

Abstract
Ulcerative colitis (UC) is debilitating and carries a high colon cancer risk. Apoptosis of inflammatory cells is a key mechanism regulating UC. We have recently shown that American ginseng (AG), and to a greater extent, a Hexane fraction of AG (HAG) can cause apoptosis and suppress mouse colitis through a p53-mediated mechanism. Here, we tested the hypothesis that HAG suppresses colitis through a p53 mechanism. We found only a limited impact of p53 in the ability of HAG to induce inflammatory cell apoptosis and suppress mouse colitis in vitro and in vivo. Finally, we asked whether HAG could cause cell cycle arrest of HCT116 colon cancer cells in vitro. Interestingly, HAG caused a G1 arrest of such cells independent of p53 status. Findings are significant because HAG suppresses colitis and associated colon cancer, and mutation in p53 is observed in most colitis-driven colon cancers. Therefore, HAG might be very effective in targeting the inflammatory cells and cancer cells since it induces apoptosis of inflammatory cells and cell cycle arrest in both p53−/− and WT p53 colon cancer cells.
1. Introduction
Inflammatory Bowel Disease (IBD) is considered an autoimmune disease that causes chronic inflammation in the gastro-intestinal tract. Ulcerative colitis (UC) is one of the IBDs' reflecting chronic relapsing inflammatory disorders of the intestine [1]. Among the cell types, epithelial cells, myeloid innate cells, effector T cells, regulatory cells, and B cells have been implicated in IBD pathogenesis [2]. Excessive effector T-cell cytokine secretion or defective regulatory T cells lead to the disease propagation of IBD [2]. The intestinal barrier performs two important tasks to keep the balance between health and disease. First, it must mediate effective absorption of fluids, nutrients, and minerals from the lumen across the epithelium and into the microcirculation and microvilli [3]. Second, the barrier must be impermeable to prevent the transfer of potentially pathogenic microbes and infectious agents [3]. Dysfunction in this intestinal barrier leads to the dysfunction in the intestinal immune system, which has been implicated as the major mechanism by which chronic inflammation occurs in colitis [4]. Hence a common feature of IBD pathogenesis is the dysregulated effector T cell response to commensal microbiota. Because the intestinal mucosa is constantly exposed to several antigens, the mucosal immune system have evolved several strategies to avoid an unnecessary and uncontrolled inflammatory reaction. Once the antigen from the commensal microbiota has been eradicated, T lymphocytes of the intestinal mucosa require a method to attenuate the local immune response [4]. One of the regulatory methods, downregulation of activated T lymphocytes via apoptosis, is a very potent and effective strategy, now considered as a key controlling mechanism of IBD [5]. Failure to regulate T-cell responses in the intestinal or colonic mucosa leads to an inappropriate and sustained injurious immunologic reaction [6, 7]. Because of their heightened activation and activity in IBD pathogenesis, effector T cells are considered an excellent target for therapeutics.
Cells undergoing apoptosis are characterized by the shrinkage and condensation of their cytoplasm, increased mitochondrial permeability, chromatin condensation into caps at the edge of the nucleus, DNA fragmentation, and the appearance of plasma membrane blebs, often referred to as “apoptotic bodies” [8]. There are two signaling pathways for the cell death by apoptosis: the intrinsic and extrinsic pathways [9, 10]. Activation of the extrinsic pathway occurs by ligand-induced cell surface receptor (e.g., tumor necrosis factor receptor 1 TNFR1, Fas, and death receptor 5) activation [11]. The intrinsic pathway is activated with growth factor deprivation, oncogene activation, or when DNA damage is detected by cellular sensors such as ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), and tumor protein 53 (p53) [11]. Inflammation-induced reactive oxygen species and nitric oxide lead to p53 stabilization and accumulation [12, 13]. This activates p53 to eliminate the damaged cells by apoptosis [12, 13]. This is an intrinsic approach for the apoptosis of damaged cells, where activated p53 plays an important role in mediating apoptosis in epithelial and inflammatory cells during the process of colitis [4, 12, 14–17]. The post-translational modification (often phosphorylation at Serine 15) tends to elevate the level of wild-type (WT) p53 during inflammation [12]. p53 plays at least 2 separate roles in the responses to therapeutic agents It is an important component of cellular checkpoints (cell cycle arrest), and it can mediate apoptosis [18]. The role of p53 in the responses of tumor cells to therapy is controversial. For example, loss of p53 function can cause resistance to 5-fluorouracil (5-FU), but increased sensitivity to DNA damaging agent such as Adriamycin [18]. This provides a clear indication that some drugs may exert an apoptosis-inducing effect through a p53-dependent while other drugs effect through a p53-independent pathway.
Cancer is one of the scenarios where too little apoptosis occurs, resulting in malignant cells that will not die and continue to proliferate. Four cellular functions are inappropriately regulated in the cancer cells: cellular proliferation, differentiation, chromosomal and genetic organization, and apoptosis (Reviewed in [19]). Abnormal cell proliferation leading to accumulation of clonal cells is seen in cancer. As mentioned this could be due to the defects with the cell cycle control. Upon sensing DNA damage, p53 is activated, resulting in either a G1 cell cycle arrest [20, 21] or apoptosis [22, 23]. Cells undergo a G1 cell cycle arrest to allow the DNA repair before replication and if the DNA damage is beyond repair, the cells tend to undergo apoptosis. Cells can also undergo a G1 arrest due to targeting cyclins and cyclin-dependent kinases. p53 mutation is observed in most cancers [24], which often tend to relax this G1-S cell cycle transition because p53 could not be activated. Hence, cancer cells lack the appropriate G1-S checkpoint regulation and controlled apoptosis.
American ginseng (AG, Panax quinquefolius) is grown in the eastern temperate forest areas of North America, from British Columbia, southern Quebec, Ontario, Minnesota, and Wisconsin in the north, to Oklahoma, the Ozark Plateau, and Georgia in the south [25]. AG is an obligate shade perennial native of North America and its root is the commonly used component. AG has been reported to have a wide range of pharmacological effects, including effects on the central nervous system, blood-sugar levels, cardiovascular system, endocrine system, immune system, and cancer [4, 26, 27]. Recently we have shown AG extract suppresses colitis by the accumulation and activation of p53 to induce apoptosis of inflammatory cells [4]. To further delineate the active anti-inflammatory and proapoptotic components present in the AG, we sub-fractionated a different fraction of AG. A Hexane Fraction of AG (HAG) showed increased anti-inflammatory and pro-apoptotic properties in the chemically induced mouse model of colitis [28]. The elevation of WT p53 levels during inflammation [12] resulting in apoptosis of inflammatory and damaged cells [4, 29, 30] led us to the notion that the active anti-inflammatory components present in HAG might suppress colitis through the p53 pathway. Here, we tested this hypothesis.
