

Abstract
The development of genetic technologies has led to the identification of several copy number variations (CNVs) in the human genome. Genome rearrangements affect dosage-sensitive gene expression in normal brain development. There is strong evidence associating human psychiatric disorders, especially autism spectrum disorders (ASDs) and schizophrenia to genetic risk factors and accumulated CNV risk loci. Deletions in 1q21, 3q29, 15q13, 17p12, and 22q11, as well as duplications in 16p11, 16p13, and 15q11-13 have been reported as recurrent CNVs in ASD and/or schizophrenia. Chromosome engineering can be a useful technology to reflect human diseases in animal models, especially CNV-based psychiatric disorders. This system, based on the Cre/loxP strategy, uses large chromosome rearrangement such as deletion, duplication, inversion, and translocation. Although it is hard to reflect human pathophysiology in animal models, some aspects of molecular pathways, brain anatomy, cognitive, and behavioral phenotypes can be addressed. Some groups have created animal models of psychiatric disorders, ASD, and schizophrenia, which are based on human CNV. These mouse models display some brain anatomical and behavioral abnormalities, providing insight into human neuropsychiatric disorders that will contribute to novel drug screening for these devastating disorders.
1. Introduction
Copy number variation (CNV) is a structural genomic variation of the human genome that may either be inherited or caused by de novo mutation. It includes translocation, inversion, duplication, triplication, and deletion. CNVs can range in size from kilobases (Kbs) to several megabases (Mbs) that have not been identified by conventional chromosomal analysis. However, recent technology of genome-wide analysis such as comparative genomic hybridization (CGH) has led to the discovery of extensive genomic structural variation [1–3]. A recent report using microarray technology revealed that as much as 12% of the human genome are variable in copy number [4]. These known CNVs are available from the interactive web-based database DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources, http://decipher.sanger.ac.uk/). The DECIPHER database is a Consortium comprised of an international network of more than 100 centers and has uploaded more than 2000 cases (current statistics can be found on the DECIPHER homepage) [5].
CNVs can be de novo or familial. De novo mutations are more likely to contribute to the development of sporadic genomic disorders [6, 7]. In psychiatric disorders, ASD and schizophrenia, extension of genome-wide association studies (GWAS) have led to the discovery of both inherited and de novo sporadic CNVs. Such CNVs resulted in altering gene dosage and dosage-sensitive gene expression, which may contribute to these disorders complexities [8]. These human genetics studies have detected several CNVs (e.g., 1q21, 3q29, 10q26, 11p14, 15q11, 15q13, 16p13, 17p12, and 22q11). This discovery suggests an important role for the strict regulation of gene dosage in ASD and schizophrenia.
To understand psychiatric disorders, animal models are needed because particular experiments in human are impossible. While it is difficult to model human psychiatric phenotypes in animals (e.g., hallucinations and delusions characteristic of schizophrenia that are human specific), animal models may contribute to the elucidation of brain anatomy, behavioral characteristics, and molecular mechanisms that reflect aspects of human phenotypes.
Although there is a strong association between genetic rearrangement and psychiatric disorders (e.g., ASD and schizophrenia), valid animal models that reflect etiology are rare. Several efforts have been made to generate mouse models of psychiatric disorders by conventional gene targeting, conditional gene targeting, and point mutation by chemical mutagens. But these techniques are not enough to reflect complex human genomic rearrangements, such as large deletions, inversions, and duplications. In this regard, the Cre/loxP-based chromosome engineering technique is useful to generate this kind of complex genomic rearrangements in the mouse genome. By using this chromosome engineering technique, we can accomplish CNV-based unbiased animal models of psychiatric disorders. In this paper, we focus on animal model of ASD (and schizophrenia) which was generated by chromosome engineering, principle of this technology, and discuss for future directions.
2. Chromosome Engineering in Mice
Genetic abnormalities such as point mutations, deletions, duplications, inversions, and translocations can be induced by exposure to X-ray radiation, chemical mutagens (e.g., N-ethyl-N-nitrosourea (ENU)), conventional gene-targeting, or chromosome engineering. X-ray causes DNA double-strand breaks, inducing genomic instability [16]. The chemical mutagen, ENU, induces single-base-pair substitutions in the genome causing mutations with partial functions [17, 18]. Animal models containing genes with point mutations can be used to reveal the gene's functional domain in vivo [19]. Both techniques are valuable but cannot predict the mutated position within the gene. Conventional gene targeting (replacement) is used to disrupt a gene (inserting markers or reporters) to determine a gene's function. Conditional gene-targeting utilizing the Cre/loxP and Flp/FRT system allows spatial and temporal control of gene expression. It has been increasingly used for gene function analysis in vivo.
Chromosome engineering is based on Cre/loxP technology, which can induce chromosome rearrangements (deletions, duplications [10, 20, 21], and inversions [22, 23]) in the mouse genome (Figure 1).
