

Abstract
The unlimited proliferation potential of cancer cells requires the maintenance of their telomeres. This is frequently accomplished by reactivation of telomerase. However, in a significant fraction of tumors an alternative lengthening of telomeres (ALT) mechanism is active. The molecular mechanism of the ALT pathway remains elusive. In particular, the role of characteristic complexes of promyelocytic leukemia nuclear bodies (PML-NBs) with telomeres, the ALT-associated PML-NBs (APBs), is currently under investigation. Here, we review recent findings on the assembly, structure and functions of APBs. It is discussed how genomic aberrations in ALT-positive cancer cells could result in the formation of APBs and in ALT activity. We conclude that they are important functional intermediates in what is considered the canonical ALT pathway and discuss deregulations of cellular pathways that contribute to the emergence of the ALT phenotype.
Keywords: alternative lengthening of telomeres, ALT, promyelocytic leukemia nuclear bodies, ALT-associated PML bodies, DNA repair, DNA recombination, cellular immortalization
Telomere Maintenance Mechanisms
The ends of linear chromosomes, the telomeres, consist in humans of 5'-TTAGGG-3' repeats that adopt a specific chromatin structure. The DNA at the telomeres is mostly double-stranded with a single-stranded terminal 3'-overhang of the G-rich strand.1 The telomere repeats can fold back on itself with the G-rich tail invading the telomeric duplex DNA to form a telomere loop (t-loop).2 This specific DNA structure is recognized by “shelterin” proteins, including TRF1, TRF2, POT1, RAP1, TIN2 and TPP1. This nucleoprotein complex protects the chromosome end from being recognized as a DNA double-strand break (DSB) and prevents triggering a DNA damage response (DDR).3,4
Due to the end-replication problem and exonucleolytic activities, the ends of linear chromosomes shorten with every cell division. Upon reaching a critical telomere length, DDR factors induce replicative senescence or apoptosis.5,6 Accordingly, the number of cell divisions becomes limited, which represents an important tumor suppressor mechanism.5,7 Cancer cells circumvent this constraint to gain an unlimited proliferation potential by acquiring a telomere maintenance mechanism (TMM).8 In about 85% of human cancers, telomere maintenance is achieved by reactivation of telomerase, a reverse transcriptase that synthesizes telomeric repeats but is normally only active in embryonic and adult stem cells.9,10 Some cancer types extend the telomeres in the absence of telomerase activity by an alternative lengthening of telomeres (ALT) pathway that operates via DNA repair and recombination processes.11 Which type of TMM is active seems to depend on the origin of the tumor. ALT is rarely found in carcinomas but frequently activated in tumors of mesenchymal and neuroepithelial origin like osteosarcomas, liposarcomas or astrocytomas.12,13
Features of the Alternative Lengthening of Telomeres Pathway
Several characteristic features are associated with ALT activity. First, the telomere length distribution is highly heterogeneous and ranges from less than 3 kb to more than 50 kb (Fig. 1).14 In contrast, in human telomerase-positive cells all telomeres typically have a similar length of around 10 kb. Second, ALT-positive cells have been described to contain several classes of extrachromosomal telomeric repeats (ECTRs) in the nucleus. ECTRs comprise double- and single-stranded circular molecules, linear telomeric DNA and t-complex molecules that consist of high molecular weight DNA with highly branched structures.15-18 These DNA fragments might be products of t-loop resolution by recombination enzymes. The function of ECTRs in the telomere lengthening process is unknown but the amount of partially single-stranded telomeric (CCCTAA)n DNA circles (C-circles) appears to correlate with ALT activity.19 Third, the occurrence of telomere sister chromatid exchanges (T-SCE) is generally increased in ALT cells.20,21 Fourth, in ALT cells promyelocytic leukemia nuclear bodies (PML-NBs) associate with some telomeres (Fig. 1).22,23 These complexes are called ALT-associated PML-NBs (APBs).23 Here, we will focus on these nuclear subcompartments rather than covering the ALT pathway in general, which has been reviewed in references 18, 24 and 25. We will summarize what is known about the structure and assembly of APBs and consider their possible functions for ALT. Since APB formation alone is presumably not sufficient to trigger ALT activity, we will discuss the additional deregulation events that might be involved in ALT initiation. Interestingly, recent evidence suggests that telomerase-independent telomere maintenance can also occur in the absence of APBs and/or other characteristic ALT features.26-28 Based on these findings, we will consider the possibility that more than one ALT mechanism exists, and how this might reconcile apparently contradicting results concerning the role of APBs in ALT-mediated telomere elongation.