2. Material and Methods
2.1. Bioassay-Guided Fractionation of Hexane Fraction of AG (HAG)
The P. quinquefolius extract has been described previously in detail by our laboratory [16]. As well, we have recently described the generation of the HAG used in the present study [28].
2.2. Chemicals and Reagents
Dextran sulfate sodium (DSS) was purchased from MP Biomedicals (Solon, OH: molecular weight, 36,000–50,000).
2.3. Cell Culture and Treatment
TK6 (p53+/+) and NH32 (p53−/−) cell lines were a kind gift from Curtis Harris (National Cancer Institute), originally derived from Dr. William Thilly's and Howard Liber's labs. TK6 cells are a lymphoblastoid cell line derived from the spleen more than 30 years ago [31]. NH32 cells are an isogenic derivative of TK6 cells in which both alleles of the p53 gene were knocked out [32]. Jurkat T cells are an immortalized line of T lymphocyte cells derived in the late 1970s from the peripheral blood of a 14-year-old male with T-cell leukemia [33]. Jurkat T cells have a defective p53 pathway due to a mutation in the COOH-terminal domain responsible for transactivation [34, 35]. TK6, NH32, and Jurkat T cells were maintained in exponentially growing suspension culture at 37°C in a humidified 5% CO2 atmosphere in RPMI 1640 supplemented with 10% heat-inactivated calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine.
CD4+/CD25− cells from C57BL/6 mice were purified from the spleens using nylon wool columns (Polysciences, Warrington, PA, USA) followed by depletion of B cells and macrophages. CD4+/CD25− T cells were then isolated using an MACS miniseparator and CD4 and CD25 microbeads according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA, USA) by depletion of CD4−CD25+ T cells (negative selection). The purity of T cells was 88.6% (Supplementary Figure 1 available online at doi:10.1155/2012/785739) as determined by flow cytometry (Cytomics FC 500, Beckman Coulter, Brea, CA, USA). Briefly, isolated CD4+/CD25− T cells were washed with PBS and 2 × 105 cells were re-suspended with 100 μL of PBS and incubated with anti-mouse CD4 antibody conjugated to Allophycocyanin (APC) (Clone: GK1.5; Isotype: rat IgG2b, kappa, eBioscience, San Diego, CA, USA) (0.125 μg/100 μL of cell suspension). The anti-mouse CD25 antibody conjugated to R-Phycoerythrin (PE) (Clone: 7D4; Isotype: rat IgM, Miltenyi Biotec, Auburn, CA, USA) was incubated during the isolation of CD4+/CD25− effector T cells according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA, USA). The purity of CD4+ T cells was determined by obtaining a dot plot of FL-4 (APC) versus FL-2 (PE) and individual histogram plots of FL-4 and FL-2 versus the number of events (Supplementary Figure 1). The isogenic control was the nonantibody treated isolated effector T cells.
Isolated CD4+/CD25− effector T cells (1 × 106) were cultured in six-well plates overnight followed by experimentation as indicated. All cells were maintained in exponentially growing suspension culture at 37°C in a humidified, 5% CO2 atmosphere in RPMI 1640 supplemented with 10% heat-inactivated calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine.
HCT-116, human epithelial colon cancer cells line, proficient in p53, and isogenic HCT-116 p53−/− cells were maintained in exponentially growing adherent culture at 37°C in a humidified 5% CO2 atmosphere in RPMI 1640 supplemented with 10% heat-inactivated calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine.
2.4. DSS Mouse Model of Colitis
We followed our previous protocol for our DSS (MP Biomedicals, Solon, OH: 36,000–50,000 mw) mouse model of colitis [16]. Briefly (Figure 1), 8–10-week-old C57BL/6 mice received either water ad libitum or 1% DSS. All mice were on an AIN93 M diet as described previously [36]. 11.9 mg/kg of HAG was dissolved in 100 μL 1 × PBS per mouse and administered daily by oral gavage (per os, PO). 11.9 mg/kg daily is the human equivalent dose of 58 mg daily [37]. Of note, currently the use of ginseng in human clinical trials can range anywhere from 200 mg to 9 g daily [38, 39]. The control group of mice was given 100 μL of 1 × PBS by oral gavage. All procedures performed were in accordance with the Guide for the care and Use of Laboratory Animals (National Research Council, Washington, DC, USA) and approved by the Animal Resource Facility, University of South Carolina, Institutional Animal Care and Use Committee. To determine whether the HAG can prevent the onset of colitis, mice were fed 1% DSS for 2.5 cycles (7 days DSS, 7 days water making 1 cycle). The vehicle or the HAG was administered daily by oral gavage 7 days prior to the first DSS exposure and continued throughout the course of the experiment. Mice were euthanized at 2.5 cycles (Figure 1). For pathology, colon tissue samples were washed with phosphate-buffered saline (PBS; Mediatech, Herndon, VA, USA), cut longitudinally, swissrolled, then formalin-fixed overnight, and paraffin-embedded.
Figure 1.
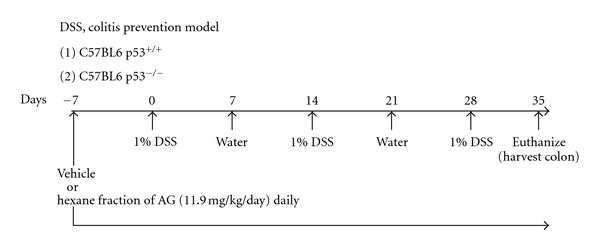
Experimental protocol for the DSS, prevention mouse model of colitis. 11.9 mg/kg/day of HAG or vehicle (1 × PBS) was given to the respective group of mice by oral gavage 7 days prior to the first DSS cycle and continued daily until the end of the experiment (2.5 cycles). The mice (both C57/BL6 p53−/− and C57/BL6 p53+/+) were euthanized on day 35.
2.5. Quantification of Inflammation to Examine Effects on Colitis
Paraffin-embedded tissues were serially sectioned, and one section from each mouse was stained with H&E. Sections were microscopically examined by two blinded investigators (D.P and X.C) for histopathologic changes using a scoring system previously used and validated by ourselves and others [4, 28, 40, 41]. Histology score was determined by multiplying the percent involvement for each of the three following histologic features by the percent area of involvement [4]: inflammation severity (0, none; 1, minimal; 2, moderate; 3, severe), inflammation extent (0, none; 1, mucosa; 2, mucosa and submucosa; 3, transmural), and extent of crypt damage (0, none; 1, one third of crypt damaged; 2, two thirds of crypt damaged; 3, crypts lost, surface epithelium intact; 4, crypts lost, surface epithelium lost). Percent area involvement was defined as: 0, 0%; 1, 1–25%; 2, 26–50%; 3, 51–75%; 4, 76–100%. Therefore, the minimal score is 0 and the maximal score is 40. Since, DSS-induced colitis in mice leads to the damage in colonic epithelial barrier and is characterized by extent and depth of inflammation, a grading or scoring system of inflammation with all these parameters provides an excellent measure of histologic assessment of DSS-induced colitis.