Figure 1.
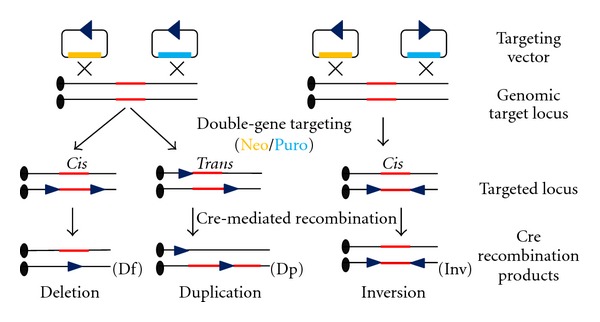
Chromosome engineering in mouse embryonic stem cells.
Targeting vectors can be targeted in two orientations that result in deletion, duplication, or inversion. Each targeting vector has a loxP site and drug selection marker, neomycin resistance (Neo), or puromycin-resistant gene (Puro). Cis and Trans indicate loxP sites.
Two loxP sites are sequentially inserted by each targeting vector into the mouse embryonic stem (ES) cell genome. Each targeting vector contains a selection marker, neomycin, or puromycin resistance gene. The vectors are manipulated by hypoxanthine phosphoribosyl transferase (HPRT) expression following Cre recombinase expression in ES cells. Transient expression of Cre recombinase induces rearrangement between loxP sites in the mouse genome. Clones which carry the desired chromosomal rearrangement are identified by various methods: drug selection by hypoxanthine-aminopterin-thymidine (HAT) media, genomic Southern blot analysis, fluorescent in situ hybridization (FISH), and CGH (comparative genomic hybridization) array. Although CGH array cannot identify structural chromosome aberrations such as balanced reciprocal translocations and inversions, this technique is a powerful tool to detect CNVs from genome.
To inactivate a target gene or locus by chromosome engineering, a gene target vector must be chosen or created. The Mutagenic Insertion and Chromosome Engineering Resource (MICER) (http://www.sanger.ac.uk/resources/mouse/micer/) was developed by Dr. Allan Bradley's group, the Wellcome Trust Sanger Institute and is useful as a gene-targeting vectors resource [24]. These ready to use targeting vectors can be accessed through the Ensembl mouse genome browser (http://www.ensembl.org/index.html). It is important to note that these targeting vectors use an insertion vector system rather than a replacement vector system. Given the same length of homologous sequence insertion vectors have a ninefold higher targeting efficiency than replacement vectors [25].
3. Animal Models Based on Human CNVs
In spite of a strong association between ASD (and schizophrenia) and CNV, animal models of CNV that reflect human genomic rearrangement are few. These animal models were generated by chromosome engineering and have several psychotic phenotypes similar to those seen in patients with genomic rearrangement (Table 1). In this section we focus on 15q11-13, 16p11.2, and 22q11 locus, which are well-known copy number variant linked to ASD (or/and schizophrenia).
Table 1.
Behavioral phenotypes of mouse models.
| Human chromosomal region | Behavioral phenotypes | Reference |
|---|---|---|
| 7q11.23 (deletion) | Increased sociability | [9] |
| Increased acoustic startle response | ||
| Cognitive deficits | ||
| Growth retarded (male) | ||
|
| ||
| 15q11-13 (duplication) | Decreased sociability | [10, 11] |
| Behavioral inflexibility | ||
| abnormal ultrasonic vocalizations | ||
| decreased spontaneous activity | ||
| Increased anxiety | ||
|
| ||
| 16p11.2 (deletion) | Hyperactive | [12] |
| difficulty adapting to change | ||
| sleeping abnormalities | ||
| repetitive or restricted behaviors | ||
|
| ||
| 16p11.2 (duplication) | Hypoactive | [12] |
|
| ||
| 17p11.2 (deletion) | cranio facial abnormalities | [13] |
| Seizures | ||
| Obesity | ||
|
| ||
| 22q11.21 (deletion) | Deficits in sensorimotor gating | [14, 15] |
| Working memory deficit | ||
| Deficit in both cued and contextual fear memory | ||
3.1. 15q11-13 Duplication Syndrome (ASD)
Human chromosome region, 15q11-13, is a complicated region that contains γ-aminobutyric acidreceptor A (GABAA receptor) clusters and several imprinting genes[26]. Paternally expressing genes include MKRN3, MAGEL2, NDN, and SNURF-SNRPN. Maternally expressing genes include UBE3A and ATP10A. In addition to these genes, this locus includes noncoding small nucleolar RNAs (snoRNAs) that are located between SNURF-SNRPN and UBE3A, which are paternally expressed and brain specific [27, 28]. Deletion or duplication of this locus causes severe neurological phenotypes. Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are affected by changes in the 15q11-13 locus. Most notably, deletion of UBE3A has been identified to lead to AS phenotypes. Major clinical features of PWS include low birth weight, short stature, small hands and feet, severe hypotonia, feeding difficulties, obesity associated with hyperphagia starting in early childhood, mild to moderate mental retardation, and learning and behavioral problems including obsessive-compulsive disorder and autism [29, 30]. AS patients exhibit developmental delay, gait ataxia, balance disorder, frequent laughter/smiling, easily excitable personality, hyperactivity, speech impairment, microcephaly, seizures, epilepsy, and abnormal EEG (electroencephalogram) [31]. Additionally, AS patients often exhibit socialization and communication deficits, which are diagnostic criteria for ASD [32, 33].
