport in neurites. Mol. Genet. Metab. 2006;87:349–358. doi: 10.1016/j.ymgme.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Chen H. McCarty D.M. Bruce A.T. Suzuki K. Oligodendrocyte-specific gene expression in mouse brain: Use of a myelin-forming cell type-specific promoter in an adeno-associated virus. J. Neurosci. Res. 1999;55:504–513. doi: 10.1002/(SICI)1097-4547(19990215)55:4<504::AID-JNR10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Colle M.A. Piguet F. Bertrand L., et al. Efficient intracerebral delivery of AAV5 vector encoding human ARSA in non-human primate. Hum. Mol. Genet. 2010;19:147–158. doi: 10.1093/hmg/ddp475. [DOI] [PubMed] [Google Scholar]
- Consiglio A. Martino S. Dolcetta D., et al. Metabolic correction in oligodendrocytes derived from metachromatic leukodystrophy mouse model by using encapsulated recombinant myoblasts. J. Neurol. Sci. 2007;255:7–16. doi: 10.1016/j.jns.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Dodiya H.B. Bjorklund T. Stansell J., III, et al. Differential transduction following basal ganglia administration of distinct pseudotyped AAV capsid serotypes in nonhuman primates. Mol. Ther. 2010;18:579–587. doi: 10.1038/mt.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust K.D. Flotte T.R. Reier P.J. Mandel R.J. Recombinant adeno-associated virus-mediated global anterograde delivery of glial cell line-derived neurotrophic factor to the spinal cord: Comparison of rubrospinal and corticospinal tracts in the rat. Hum. Gene Ther. 2008;19:71–82. doi: 10.1089/hum.2007.104. [DOI] [PubMed] [Google Scholar]
- Foust K.D. Nurre E. Montgomery C.L., et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N. Suzuki K. Vanier M.T., et al. Targeted disruption of the mouse sphingolipid activator protein gene: A complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- Hadaczek P. Forsayeth J. Mirek H., et al. Transduction of nonhuman primate brain with adeno-associated virus serotype 1: Vector trafficking and immune response. Hum. Gene Ther. 2009;20:225–237. doi: 10.1089/hum.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig A.K. Levy B. Ogilvie J.M., et al. Intravitreal gene therapy reduces lysosomal storage in specific areas of the CNS in mucopolysaccharidosis VII mice. J. Neurosci. 2003;23:3302–3307. doi: 10.1523/JNEUROSCI.23-08-03302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B. Saftig P. Hartmann D., et al. Phenotype of arylsulfatase A-deficient mice: Relationship to human metachromatic leukodystrophy. Proc. Natl. Acad. Sci. U.S.A. 1996;93:14821–14826. doi: 10.1073/pnas.93.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis E.R., II Kadoya K. Hirsch M., et al. Efficient retrograde neuronal transduction utilizing self-complementary AAV1. Mol. Ther. 2008;16:296–301. doi: 10.1038/sj.mt.6300367. [DOI] [PubMed] [Google Scholar]
- Isaac G. Pernber Z. Gieselmann V., et al. Sulfatide with short fatty acid dominates in astrocytes and neurons. FEBS J. 2006;273:1782–1790. doi: 10.1111/j.1742-4658.2006.05195.x. [DOI] [PubMed] [Google Scholar]
- Kaspar B.K. Erickson D. Schaffer D., et al. Targeted retrograde gene delivery for neuronal protection. Mol. Ther. 2002;5:50–56. doi: 10.1006/mthe.2001.0520. [DOI] [PubMed] [Google Scholar]
- Kaspar B.K. Llado J. Sherkat N., et al. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kells A.P. Hadaczek P. Yin D., et al. Efficient gene therapy-based method for the delivery of therapeutics to primate cortex. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2407–2411. doi: 10.1073/pnas.0810682106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R.L. Dayton R.D. Tatom J.B., et al. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: Effects of serotype, promoter and purification method. Mol. Ther. 2008;16:89–96. doi: 10.1038/sj.mt.6300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurai T. Hisayasu S. Kitagawa R., et al. AAV1 mediated co-expression of formylglycine-generating enzyme and arylsulfatase A efficiently corrects sulfatide storage in a mouse model of metachromatic leukodystrophy. Mol. Ther. 2007;15:38–43. doi: 10.1038/sj.mt.6300012. [DOI] [PubMed] [Google Scholar]
- Lawlor P.A. Bland R.J. Mouravlev A., et al. Efficient gene delivery and selective transduction of glial cells in the mammalian brain by AAV serotypes isolated from nonhuman primates. Mol. Ther. 2009;17:1692–1702. doi: 10.1038/mt.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca T. Givogri M.I. Perani L., et al. Axons mediate the distribution of arylsulfatase A within the mouse hippocampus upon gene delivery. Mol. Ther. 2005;12:669–679. doi: 10.1016/j.ymthe.2005.06.438. [DOI] [PubMed] [Google Scholar]