2.6. Western Blot Analysis and Antibodies
Western blots were carried out as described previously [42]. Antibodies used include: p53 (mouse monoclonal, DO-1, diluted 1 in 500, cat# OP43T; Calbiochem, Gibbstown, NJ, USA) and GAPDH (Rabbit monoclonal, diluted 1 in 1000, cat# 5174 P; Cell Signaling Technology, Danvers, MA, USA). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ, USA). Both secondary antibodies were diluted at 1 : 2000. All antibodies were diluted in 5% milk/PBST (0.1% Tween 20 in 1 × PBS). Western blot signal was detected by Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA) and developed onto Hyperfilm (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Briefly, after treating blot with the chemiluminescent substrate (Pierce ECL) for a minute, the blot was exposed to the hyperfilm in the dark (Exposure time was optimized based on the band signal obtained) and the film was developed in an automatic X-ray film processor (Futura Classic E automatic X-ray film processor, Fisher Industry, Geneva, IL, USA).
2.7. Annexin V Assay
CD4+/CD25− effector T cells were seeded at 1 × 106 per well into six-well dishes for 24 h in triplicate (n = 3). Fresh medium or fresh medium containing freshly dissolved indicated concentrations of HAG (0–300 μg/mL) was added for 24 h, as indicated. Cells were then harvested for Annexin V according to instructions provided by the kit manufacturer (BD Biosciences, San Diego, CA, USA). Annexin V/propidium iodide (PI) staining was examined using a Beckman Coulter Cytomics FC500 flow cytometer.
2.8. Cell Cycle Analysis
1 × 106 cells/wells of HCT-116 WT and HCT-116 p53−/− cells were incubated in 1.0% NBCS supplemented RPMI-1640 media for 24 hrs in 6-well culture plate. The media was changed and the cells were treated with Hexane fraction of AG (0–500 μg/mL). The cells were harvested after 24 hrs of treatment and cell cycle assay was performed by labeling the dsDNA of the cells with DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich, MO, USA). Briefly, the harvested cells were fixed by gently vortexing and adding 70% ethanol dropwise. The fixed cells were incubated at 4°C for at least 30 minutes. The cells were washed with PBS/1% BSA and stained with 1 μg/mL of DAPI (in PBS/0.1% Triton X-100) for 10 min. at room temperature. 20,000 cells/events were directly analyzed by BD-LSR-II flow-cytometer (BD Biosciences, San Jose, CA, USA). Based on the DNA content, the different phases of the cell cycle was determined by using BD FACSDiva software (BD Biosciences, San Jose, CA, USA). Experiments were repeated three times.
2.9. Flow-Cytometric TUNEL (Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling) Assay
TK6 (p53 WT), NH32 (isogenic p53−/−), and Jurkat T (dysfunctional p53) cells were incubated in 0.1% NBCS supplemented RPMI-1640 media for 24 hrs. The media was changed and the cells were treated with Hexane fraction of AG (0–500 μg/mL) as indicated in Figure 2. Cells were harvested after 24 hours of treatment and TUNEL assay was performed as described by vendor (Roche Diagnostics, IN) in triplicate (n = 3). Briefly, 1 × 106 cells were fixed using a 100 μL of fixation solution (2% paraformaldehyde) and permeabilized using a permeabilization solution (0.1% Triton X-100 in 0.1% sodium citrate). Cells were washed and incubated with TUNEL reaction mixture (label solution and enzyme solution) (Roche Diagnostics, IN, USA). Apoptosis in the samples were analyzed by flow cytometry (Beckman Coulter, CA, USA). The fluorescence was evaluated using the excitation wavelength of 488 nm and detected in the range of 515–565 nm (green, FL-1 channel). The dot plot of FS Versus FL-1 and histogram plot of (number of event) Versus (FL-1 channel) were plotted to obtain a percentage increase in the apoptosis of the Hexane fraction of AG treated cells. Positive control for apoptosis is the fixed and permeabilized cells treated with DNase I recombinant (3 U/mL in 50 mM Tris-HCL, pH7.5, and 1 mg/mL BSA) (Invitrogen, CA, USA) to induce DNA strand breaks prior to labeling (following the vendors protocol). Negative control for apoptosis is the nontreated, healthy cells. Isogenic enzyme control is the fixed and permeabilized cells with the labeling solution but without the terminal transferase enzyme.
Figure 2.
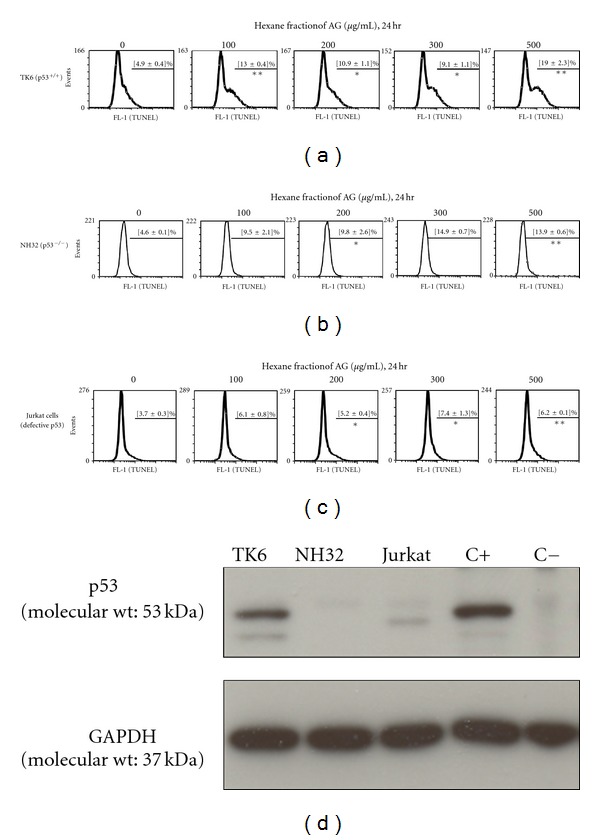
The Hexane fraction of AG (HAG) drives apoptosis (TUNEL+) of lymphoblasts marginally better in p53+/+ cells compared with p53−/− cells. The TK6 human lymphoblastoid line, NH32 isogenic p53 knockout cells, and Jurkat T cells (dysfunctional p53 pathway) were cultured in RPMI-1640 + 10% NBCS. Cells were exposed to HAG (in triplicate, n = 3 per treatment) in RPMI-1640 + 0.1% NBCS for 24 hr at indicated doses and harvested for TUNEL apoptotic assay. The percentage of the cells (Mean ± S.E.) staining positive for TUNEL, indicating apoptosis are given (n = 3): (a) TK6 cells; (b) NH32 cells; (c) Jurkat T cells. A minimum of 20,000 events was counted by flow cytometry from each treatment. (d) Confirmation of p53 protein status of the TK6, NH32, and Jurkat T cells. Cell lysates were analyzed by western blot analysis. C+ indicates the positive control for p53, which is an archived HCT-116 cell lysate with wild-type p53. C−, indicates the negative control for p53, which is an archived HCT-116 p53−/− cell lysate with p53 knockout. Results indicate HAG induces apoptosis in lymphoblasts through a limited p53 activity. Significant differences are indicated, where *P-value <0.05 and **P-value <0.005, when compared to the control (0 μg/mL) treatment.