Duplication of the 15q11-13 locus was first reported as a partial trisomy of chromosome 15 [34], and then two individuals with autistic disorder were reported [35]. This locus has been known as the most frequent cytogenetic abnormality in ASD [36, 37]. Generally patients with 15q11-13 duplication show hypotonia, delay in motor skills and language development, epilepsy, and cognitive and learning problems. Recently, Michelson et al. reported a patient with severe intractable epilepsy who has familial partial trisomy 15q11-13 inherited from a mother who has schizophrenia [38]. Autistic phenotype associated with 15q11-13 duplication, usually believed that maternal origin, UBE3A is involved [39–46]. Although maternal locus supposed to critical, paternally inherited patients had also developmental delay [44, 46–49]. Clinical reports have been accumulating but no mechanism has been addressed.
To address this question, Nakatani et al. generated a mouse model of human 15q11-13 duplication [10]. This mouse was generated by chromosomal engineering based on the Cre/loxP system, and it has a 6.3 Mb duplicated locus in mouse chromosome 7c which is highly similar to human 15q11-13 (Figure 2(a)).
Figure 2.

Schematic representation in studies applying chromosome engineering. Published in (a) Nakatani et al. [10], (b) Horev et al. [12], (c) Stark et al. [14]. The paternally, maternally expressed, and nonimprinting genes were labeled with blue, red, and black, respectively.
Gene expression analysis revealed that paternally expressed genes, both Ndn and Snrpn, were twofold higher in paternally inherited mice (patDp/+) than wild-type (WT) mice. Similarly, maternally expressed gene Ube3a was twofold higher in maternally inherited mice (matDp/+) than WT mice. Histological analysis revealed no gross brain abnormalities. Monoamine levels in patDp/+ adult mice, serotonin (5-HT), and their metabolites 5-hydroxyindoleacetic acid (5-HIAA) were significantly downregulated in the midbrain and olfactory bulb. Also 5-HT content in developmental stage from postnatal 1 to 3 weeks in patDp/+ brain regions (cortex, hippocampus, cerebellum, midbrain, hypothalamus, pons, and medulla) was downregulated. This indicates 5-HT signaling during the developmental stage was significantly impaired in the brains of patDp/+ mice [10]. 5-HT influences not only mental condition (mood, social behavior, appetite, aggression and sleep) but also normal development of the central nervous system [50–52]. In addition to this, abnormal 5-HT levels have been found in ASD patient blood cells. For these reasons, 5-HT is one of the drug targets for ASD therapy. Treatment with serotonin reuptake inhibitors (SSRIs) have shown moderate success in recovering behaviors [53].
Behavioral tests revealed that patDp/+ mice display autistic behaviors such as less social interaction in the three-chamber social interaction test [54], abnormal ultrasonic vocalizations (USVs) [55] in postnatal developing pups separated from their dams, and behavioral inflexibility in the Morris Water Maze and Barnes Maze [10]. The phenotypes seen in patDp/+ mice indicate that these mice have impaired behaviors that include social interaction, communication, restricted interest, and resistance to change. These deficits correspond to human autistic phenotypes [56, 57]. Furthermore, patDp/+ mice showed anxiety-related phenotypes: decreased locomotor and exploratory activities in the open field and Y-maze test, and long latencies in novelty suppressed feeding test [11]. These anxiety-related phenotypes frequently accompany autistic symptoms in humans [58, 59]. Also the marble burying test, which is a useful test for the study of anxiety, obsessive-compulsive disorder (OCD), and neophobia, found that the number of buried marbles was significantly low in patDp/+ mice [11].
3.2. 16p11.2 Deletion/Duplication Syndrome (Deletion: ASD, Duplication: Schizophrenia)
Deletion or duplication of the chromosome 16p11.2 locus was observed in nearly 1% of multiplex families with ASD [60]. Meta-analysis of patients with ASD and/or developmental delay estimated that 16p11.2 locus deletion is associated with a 38.7-fold increase in the odds of ASD/developmental delay. On the other hand, 16p11.2 locus duplication is associated with a 20.7-fold increase in the odds of ASD/developmental delay [60–63]. In addition to these, 16p11.2 deletion is associated with obesity [64], and duplication is associated with schizophrenia [65] as well as ASD [60, 66]. Notably, a brain anatomical abnormality (abnormal head size) has been reported to be associated with this locus. For instance, patients with the 16p11.2 deletion had statistically significant macrocephaly and those with duplication had microcephaly [67].