- Martino S. Consiglio A. Cavalieri C., et al. Expression and purification of a human, soluble arylsulfatase A for metachromatic leukodystrophy enzyme replacement therapy. J. Biotechnol. 2005;117:243–251. doi: 10.1016/j.jbiotec.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Matthes F. Wolte P. Bockenhoff A., et al. Transport of arylsulfatase A across the blood–brain barrier in vitro. J. Biol. Chem. 2011;286:17487–17494. doi: 10.1074/jbc.M110.189381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzner U. Harzer K. Learish R.D., et al. Long-term expression and transfer of arylsulfatase A into brain of arylsulfatase A-deficient mice transplanted with bone marrow expressing the arylsulfatase A cDNA from a retroviral vector. Gene Ther. 2000;7:1250–1257. doi: 10.1038/sj.gt.3301232. [DOI] [PubMed] [Google Scholar]
- Matzner U. Lullmann-Rauch R. Stroobants S., et al. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol. Ther. 2009;17:600–606. doi: 10.1038/mt.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini M.A. Lee E.B. Heuer G.G. Wolfe J.H. Distribution of a lysosomal enzyme in the adult brain by axonal transport and by cells of the rostral migratory stream. J. Neurosci. 2002;22:6437–6446. doi: 10.1523/JNEUROSCI.22-15-06437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters C. Steward C.G. Hematopoietic cell transplantation for inherited metabolic diseases: An overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003;31:229–239. doi: 10.1038/sj.bmt.1703839. [DOI] [PubMed] [Google Scholar]
- Platt F.M. Lachmann R.H. Treating lysosomal storage disorders: Current practice and future prospects. Biochim. Biophys. Acta. 2009;1793:737–745. doi: 10.1016/j.bbamcr.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Sangalli A. Taveggia C. Salviati A., et al. Transduced fibroblasts and metachromatic leukodystrophy lymphocytes transfer arylsulfatase A to myelinating glia and deficient cells in vitro. Hum. Gene Ther. 1998;9:2111–2119. doi: 10.1089/hum.1998.9.14-2111. [DOI] [PubMed] [Google Scholar]
- Sevin C. Benraiss A. Van Dam D., et al. Intracerebral adeno-associated virus-mediated gene transfer in rapidly progressive forms of metachromatic leukodystrophy. Hum. Mol. Genet. 2006;15:53–64. doi: 10.1093/hmg/ddi425. [DOI] [PubMed] [Google Scholar]
- Sevin C. Verot L. Benraiss A., et al. Partial cure of established disease in an animal model of metachromatic leukodystrophy after intracerebral adeno-associated virus-mediated gene transfer. Gene Ther. 2007;14:405–414. doi: 10.1038/sj.gt.3302883. [DOI] [PubMed] [Google Scholar]
- Sondhi D. Hackett N.R. Peterson D.A., et al. Enhanced survival of the LINCL mouse following CLN2 gene transfer using the rh.10 rhesus macaque-derived adeno-associated virus vector. Mol. Ther. 2007;15:481–491. doi: 10.1038/sj.mt.6300049. [DOI] [PubMed] [Google Scholar]
- Sondhi D. Peterson D.A. Edelstein A.M., et al. Survival advantage of neonatal CNS gene transfer for late infantile neuronal ceroid lipofuscinosis. Exp. Neurol. 2008;213:18–27. doi: 10.1016/j.expneurol.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymans J.M. Vandenberghe L.H. Haute C.V., et al. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum. Gene Ther. 2007;18:195–206. doi: 10.1089/hum.2006.178. [DOI] [PubMed] [Google Scholar]
- Tylki-Szymanska A. Czartoryska B. Bunge S., et al. Clinical, biochemical and molecular findings in a two-generation Morquio A family. Clin. Genet. 1998;53:369–374. doi: 10.1111/j.1399-0004.1998.tb02747.x. [DOI] [PubMed] [Google Scholar]
- Von Figura K. Gieselmann V. Jaeken J. Metachromatic leukodystrophy. In: Scriver C., editor; Beaudet A., editor; Sly W., editor; Valle D., editor. The Metabolic, Molecular Bases of Inherited Disease. 8th. McGraw-Hill; New York: 2001. pp. 3695–3724. [Google Scholar]
- Wang C. Wang C.M. Clark K.R. Sferra T.J. Recombinant AAV serotype 1 transduction efficiency and tropism in the murine brain. Gene Ther. 2003;10:1528–1534. doi: 10.1038/sj.gt.3302011. [DOI] [PubMed] [Google Scholar]
- Wittke D. Hartmann D. Gieselmann V. Lullmann-Rauch R. Lysosomal sulfatide storage in the brain of arylsulfatase A-deficient mice: Cellular alterations and topographic distribution. Acta Neuropathol. 2004;108:261–271. doi: 10.1007/s00401-004-0883-6. [DOI] [PubMed] [Google Scholar]