2.10. Statistical Analysis
Statistical analysis was done using one-way ANOVA with Scheffe's post hoc test for comparison of endpoint data between mouse groups, as well as in vitro endpoints. The results were analyzed using the Stat-view II statistical program (Abacus Concepts, Inc., Piscataway, NJ, USA) and Microsoft Excel (Microsoft, Bellevue, WA, USA) for Macintosh computers. The P value chosen for significance in this study was 0.05.
3. Results
In UC, intestinal immune responses are often characterized by activation of lamina propria T lymphocytes (LPL) with potent effector functions [43]. Among its regulatory mechanisms, down-regulation of activated T lymphocytes via apoptosis is a very potent and effective strategy, now considered as a key controlling mechanism of UC [5]. To this end, lymphoblastoid cell lines TK6 (WT p53), NH32 (isogenic to TK6, but p53−/−), and Jurkat T cells (which have a dysfunctional p53 pathway) were tested for the apoptosis inducing property of HAG. Previously, we showed the whole AG extract induces apoptosis of TK6, but not NH32 cells [4]. Interestingly, here, we show HAG was able to induce apoptosis (TUNEL+) not only in TK6 cells but (albeit to a lesser extent) also in NH32 cells (Figures 2(a) and 2(b)). HAG induced a 3.9-fold increase in apoptosis of TK6 cells at 500 μg/mL of HAG when compared to the untreated cells. The isogenic p53−/− NH32 cells underwent apoptosis upto 3-fold at 500 μg/mL of HAG when compared to the non-treated cells (Figures 2(a) and 2(b)). Also, at 100 μg/mL, the HAG induced 2.6-fold increase in apoptosis in the TK6 cells, but a 2-fold increase in apoptosis in the NH32 cells. Interestingly, the Jurkat T-cell line (which has a defective p53 pathway) was somewhat resistant to HAG-induced apoptosis (Figure 2(c)), which is consistent with what we have previously observed with the whole AG extract [4]. Figure 2(d) provides confirmation of the p53 status of each of the cell lines used in these experiments.
Overly aggressive CD4+/CD25− T cells are thought to contribute to colitis, and defects in mucosal T-cell apoptosis are likely to be critical in the pathogenesis of colitis [4, 6, 7]. Recently we have shown HAG induces apoptosis of CD4+/CD25− T cells derived from C57/BL6 WT mice during the suppression of colitis [28]. In the present paper, consistent with results from TK6 (p53+/+) and NH32 (p53−/−) cells, HAG induced apoptosis of the CD4+/CD25− T cells isolated from both WT and p53−/− C57/BL6 mice (Table 1). However, the induction of apoptosis was suppressed in the absence of p53. There was 2.6-fold increase From (5.1 ± 0.9%) to (13.2 ± 2.8%) in apoptosis (mean ± S.E.) of effector T cells isolated from p53−/− C57/BL6 mice and a 4.6-fold increase From (4.5 ± 0.5%) to (20.7 ± 8.9%) in the apoptosis was observed in CD4+/CD25− T cells isolated from WT C57/BL6 mice upon treatment with increasing concentration of HAG (0–300 μg/mL).
Table 1.
Apoptosis (Annexin V positive; propidium iodide negative) of CD4+/CD25− effector T cells from the spleen of p53−/− mice and p53+/+ mice by the increasing concentration of Hexane fraction of AG for 24 hrs.
| Dose: hexane fraction of AG (24 hr) | Percentage of early apoptotic cells | |
|---|---|---|
| C57/BL6 p53−/− Mice | C57/BL6 p53+/+ Mice | |
| 0 μg/mL | 4.1% | 4.9% |
| 100 μg/mL | 5.9% | 9.8% |
| 200 μg/mL | 9.1% | 15.7% |
| 300 μg/mL | 18.6% | 38.5% |
We have recently shown that HAG suppresses chemically (DSS mouse model) induced colitis in the WT p53 C57/BL6 mice [28]. Previously we have also shown that AG whole extract suppresses chemically induced colitis only in the WT p53 C57/BL6 mice and not in the p53−/− C57/BL6 mice [4]. This led us to the conclusion that the suppression of colitis by AG extract is p53 dependent. We tested the same hypothesis for the HAG. As shown in the Figure 3, the HAG was able to suppress the DSS-induced colitis in both the p53−/− and WT p53 mice. However, consistent with our results on apoptosis of inflammatory cells in vitro, the suppression was more prominent in WT mice than in p53−/− mice. The H&E stained swissrolled colon inflammation score of WT C57/BL6 mice was reduced from 15 ± 0.86 to 4.4 ± 0.32 (average ± S.E.) (a reduction of 71%) in the HAG-treated mice (Figure 3). Similar results were obtained with the p53−/− C57/BL6 mice, where the score dropped from 14.5 ± 2.12 to 7.8 ± 1.78 (a reduction of 46%) in the HAG-treated mice. This result is consistent with the notion that the HAG seems to act not only through a p53 pathway but also through a p53-independent pathway while suppressing colitis.
Figure 3.