A mouse model of human 16p11.2 deletion (df/+) as well as duplication (dp/+) has been reported [12] (Figure 2(b)). This mouse model was generated by Cre/loxP-based chromosome engineering. This locus includes 27 genes, SPN, QPRT, c16orf54, KIF22, MAZ, PRRT2, c16orf53, MVP, CDIPT, SEZ6L2, ASPHD1, KCTD13, LOC124446, HIRIP3, CCDC95, DOC2A, FAM57B, ALDOA, PPP4C, YPEL3, GDPD3, MAPK3, and CORO1A.
Young df/+ mice (before weaning) tend to be smaller than WT siblings, but as adults they are almost the same size as WT siblings and look healthy. Interestingly, 16p11.2 CNV mice, df/+ and dp/+ mice have opposite phenotypes. In a novel environmental cage, df/+ mice displayed longer distance traveled and time spent walking as compared with WT mice. In contrast, dp/+ mice traveled a shorter distance and spent less time walking as compared with WT mice. Additionally, df/+ mice were significantly active in both dark and light period. These results indicate that 16p11.2 locus affects not only physical activity but also diurnal activity and sleeping related symptoms. Also, a brain anatomical study using Magnetic resonance imaging (MRI) identified several regional changes in 16p11.2 CNV mice. For instance, df/+ mice showed increased volume of several brain regions (percentage of total brain volume): forebrain, superior colliculus, fornix, hypothalamus, mammillothalamic tract, medial septum, midbrain, and periaqueductal grey. These brain regional volumetric changes were more significant between df/+ and dp/+ than betweendf/+ and WT mice.
3.3. 22q11.2 Deletion Syndrome (22q11.2DS, DiGeorge Syndrome (DGS), Velo-Cardio-Facial Syndrome (VCFS)) (Schizophrenia)
Microdeletion of chromosome 22q11 is found in 1 out of every 4000 live births, making it one of the most common interstitial deletions [68]. This 22q11.2 microdeletion causes craniofacial, cardiovascular abnormalities, immunodeficiency, hypocalcaemia, short stature, and cognitive dysfunctions [69–71]. Microdeletion of this region accounts for 1-2% of the cases of people with schizophrenia [72, 73]. Also, this locus accounts for up to 1-2% of cases of sporadic schizophrenia [74–76]. Some neuroanatomical changes have been reported in patients with 22q11.2DS as well. Volumetric reduction in total brain volume includes cortical regions (e.g., frontal, parietal, temporal, and occipital lobes), hippocampus, and cerebellum [77–86]. However, inconsistency in these neuroimaging reports may be due to the small numbers of subjects used and differences in methodology [72]. Yet these neuroanatomical reports are informative because some abnormalities are consistent with phenotypes of those who have non-22q11.2 DS-associated schizophrenia [76].
The majority of deletions in this locus are 3 Mb deletions (−90% of the cases), but 1.5 Mb deletions (<10% of the cases) contain 28 known genes which include critical genes and increased risk of mental disorders [73, 87].
The mouse chromosome 16 region is conserved with human 22q11.2. Animal models of the human 22q11.2 deletion were generated by 2 groups, and both groups used chromosome engineering [14, 88] (Figure 2(c)). These mouse models, Df(16)A+/− [14] and LgDel/+ [88], include 1.5 Mb critical regions, and both of them display several behavioral abnormalities, such as deficits in working memory, sensorimotor gating, and fear conditioning [14, 89–92]. Working memory deficits are becoming one of the main features of patients with schizophrenia, thus these animal models are supposed to reflect some aspects of 22q11.2 DS syndrome phenotype. In addition to behavioral abnormalities in this mouse, diminished 22q11 locus dosage disrupts cortical neurogenesis, interneuron migration [93], dendritic complexity, and formation of excitatory synapses [94]. Although, several interesting phenotypes have been reported in this mutant mouse, there are no studies published about brain structural abnormalities even though several brain abnormalities have reported in human studies. These brain anatomical changes and molecular mechanisms that underlie these phenotypes will be interesting to elucidate and will be addressed by using brain imaging techniques.
4. Future Perspectives
Application of new technologies, such as Comparative Genomic Hybridization (CGH) and next-generation sequencing, will reveal more additional genomic rearrangements related to psychiatric disorders. Thus, to analyze both phenotypes and underlying molecular mechanisms that originate from genetic rearrangements, animal models will be a powerful tool. In this context, chromosome engineering will be a valuable tool. Recently Ruf et al. [95] reported that they generated several hundred mice and embryos which have one loxP and LacZ site at a random genomic positions that inserted by sleeping beauty-based transposition system. These lines are mapped in Transposon and Recombinase Associated Chromosomal Engineering Resource database (TRACER, http://tracerdatabase.embl.de/fmi/iwp/res/iwp_home.html). This database is useful in creating chromosome rearrangements in vivo.
A logical next step is to identify responsible gene(s) in CNV. It is an orthodox approach to narrow down the region by systematically insertion of loxP combining the existed lines such as above TRACER. Generating Bacterial Artificial Chromosome (BAC) transgenic mice is another way to identify critical genes. BAC transgenes inserted to the genome faithfully recapitulate chromosomal endogenous gene expression, since BAC transgenic mice may appropriate animal model of gene duplication. Also transient overexpressing (or knockdown) each transcript in developmental brain is possible strategy. Recently Golzio et al. [96] identified a responsible gene KCTD13 in 16p11.2 locus which causes brain malformation by using zebrafish. Use of these technologies in generating valid and etiology-based animal model of psychiatric disorders will contribute to the development of drugs against disorders and elucidation of molecular mechanisms that underlie these psychiatric disorders.
Acknowledgments
The authors thank Geetha Kannan for help with paper preparation. The authors appreciate the help of colleagues who contributed to original work that reported in this paper and all members of our laboratory for useful discussions and comments. This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (T. Takumi, J. Nomura) and by research grants from the Takeda Science Foundation (T. Takumi), the Asahi Glass Foundation (T. Takumi), and the Naito Foundation (T. Takumi).
References
- 1.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nature Genetics. 2004;36(9):949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 3.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nature Genetics. 2007;39(1):S16–S21. doi: 10.1038/ng2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. American Journal of Human Genetics. 2009;84(4):524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarroll SA, Kuruvilla FG, Korn JM, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genetics. 2008;40(10):1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 7.Merikangas AK, Corvin AP, Gallagher L. Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends in Genetics. 2009;25(12):536–544. doi: 10.1016/j.tig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Crespi BJ, Thiselton DL. Comparative immunogenetics of autism and schizophrenia. Genes, Brain and Behavior. 2011;10(7):689–701. doi: 10.1111/j.1601-183X.2011.00710.x. [DOI] [PubMed] [Google Scholar]
- 9.Li HH, Roy M, Kuscuoglu U, et al. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Molecular Medicine. 2009;1(1):50–65. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakatani J, Tamada K, Hatanaka F, et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137(7):1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamada K, Tomonaga S, Hatanaka F, et al. Decreased exploratory activity in a mouse model of 15q duplication syndrome; implications for disturbance of serotonin signaling. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0015126.e15126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horev G, Ellegood J, Lerch JP, et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(41):17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walz K, Caratini-Rivera S, Bi W, et al. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Molecular and Cellular Biology. 2003;23(10):3646–3655. doi: 10.1128/MCB.23.10.3646-3655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark KL, Xu B, Bagchi A, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nature Genetics. 2008;40(6):751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- 15.Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464(7289):763–767. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L, Snyder AR, Morgan WF. Radiation-induced genomic instability and its implications for radiation carcinogenesis. Oncogene. 2003;22(37):5848–5854. doi: 10.1038/sj.onc.1206697. [DOI] [PubMed] [Google Scholar]
- 17.Gondo Y. Trends in large-scale mouse mutagenesis: from genetics to functional genomics. Nature Reviews Genetics. 2008;9(10):803–810. doi: 10.1038/nrg2431. [DOI] [PubMed] [Google Scholar]
- 18.Gondo Y, Murata T, Makino S, Fukumura R, Ishitsuka Y. Mouse mutagenesis and disease models for neuropsychiatric disorders. Current Topics of Behavioral Neurosciences. 2011;7:1–35. doi: 10.1007/7854_2010_106. [DOI] [PubMed] [Google Scholar]
- 19.Sayah DM, Khan AH, Gasperoni TL, Smith DJ. A genetic screen for novel behavioral mutations in mice. Molecular Psychiatry. 2000;5(4):369–377. doi: 10.1038/sj.mp.4000742. [DOI] [PubMed] [Google Scholar]
- 20.Liu P, Zhang H, McLellan A, Vogel H, Bradley A. Embryonic lethality and tumorigenesis caused by segmental aneuploidy on mouse chromosome 11. Genetics. 1998;150(3):1155–1168. doi: 10.1093/genetics/150.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walz K, Caratini-Rivera S, Bi W, et al. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Molecular and Cellular Biology. 2003;23(10):3646–3655. doi: 10.1128/MCB.23.10.3646-3655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng B, Sage M, Cai WW, et al. Engineering a mouse balancer chromosome. Nature Genetics. 1999;22(4):375–378. doi: 10.1038/11949. [DOI] [PubMed] [Google Scholar]