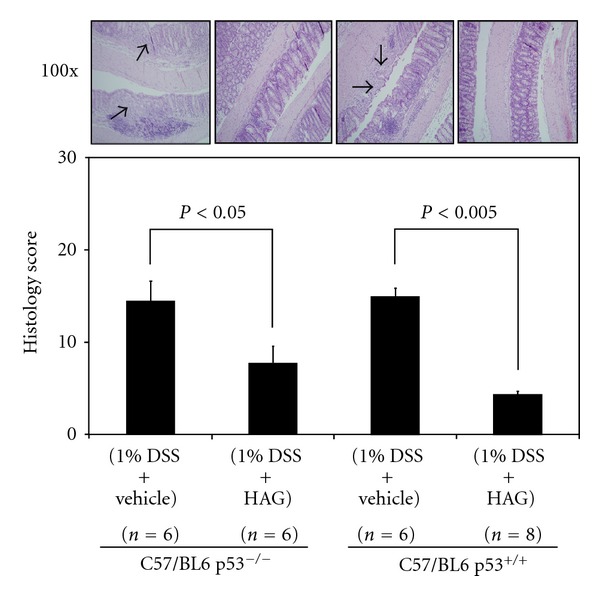
Effect of the HAG on the colon histology score of the acute DSS colitis model. Histological Inflammation score was determined from the H&E stained colon of each group of mice treated with HAG after 2.5 cycles of DSS. Values represent mean ± S.E. Representative H&E stained colon (100x magnified) is shown for each group. Arrows point to areas of inflammation and ulceration. Significant differences are indicated.
Recently we have shown that HAG reduces colon cancer associated with colitis in mice [28]. The mitogenic stimuli triggered signal transduction pathways eventually converge on the cell cycle checkpoint that controls the G1 to S phase transition and activate appropriate cyclin-dependent kinases [19]. This anomaly increases proliferation of the mutated cells to increase the cancer growth and progression. Thus, one way to cause cancer to subside is to prevent this abnormal cancer-cell growth by inducing cell cycle arrest. We tested this hypothesis in vitro by using two different cell lines, HCT-116 p53 WT and HCT-116 p53−/−, and treated these cells with increasing concentrations of HAG for 24 hr. In three repeated experiments, HAG was able to increase a G1 cell cycle arrest in both the WT and p53−/− HCT-116 cells. Representative results are given in Figure 4(a). HAG increased the G1 cell cycle arrest of HCT-116 p53 WT cells 1.7-fold (34% to 56.7%) at 100 μg/mL, HAG treatment and the percentage of cells in G1 remained above 50% with the other concentrations (up to 500 μg/mL) (Figure 4(a)). Similarly there was a 1.5-fold increase (34.1% to 51.2%) of HCT-116 p53−/− cells in G1 phase after treatment with 100 μg/mL of HAG (Figure 4(a)), and this checkpoint remaining with the remaining concentrations. Figure 4(b) provides confirmation of the p53 status of HCT-116 cells used in this experiment.
Figure 4.
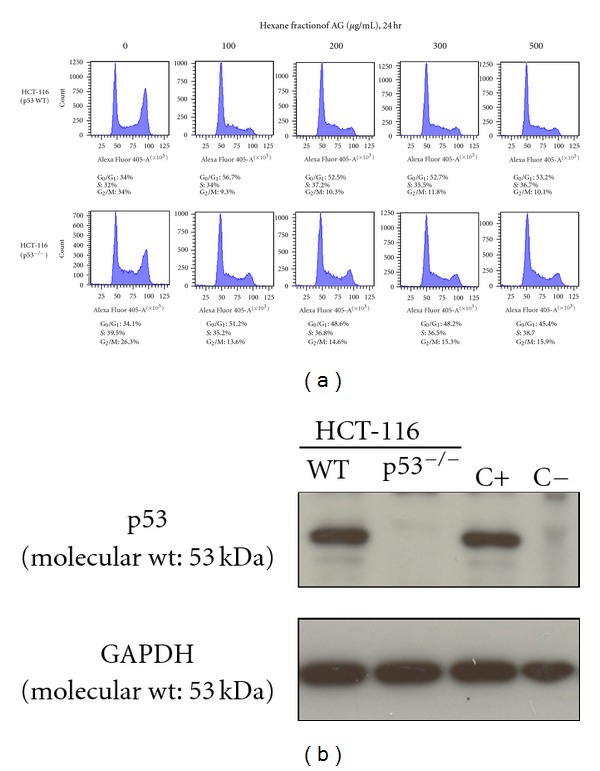
The HAG induces a G1 checkpoint in colon cancer cells in both p53+/+ and p53−/− colon cancer cells. (a) HCT-116 WT and p53−/− cells were exposed to HAG in RPMI-1640 + 1% NBCS for 24 hr at indicated doses and harvested for cell cycle assay with DAPI stain. Representative percentage of cells in G1, S, and G2/M-phases are indicated for each treatment. A minimum of 20,000 events was counted by flow cytometry from each treatment. (b) Confirmation of p53 protein status of the HCT-116 cells. Cell lysates were analyzed by western blot analysis. C+ indicates the positive control for p53, which is an archived HCT-116 cell lysate with wild-type p53. C− indicates the negative control for p53, which is an archived HCT-116 p53−/− cell lysate with p53 knockout.
4. Discussion
Our HAG is extracted from AG whole extract by non-polar solvent n-Hexane. Mostly polyacetylenes (panaxynol, panaxydol, and panaxydiol) and fatty acids (FA) present in the AG whole extract were extracted in this fraction [28]. Since HAG is more effective at suppressing colitis than the whole AG extract [28] and based on our previous study, where AG whole extract suppressed colitis through p53-mediated apoptosis of inflammatory cells [4], we hypothesized that HAG also induces apoptosis of inflammatory cells through a p53-mediated mechanism. We pursued our study with HAG and its apoptotic properties with different inflammatory cells. Interestingly, we found that, unlike the whole AG extract, HAG was able to induce modest apoptosis of p53−/− CD4+/CD25− effector T cells and p53−/− lymphoblastoid cells in addition to their p53 WT counterpart cells (Figure 2, Table 1). This indicates that HAG mechanistically acts differently than the AG whole extract, where it shows a modest involvement of p53-dependent apoptosis. This still raises the question, what is the most active component present in the HAG? Recently we have identified the different components present in the HAG, with FAs comprising 43% w/w, polyacetylenes 26.52% w/w (7.39% Panaxydiol, 8.92% Panaxydol, and 10.21% Panaxynol), and less than 0.1% w/w ginsenosides [28]. This indicates that specific FA ingredients or specific polyacetylenes, or both are responsible for the apoptotic property of HAG through both p53-dependent and p53-independent mechanisms.
Interestingly, Wong et al. have shown that Asian ginseng extracted with ethanol induces a G2-M arrest and apoptosis via modulation of MAPK and p53 pathway in LLC-1 cells (Mouse Lewis lung carcinoma cells) [44]. The AG whole extract is also extracted with ethanol and the ginsenoside composition is for the most part, similar in Asian and American ginseng. Separate studies from Kim et al. [45, 46] have shown that the ginsenosides-Rs3 and -Rs4 selectively elevate protein levels of p53 and p21WAF1, and downregulate the activities of the cyclin-dependent kinases, resulting in cell cycle arrest at the G1/S boundary and apoptosis of SK-HEP-1 cells (immortalized human hepatoma cells) [45, 46]. All these observations suggest that ginsenosides may be a key player in modulating apoptosis and cell cycle through the p53 pathway and are consistent with the findings here and our previous findings that our whole AG extract has 10.1% ginsenoside (w/w) compared to the HAG which has 0.074% ginsenoside (w/w) content [28].