- 23.Nishijima I, Mills A, Qi Y, Mills M, Bradley A. Two new balancer chromosomes on mouse chromosome 4 to facilitate functional annotation of human chromosome lp. Genesis. 2003;36(3):142–148. doi: 10.1002/gene.10207. [DOI] [PubMed] [Google Scholar]
- 24.Adams DJ, Biggs PJ, Cox T, et al. Mutagenic insertion and chromosome engineering resource (MICER) Nature Genetics. 2004;36(8):867–871. doi: 10.1038/ng1388. [DOI] [PubMed] [Google Scholar]
- 25.Hasty P, Rivera-Perez J, Chang C, Bradley A. Target frequency and integration pattern for insertion and replacement vectors in embryonic stem cells. Molecular and Cellular Biology. 1991;11(9):4509–4517. doi: 10.1128/mcb.11.9.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takumi T. The neurobiology of mouse models syntenic to human chromosome 15q. Journal of Neurodevelopmental Disorders. 2011;3(3):270–281. doi: 10.1007/s11689-011-9088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Runte M, Hüttenhofer A, Groß S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Human Molecular Genetics. 2001;10(23):2687–2700. doi: 10.1093/hmg/10.23.2687. [DOI] [PubMed] [Google Scholar]
- 28.Cavaillé J, Buiting K, Kiefmann M, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14311–14316. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cassidy SB, Dykens E, Williams CA. Prader-Willi and Angelman syndromes: sister imprinted disorders. American Journal of Medical Genetics. 2000;97(2):136–146. doi: 10.1002/1096-8628(200022)97:2<136::aid-ajmg5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 30.Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. Journal of Intellectual Disability Research. 2000;44(1):25–30. doi: 10.1046/j.1365-2788.2000.00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams CA, Beaudet AL, Clayton-Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. American Journal of Medical Genetics. 2006;140(5):413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 32.Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M. Autism in Angelman syndrome: s population-based study. Pediatric Neurology. 1996;14(2):131–136. doi: 10.1016/0887-8994(96)00011-2. [DOI] [PubMed] [Google Scholar]
- 33.Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clinical Genetics. 2004;66(6):530–536. doi: 10.1111/j.1399-0004.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 34.Gillberg C, Steffenburg S, Wahlstrom J, et al. Autism associated with marker chromosome. Journal of the American Academy of Child and Adolescent Psychiatry. 1991;30(3):489–494. doi: 10.1097/00004583-199105000-00022. [DOI] [PubMed] [Google Scholar]
- 35.Baker P, Piven J, Schwartz S, Patil S. Brief report: duplication of chromosome 15q11-13 in two individuals with autistic disorder. Journal of Autism and Developmental Disorders. 1994;24(4):529–535. doi: 10.1007/BF02172133. [DOI] [PubMed] [Google Scholar]
- 36.Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiology of Disease. 2010;38(2):181–191. doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vorstman JAS, Staal WG, Van Daalen E, Van Engeland H, Hochstenbach PFR, Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Molecular Psychiatry. 2006;11(1):18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- 38.Michelson M, Eden A, Vinkler C, et al. Familial partial trisomy 15q11-13 presenting as intractable epilepsy in the child and schizophrenia in the mother. European Journal of Paediatric Neurology. 2011;15(3):230–233. doi: 10.1016/j.ejpn.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Cook EH, Lindgren V, Leventhal BL, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. American Journal of Human Genetics. 1997;60(4):928–934. [PMC free article] [PubMed] [Google Scholar]
- 40.Cook EH, Courchesne RY, Cox NJ, et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. American Journal of Human Genetics. 1998;62(5):1077–1083. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schroer RJ, Phelan MC, Michaelis RC, et al. Autism and maternally derived aberrations of chromosome 15q. American Journal of Medical Genetics. 1998;76(4):327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 42.Bolton PF, Dennis NR, Browne CE, et al. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. American Journal of Medical Genetics. 2001;105(8):675–685. doi: 10.1002/ajmg.1551. [DOI] [PubMed] [Google Scholar]
- 43.Browne CE, Dennis NR, Maher E, et al. Inherited interstitial duplications of proximal 15q: genotype-phenotype correlations. American Journal of Human Genetics. 1997;61(6):1342–1352. doi: 10.1086/301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mao R, Jalal SM, Snow K, Michels VV, Szabo SM, Babovic-Vuksanovic D. Characteristics of two cases with dup(15)(q11.2-q12): one of maternal and one of paternal origin. Genetics in Medicine. 2000;2(2):131–135. doi: 10.1097/00125817-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Repetto GM, White LM, Bader PJ, Johnson D, Knoll JHM. Interstitial duplications of chromosome region 15qllql3: clinical and molecular characterization. American Journal of Medical Genetics. 1998;79(2):82–89. doi: 10.1002/(sici)1096-8628(19980901)79:2<82::aid-ajmg2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 46.Roberts SE, Dennis NR, Browne CE, et al. Characterisation of interstitial duplications and triplications of chromosome 15q11-q13. Human Genetics. 2002;110(3):227–234. doi: 10.1007/s00439-002-0678-6. [DOI] [PubMed] [Google Scholar]
- 47.Bolton PF, Veltman MWM, Weisblatt E, et al. Chromosome 15q11-13 abnormalities and other medical conditions in individuals with autism spectrum disorders. Psychiatric Genetics. 2004;14(3):131–137. doi: 10.1097/00041444-200409000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Mohandas TK, Park JP, Spellman RA, et al. Paternally derived de novo interstitial duplication of proximal 15q in a patient with developmental delay. American Journal of Medical Genetics. 1999;82(4):294–300. [PubMed] [Google Scholar]
- 49.Veltman MWM, Thompson RJ, Craig EE, et al. A paternally inherited duplication in the Prader-Willi/Angelman Syndrome Critical Region: a case and family study. Journal of Autism and Developmental Disorders. 2005;35(1):117–127. doi: 10.1007/s10803-004-1039-1. [DOI] [PubMed] [Google Scholar]
- 50.McDougle CJ, Erickson CA, Stigler KA, Posey DJ. Neurochemistry in the pathophysiology of autism. Journal of Clinical Psychiatry. 2005;66(10):9–18. [PubMed] [Google Scholar]
- 51.Bonnin A, Levitt P. Placental source for 5-HT that tunes fetal brain development. Neuropsychopharmacology. 2012;37(1):299–300. doi: 10.1038/npp.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonnin A, Goeden N, Chen K, et al. A transient placental source of serotonin for the fetal forebrain. Nature. 2011;472(7343):347–350. doi: 10.1038/nature09972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West L, Brunssen SH, Waldrop J. Review of the evidence for treatment of children with autism with selective serotonin reuptake inhibitors. Journal for Specialists in Pediatric Nursing. 2009;14(3):183–191. doi: 10.1111/j.1744-6155.2009.00196.x. [DOI] [PubMed] [Google Scholar]
- 54.Yang M, Silverman JL, Crawley JN. Automated three-chambered social approach task for mice. Current Protocols in Neuroscience. 2011;(supplement 56) doi: 10.1002/0471142301.ns0826s56.8.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fischer J, Hammerschmidt K. Ultrasonic vocalizations in mouse models for speech and socio-cognitive disorders: insights into the evolution of vocal communication. Genes, Brain and Behavior. 2011;10(1):17–27. doi: 10.1111/j.1601-183X.2010.00610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nature Reviews Neuroscience. 2010;11(7):490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roullet FI, Crawley JN. Mouse models of autism: testing hypotheses about molecular mechanisms. Current Topics in Behavioral Neurosciences. 2011;7:187–212. doi: 10.1007/7854_2010_113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Green SA, Ben-Sasson A, Soto TW, Carter AS. Anxiety and sensory over-responsivity in toddlers with Autism spectrum disorders: bidirectional effects across time. Journal of Autism and Developmental Disorders. 2011;42(6):1112–1119. doi: 10.1007/s10803-011-1361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patterson PH. Modeling autistic features in animals. Pediatric Research. 2011;69(5):34R–40R. doi: 10.1203/PDR.0b013e318212b80f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. New England Journal of Medicine. 2008;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 61.Kumar RA, Marshall CR, Badner JA, et al. Association and mutation analyses of 16p11.2 autism candidate genes. PLoS ONE. 2009;4(2):p. e4582. doi: 10.1371/journal.pone.0004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in Autism spectrum disorder. American Journal of Human Genetics. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walsh KM, Bracken MB. Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genetics in Medicine. 2011;13(5):377–384. doi: 10.1097/GIM.0b013e3182076c0c. [DOI] [PubMed] [Google Scholar]
- 64.Bachmann-Gagescu R, Mefford HC, Cowan C, et al. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genetics in Medicine. 2010;12(10):641–647. doi: 10.1097/GIM.0b013e3181ef4286. [DOI] [PubMed] [Google Scholar]
- 65.McCarthy SE, Makarov V, Kirov G, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nature Genetics. 2009;41(11):1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernandez BA, Roberts W, Chung B, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. Journal of Medical Genetics. 2010;47(3):195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- 67.Shinawi M, Liu P, Kang SHL, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. Journal of Medical Genetics. 2010;47(5):332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scambler PJ. The 22q11 deletion syndromes. Human Molecular Genetics. 2000;9(16):2421–2426. doi: 10.1093/hmg/9.16.2421. [DOI] [PubMed] [Google Scholar]
- 69.Shprintzen RJ, Goldberg RB, Lewin ML. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate Journal. 1978;15(1):56–62. [PubMed] [Google Scholar]