Interestingly, ginsenoside Rd, which is present in AG whole extract (extracted with aqueous ethanol) and absent in the HAG, has been shown to attenuate the inflammatory response to TNBS− (2,4,6-trinotrobenzenesulfonic acid-) induced colitis by down-regulating multiple pro-inflammatory cytokines through the modulation of JNK (c-Jun N-terminal kinase) and p38 activation [47]. Similarly another ginsenoside not present in HAG, Rb1, and its metabolite compound K after oral administration blocked TNBS-induced expression of iNOS (inducible-nitric oxide synthase), Cox-2 (cyclooxygenase-2), and NF-κβ (nuclear transcription factor κβ) activation in mice [48]. Ginsenoside Rb1 and its metabolite compound inhibited the activation of key inflammatory mediators IRAK-1 (interleukin-1 receptor associated kinase-1), IKK-β (inhibitor of NF-κβ kinase), NF-κβ, and MAPK (mitogen-activated protein kinases) while suppressing colitis [48]. These studies suggest that different ginsenosides may help attenuate inflammation, and our finding that n-Hexane does not extract these ginsenosides from ginseng should not rule out that ginsenosides are not the active components of ginseng. Taken together, these observations suggest that multiple components of AG, including ginsenosides, FAs, and polyacetylenes, are responsible for the activity of AG in the suppression of colitis in animals. Further studies are necessary to delineate the most active ingredient(s)/molecule(s).
Moon et al. have shown that during the G1/S cell cycle arrest, protein levels of p21WAF1, p16INK4A, p53, pRb (retinoblastoma protein), and E2F-1 (transcription factor E2F1) were not changed after exposure to the polyacetylene, panaxydol (isolated from P. ginseng), in the human malignant melanoma cell line, SK-MEL-1 [49]. Human promyelocytic leukemia HL60 cells do not express the p53 protein due to a large deletion in the gene [50]. The polyacetylenes, panaxynol, and panaxydol (isolated from the lipophilic fractions of Panax notoginseng) have been shown to inhibit the proliferation of HL60 cells in a time− and dose-dependent manner via an apoptotic pathway [51]. These studies indicate that polyacetylenes may act independently of p53 while inducing apoptosis of certain cell lines. HAG-induced apoptosis indicated by cleaved PARP in TK6 cells, appears to change the protein levels of WT p53 and activated form of p53 (phospho-Ser15) very little [28]. After 24 hr of HAG treatment (300 μg/mL), we observed a 2.6-fold increase in apoptosis of p53−/− CD4+/CD25− T cells and a 4.6-fold increase in apoptosis of p53+/+ CD4+/CD25− T cells when compared to untreated cells (Table 1). Figures 2(a), 2(b) and 2(c), and our recent study [28] are consistent and indicate that p53 has a limited role in inducing HAG-mediated apoptosis of inflammatory cells both in vitro and ex vivo. A clearly increased p53, phospho-p53 Ser-15, and cleaved PARP protein levels along with the increased apoptosis were reported with the AG whole extract, suggesting p53 plays a key role in apoptosis of inflammatory cells induced by the whole AG extract [4]. However, an increased cleaved PARP protein expression and only a slight change in the p53, phospho-p53 (Ser-15), and p53-upregulated mediator of Apoptosis (PUMA) protein expression of TK6 cells after treatment with HAG [28] are consistent with our current results that HAG induces apoptosis but this induction of apoptosis is likely mediated through both p53-dependent and p53-independent mechanisms.
The other high volume components present in HAG are FAs, where 19% w/w is linoleic acid, a polyunsaturated fatty acid [28]. Kwon et al. have reported that linoleic acid treatment resulted in a concentration-dependent growth inhibition of AGS cells (human gastric adenocarcinoma cells) by inducing apoptosis in a p53-independent manner with an elevated Fas and Fas ligand expression [52]. This, then, is consistent with our results regarding a limited role for p53 in the cells undergoing apoptosis after treatment with HAG.
We further tested our compound in the in vivo animal study. As indicated (Figure 1), both the p53−/− and p53 WT C57/BL6 mice were subjected to 2.5 cycles of 1% DSS to induce colitis. In this preventive DSS model, 75 ppm (11.9 mg/kg) of HAG or vehicle (1 × PBS) were administered to the mice daily (PO) and continued throughout the course of the experiment. In our previous published studies, AG whole extract was effective in suppressing colitis only in the p53 WT C57/BL6 mice where whole AG extract prevented colon epithelial cells from the DNA damage due to DSS and induced apoptosis of lymphocytes in vivo [4]. In the present study, with the same protocol, the HAG was able to suppress DSS-induced colonic inflammation to approximately one-third (from 15 ± 0.86 to 4.4 ± 0.32). Interestingly, the HAG also showed a modest suppression of inflammation to about one-half (from 14.5 ± 2.12 to 7.8 ± 1.78) in the p53−/− C57/BL6 mice (Figure 3). This provides evidence that the HAG is anti-inflammatory and p53 plays a limited role in the HAG-mediated suppression of colitis.
Apoptosis is an ordered cellular process that occurs in various physiological and pathological conditions. Two major problems are associated with apoptosis; too much apoptosis is associated with various degenerative diseases and too little to no apoptosis is associated with carcinogenesis [53]. Hence one way to tackle the cancerous cells is to induce apoptosis of such cells. Colon cancer is associated with long standing UC and hence inflammation seems to drive the progression of cancer. Interestingly, both HCT-116 p53+/+ and p53−/− colon cancer cells were relatively resistant to apoptosis induced by HAG (data not shown). However, there was a G1 cell cycle checkpoint with HAG treatment (Figure 4(a)). It is unlikely that the HAG causes DNA damage to cause this checkpoint. It is more likely that one or more ingredients in HAG target modulators of the cell cycle (e.g., cyclins or cyclin-dependent kinases). Experiments are ongoing to examine this possibility. Consistent with our findings, Kang et al. have shown that lipid soluble ginseng extract (red ginseng extracted with n-Hexane) induces a cell cycle arrest at the G1 phase in NCI-H460 cells (human lung cancer cells) [54]. These separate observations suggest that the HAG may halt the progression of cell cycle at G1 phase in multiple cancer cell lines.
5. Conclusion
We have shown here that the HAG can induce apoptosis in lymphoblastoid cells and CD4+/CD25− effector T cells and cause a G1 checkpoint in colon cancer cell lines. It also suppressed chemically induced colitis in C57/BL6 mice. In all the above observations, we simultaneously tested the pro-apoptotic and anti-inflammatory properties of HAG with the WT p53 and its p53−/− counterpart cells and mice. It is evident that HAG can perform its pro-apoptotic, anti-inflammatory, and cell cycle arrest activities even in the absence of p53. However, the HAG has a more robust effect in the presence of p53. These observations suggest that there is a role of p53 in the HAG-mediated apoptosis of inflammatory cells and suppression of colitis; but this role is limited. This entails that the HAG affects other pathways independent of p53 in the suppression of colitis. The reasoning for this can only be speculated. For example, is there one component in the whole AG extract that is particularly powerful in inducing p53-mediated apoptosis that is missing from the HAG? Does this component inhibit the ability of components only seen in the HAG fraction to drive p53-independent apoptosis and suppress p53-independent colitis? This is significant because the HAG suppresses colitis and associated colon cancer [28]. Because mutation in p53 is observed in most cancers, including colitis-driven colon cancer, the HAG may be particularly effective in targeting both p53+/+ and p53−/− inflammatory cells and cancer cells and hence particularly effective in inhibiting the colitis-to-cancer sequence.
Disclosure
None of the author of this paper have a relation with the commercial identities used in this paper.
Acknowledgments
This work was supported by the Center for CAM Research on Autoimmune and Inflammatory Diseases, NIH Grant 1P01AT003961-01A1 (P. S. Nagarkatti, L. J. Hofseth, M. Nagarkatti), and the COBRE funded University of South Carolina Center for Colon Cancer Research, NIH Grant P20RR17698-01 (Franklin Berger, Director). Thanks are also due to the P20RR17698-01 Statistical Core (Dr. Edsel Pena, Director), and 1P01AT003961-01A1 Immunotoxicology Core (Dr. Narendra Singh, Director), P20RR17698-01 Mouse Core (Dr. Marj Pena, Director), and P20RR17698-01 Imaging/Histology Core.
References
- 1.Maynard CL, Weaver CT. Intestinal effector T cells in health and disease. Immunity. 2009;31(3):389–400. doi: 10.1016/j.immuni.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eri R, McGuckin MA, Wadley R. T cell transfer model of colitis: a great tool to assess the contribution of T cells in chronic intestinal inflammation. Methods in Molecular Biology. 2012;844:261–275. doi: 10.1007/978-1-61779-527-5_19. [DOI] [PubMed] [Google Scholar]
- 3.Siggers RH, Hackam DJ. The role of innate immune-stimulated epithelial apoptosis during gastrointestinal inflammatory diseases. Cellular and Molecular Life Sciences. 2011;68(22):3623–3634. doi: 10.1007/s00018-011-0821-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin Y, Hofseth AB, Cui X, et al. American ginseng suppresses colitis through p53-mediated apoptosis of inflammatory cells. Cancer Prevention Research. 2010;3(3):339–347. doi: 10.1158/1940-6207.CAPR-09-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine AD. Apoptosis: implications for inflammatory bowel disease. Inflammatory Bowel Diseases. 2000;6(3):191–205. doi: 10.1097/00054725-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nature Clinical Practice Gastroenterology and Hepatology. 2006;3(7):390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 7.Neuman MG. Immune dysfunction in inflammatory bowel disease. Translational Research. 2007;149(4):173–186. doi: 10.1016/j.trsl.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. International Review of Cytology. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 9.Brenner D, Mak TW. Mitochondrial cell death effectors. Current Opinion in Cell Biology. 2009;21(6):871–877. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Özören N, El-Deiry WS. Cell surface death receptor signaling in normal and cancer cells. Seminars in Cancer Biology. 2003;13(2):135–147. doi: 10.1016/s1044-579x(02)00131-1. [DOI] [PubMed] [Google Scholar]
- 11.Dirisina R, Katzman RB, Goretsky T, Managlia E, Mittal N, Williams DB, et al. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology. 2011;141(3):1036–1045. doi: 10.1053/j.gastro.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofseth LJ, Saito S, Hussain SP, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(1):143–148. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nature Reviews Cancer. 2003;3(4):276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 14.Lashner BA, Bauer WM, Rybicki LA, Goldblum JR. Abnormal p53 immunohistochemistry is associated with an increased colorectal cancer-related mortality in patients with ulcerative colitis. The American Journal of Gastroenterology. 2003;98(6):1423–1427. doi: 10.1111/j.1572-0241.2003.07573.x. [DOI] [PubMed] [Google Scholar]
- 15.Alkim C, Savas B, Ensari A, et al. Expression of p53, VEGF, microvessel density, and cyclin-D1 in noncancerous tissue of inflammatory bowel disease. Digestive Diseases and Sciences. 2009;54(9):1979–1984. doi: 10.1007/s10620-008-0554-x. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y, Kotakadi VS, Ying L, et al. American ginseng suppresses inflammation and DNA damage associated with mouse colitis. Carcinogenesis. 2008;29(12):2351–2359. doi: 10.1093/carcin/bgn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kotakadi VS, Jin Y, Hofseth AB, et al. Ginkgo biloba extract EGb 761 has anti-inflammatory properties and ameliorates colitis in mice by driving effector T cell apoptosis. Carcinogenesis. 2008;29(9):1799–1806. doi: 10.1093/carcin/bgn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunz F, Hwang PM, Torrance C, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. The Journal of Clinical Investigation. 1999;104(3):263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreeff M, Goodrich DW, Pardee AB. Cell proliferation, differentiation, and apoptosis. In: Bast RC, Kufe DW, Pollock PE, Weicheelbaum RR, Holland JF, Frei E, et al., editors. Holland-Frei Cancer Medicine. 5th edition. Ontario, Canada: BC Decker; 2000. [Google Scholar]
- 20.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Research. 1991;51(23, part 1):6304–6311. [PubMed] [Google Scholar]
- 21.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(16):7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke AR, Purdie CA, Harrison DJ, et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362(6423):849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 23.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362(6423):847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 24.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Research. 1994;54(18):4855–4878. [PubMed] [Google Scholar]