- 70.Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. Journal of Medical Genetics. 1997;34(10):798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. The Lancet. 2007;370(9596):1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 72.Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nature Reviews Neuroscience. 2010;11(6):402–416. doi: 10.1038/nrn2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karayiorgou M, Morris MA, Morrow B, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(17):7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karayiorgou M, Gogos JA, Galke BL, et al. Genotype and phenotype analysis at the 22q11 schizophrenia susceptibility locus. Cold Spring Harbor Symposia on Quantitative Biology. 1996;61:835–843. [PubMed] [Google Scholar]
- 75.Xu B, Roos JL, Levy S, Van Rensburg EJ, Gogos JA, Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nature Genetics. 2008;40(7):880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 76.Drew LJ, Crabtree GW, Markx S, et al. The 22q11.2 microdeletion: fifteen years of insights into the genetic and neural complexity of psychiatric disorders. International Journal of Developmental Neuroscience. 2011;29(3):259–281. doi: 10.1016/j.ijdevneu.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eliez S, Schmitt JE, White CD, Reiss AL. Children and adolescents with velocardiofacial syndrome: a volumetric MRI study. American Journal of Psychiatry. 2000;157(3):409–415. doi: 10.1176/appi.ajp.157.3.409. [DOI] [PubMed] [Google Scholar]
- 78.Tan GM, Arnone D, McIntosh AM, Ebmeier KP. Meta-analysis of magnetic resonance imaging studies in chromosome 22q11.2 deletion syndrome (velocardiofacial syndrome) Schizophrenia Research. 2009;115(2-3):173–181. doi: 10.1016/j.schres.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 79.Van Amelsvoort T, Daly E, Robertson D, et al. Structural brain abnormalities associated with deletion at chromosome 22q11: quantitative neuroimaging study of adults with velo-cardio-facial syndrome. British Journal of Psychiatry. 2001;178:412–419. doi: 10.1192/bjp.178.5.412. [DOI] [PubMed] [Google Scholar]
- 80.Bearden CE, Van Erp TGM, Dutton RA, et al. Alterations in midline cortical thickness and gyrification patterns mapped in children with 22q11.2 deletions. Cerebral Cortex. 2009;19(1):115–126. doi: 10.1093/cercor/bhn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bearden CE, Van Erp TGM, Dutton RA, et al. Mapping cortical thickness in children with 22q11.2 deletions. Cerebral Cortex. 2007;17(8):1889–1898. doi: 10.1093/cercor/bhl097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shashi V, Kwapil TR, Kaczorowski J, et al. Evidence of gray matter reduction and dysfunction in chromosome 22q11.2 deletion syndrome. Psychiatry Research. 2010;181(1):1–8. doi: 10.1016/j.pscychresns.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bish JP, Pendyal A, Ding L, et al. Specific cerebellar reductions in children with chromosome 22q11.2 deletion syndrome. Neuroscience Letters. 2006;399(3):245–248. doi: 10.1016/j.neulet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 84.Kates WR, Miller AM, AbdulSabur N, et al. Temporal lobe anatomy and psychiatric symptoms in velocardiofacial syndrome (22q11.2 deletion syndrome) Journal of the American Academy of Child and Adolescent Psychiatry. 2006;45(5):587–595. doi: 10.1097/01.chi.0000205704.33077.4a. [DOI] [PubMed] [Google Scholar]
- 85.Gothelf D, Schaer M, Eliez S. Genes, brain development and psychiatric phenotypes in velo-cardio-facial syndrome. Developmental Disabilities Research Reviews. 2008;14(1):59–68. doi: 10.1002/ddrr.9. [DOI] [PubMed] [Google Scholar]
- 86.Gothelf D, Eliez S, Thompson T, et al. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nature Neuroscience. 2005;8(11):1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- 87.Carlson C, Sirotkin H, Pandita R, et al. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. American Journal of Human Genetics. 1997;61(3):620–629. doi: 10.1086/515508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Merscher S, Funke B, Epstein JA, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 89.Stark KL, Burt RA, Gogos JA, Karayiorgou M. Analysis of prepulse inhibition in mouse lines overexpressing 22q11.2 orthologues. International Journal of Neuropsychopharmacology. 2009;12(7):983–989. doi: 10.1017/S1461145709000492. [DOI] [PubMed] [Google Scholar]
- 90.Long JM, LaPorte P, Merscher S, et al. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7(4):247–257. doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 91.Paylor R, McIlwain KL, McAninch R, et al. Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Human Molecular Genetics. 2001;10(23):2645–2650. doi: 10.1093/hmg/10.23.2645. [DOI] [PubMed] [Google Scholar]
- 92.Paylor R, Glaser B, Mupo A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(20):7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(38):16434–16445. doi: 10.1073/pnas.0905696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mukai J, Dhilla A, Drew LJ, et al. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nature Neuroscience. 2008;11(11):1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ruf S, Symmons O, Uslu VV, et al. Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nature Genetics. 2011;43(4):379–387. doi: 10.1038/ng.790. [DOI] [PubMed] [Google Scholar]
- 96.Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485(7398):363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]