- 25.Assinewe VA, Baum BR, Gagnon D, Arnason JT. Phytochemistry of wild populations of Panax quinquefolius L. (North American ginseng) Journal of Agricultural and Food Chemistry. 2003;51(16):4549–4553. doi: 10.1021/jf030042h. [DOI] [PubMed] [Google Scholar]
- 26.Court WE. Ginseng: the history of an insignificant plant. Pharmaceutical Historian. 2000;30(3):38–44. [PubMed] [Google Scholar]
- 27.Li B, Wang CZ, He TC, Yuan CS, Du W. Antioxidants potentiate American ginseng-induced killing of colorectal cancer cells. Cancer Letters. 2010;289(1):62–70. doi: 10.1016/j.canlet.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poudyal D, Mai Le P, Davis T, Hofseth AB, Chumanevich A, Chumanevich AA, et al. A Hexane fraction of American ginseng suppresses mouse colitis and associated colon cancer: anti-inflammatory and proapoptotic mechanisms. Cancer Prevention Research. 2012;5(4):685–696. doi: 10.1158/1940-6207.CAPR-11-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hussain SP, Harris CC. p53 biological network: at the crossroads of the cellular-stress response pathway and molecular carcinogenesis. Journal of Nippon Medical School. 2006;73(2):54–64. doi: 10.1272/jnms.73.54. [DOI] [PubMed] [Google Scholar]
- 30.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31(1):37–49. doi: 10.1093/carcin/bgp272.bgp272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skopek TR, Liber HL, Penman BW, Thilly WG. Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: possibility for a rapid human cell mutation assay. Biochemical and Biophysical Research Communications. 1978;84(2):411–416. doi: 10.1016/0006-291x(78)90185-7. [DOI] [PubMed] [Google Scholar]
- 32.Chuang YYE, Chen Q, Liber HL. Radiation-induced mutations at the autosomal thymidine kinase locus are not elevated in p53-null cells. Cancer Research. 1999;59(13):3073–3076. [PubMed] [Google Scholar]
- 33.Schneider U, Schwenk HU, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. International Journal of Cancer. 1977;19(5):621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 34.Cheng J, Haas M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Molecular and Cellular Biology. 1990;10(10):5502–5509. doi: 10.1128/mcb.10.10.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laumann R, Jucker M, Tesch H. Point mutations in the conserved regions of the p53 tumour suppressor gene do not account for the transforming process in the Jurkat acute lymphoblastic leukemia T-Cells. Leukemia. 1992;6(3):227–228. [PubMed] [Google Scholar]
- 36.Cui X, Jin Y, Hofseth AB, et al. Resveratrol suppresses colitis and colon cancer associated with colitis. Cancer Prevention Research. 2010;3(4):549–559. doi: 10.1158/1940-6207.CAPR-09-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. The FASEB Journal. 2008;22(3):659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 38.Predy GN, Goel V, Lovlin R, Donner A, Stitt L, Basu TK. Efficacy of an extract of North American ginseng containing poly-furanosyl-pyranosyl-saccharides for preventing upper respiratory tract infections: a randomized controlled trial. Canadian Medical Association Journal. 2005;173(9):1043–1048. doi: 10.1503/cmaj.1041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vuksan V, Stavro MP, Sievenpiper JL, et al. American ginseng improves glycemia in individuals with normal glucose tolerance: effect of dose and time escalation. Journal of the American College of Nutrition. 2000;19(6):738–744. doi: 10.1080/07315724.2000.10718073. [DOI] [PubMed] [Google Scholar]
- 40.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clinical and Experimental Immunology. 1998;114(3):385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morteau O, Morham SG, Sellon R, et al. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. The Journal of Clinical Investigation. 2000;105(4):469–478. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ying L, Marino J, Hussain SP, et al. Chronic inflammation promotes retinoblastoma protein hyperphosphorylation and E2F1 activation. Cancer Research. 2005;65(20):9132–9136. doi: 10.1158/0008-5472.CAN-05-1358. [DOI] [PubMed] [Google Scholar]
- 43.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115(1):182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 44.Wong VK, Cheung SS, Li T, et al. Asian ginseng extract inhibits in vitro and in vivo growth of mouse lewis lung carcinoma via modulation of ERK-p53 and NF-κB signaling. Journal of Cellular Biochemistry. 2010;111(4):899–910. doi: 10.1002/jcb.22778. [DOI] [PubMed] [Google Scholar]
- 45.Kim SE, Lee YH, Park JH, Lee SK. Ginsenoside-Rs4, a new type of ginseng saponin concurrently induces apoptosis and selectively elevates protein levels of p53 and p21(WAF1) in human hepatoma SK-HEP-1 cells. European Journal of Cancer. 1999;35(3):507–511. doi: 10.1016/s0959-8049(98)00415-8. [DOI] [PubMed] [Google Scholar]
- 46.Kim SE, Lee YH, Park JH, Lee SK. Ginsenoside-Rs3, a new diol-type ginseng saponin, selectively elevates protein levels of p53 and p21WAF1 leading to induction of apoptosis in SK-HEP-1 cells. Anticancer Research. 1999;19(1):487–491. [PubMed] [Google Scholar]
- 47.Yang XL, Guo TK, Wang YH, Huang YH, Liu X, Wang XX, et al. Ginsenoside Rd attenuates the inflammatory response via modulating p38 and JNK signaling pathways in rats with TNBS-induced relapsing colitis. International Immunopharmacology. 2012;12(2):408–414. doi: 10.1016/j.intimp.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 48.Joh EH, Lee IA, Jung IH, Kim DH. Ginsenoside Rb1 and its metabolite compound K inhibit IRAK-1 activation—the key step of inflammation. Biochemical Pharmacology. 2011;82(3):278–286. doi: 10.1016/j.bcp.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Moon J, Yu SJ, Kim HS, Sohn J. Induction of G1 cell cycle arrest and p2KIP1 increase by panaxydol isolated from Panax ginseng. Biochemical Pharmacology. 2000;59(9):1109–1116. doi: 10.1016/s0006-2952(00)00235-5. [DOI] [PubMed] [Google Scholar]
- 50.Wolf D, Rotter V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(3):790–794. doi: 10.1073/pnas.82.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan Z, Yang R, Jiang Y, et al. Induction of apoptosis in human promyelocytic leukemia HL60 cells by panaxynol and panaxydol. Molecules. 2011;16(7):5561–5573. doi: 10.3390/molecules16075561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwon JI, Kim GY, Park KY, Ryu CH, Choi YH. Induction of apoptosis by linoleic acid is associated with the modulation of Bcl-2 family and Fas/FasL system and activation of caspases in AGS human gastric adenocarcinoma cells. Journal of Medicinal Food. 2008;11(1):1–8. doi: 10.1089/jmf.2007.073. [DOI] [PubMed] [Google Scholar]
- 53.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. Journal of Experimental & Clinical Cancer Research. 2011;30:p. 87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang MR, Kim HM, Kang JS, et al. Lipid-soluble ginseng extract induces apoptosis and G0/G1 cell cycle arrest in NCI-H460 human lung cancer cells. Plant Foods for Human Nutrition. 2011;66(2):101–106. doi: 10.1007/s11130-011-0232-6. [DOI] [PubMed] [Google Scholar]