Figure 1. Characteristic features of ALT-positive cells. (A) Telomeric repeats of metaphase chromosomes were stained with Cy3-labeled PNA probes (red). ALT-negative human lymphocytes show a homogeneous distribution of telomere repeats lengths.65 (B) Telomere FISH reveals a high variation in telomere repeat lengths in ALT-positive U2OS cells with some chromosomal ends displaying long telomeric repeats as inferred from bright FISH signals, whereas others lack any signal indicating that telomeric repeats are very short. (C) Immunofluorescence of the PML protein (green) and the telomere repeat binding factor TRF2 (red) in a U2OS cell nucleus demonstrates the presence of PML-NBs at some telomeres (indicated by arrows), which are defined as ALT-associated PML-NBs or APBs. DNA was counterstained with DAPI. The scale bars are 10 μm.
Structure and Dynamics of PML Nuclear Bodies
PML-NBs, also known as PML oncogenic domains (PODs), nuclear domain-10 (ND10) or Kremer (Kr) bodies, are mobile structures in the cell nucleus that form distinct subcompartments of 0.2 to 1 ∝m in diameter.22,29 Depending on cell type, cell cycle phase, differentiation stage and various stimuli, the number of PML-NBs in the cell varies between 5 and 30.30 Apart from a few disease-related cases discussed below, normal PML-NBs do not contain any nucleic acids but were found to be associated with the surrounding chromatin fibers and with newly synthesized RNA at their periphery in some instances.31
The main structural components of PML-NBs are the PML and SP100 protein. They assemble into a hollow sphere with a shell thickness of 50–100 nm independent of the diameter of the whole PML-NB.22 In addition to the PML and SP100 marker proteins, an ever-growing number of factors, by now about 100, are found to associate in a transient manner with PML-NBs.32 Accordingly, PML-NBs have been implicated in a remarkably large number of nuclear activities as discussed in reference 32.
Seven different PML isoforms exist in humans with an identical N-terminus containing the so-called RBCC/TRIM motif consisting of a RING protein domain, B-boxes and a coiled-coil domain/tripartite motif. The RBCC/TRIM motif is essential for PML-NB formation as it is required for the multimerization of PML protein. The central role of PML in the formation of nuclear bodies was shown in cells lacking this protein. These cells are not only devoid of PML-NBs, but also other proteins normally residing in these compartments display an aberrant localization within the nucleus.33,34 Despite sharing an identical N-terminus, the PML isoforms show different behaviors when evaluating their dynamic exchange between a PML-NB and the surrounding nucleoplasm (Fig. 2).35 Fluorescence recovery after photobleaching (FRAP) experiments identified PML-V as the most stably bound component of PML-NBs with an average residence time of 48 min, whereas the other isoforms displayed faster exchange rates on the minute time scale. The second constitutive structural component of PML-NBs, SP100, showed a short residence time of less than 1 min in the nuclear bodies. Also other components of PML-NBs are not bound tightly. Instead, as demonstrated exemplarily in FRAP experiments for DAXX (death-associated protein 6) and BLM (Bloom syndrome protein), they can move rapidly in and out of PML-NBs (Fig. 2).35

Figure 2. Dynamic structure of a PML nuclear body. A PML-NB consists of a spherical shell composed of PML and SP100 proteins, which are stabilized by non-covalent interactions of the posttranslational modification SUMO with SIM domains. The SUMO2/3 variant, which is able to form chains, is also found in the interior of the subcompartment in contrast to SUMO1, which is enriched in the PML-SP100 shell.22 The SUMO modifications promote binding of proteins that contain SIMs. Photobleaching experiments revealed that the PML-NB constituents are very dynamic with the exception of the PML-V isoform, which displays a high average residence time of ~50 min at NBs indicating that this isoform plays a role as a structural scaffold.35 PML-NB associated proteins like DAXX and BLM show a rapid turnover and can reach the interior with only a relatively small reduction in diffusive mobility. The crucial role of SUMO modifications becomes apparent during mitosis when the shell structure breaks down upon desumoylation of the PML-NB constituents. The PML protein forms unspecific aggregates by interactions of its RBCC domain.48,63
Both PML and SP100 can be posttranslationally modified by the small ubiquitin-related modifier (SUMO) and contain SUMO interacting motifs (SIMs).36 Sumoylation is mediated by a sequence of enzymatic reactions similar to the ubiquitin pathway, involving the E1 activating enzyme complex, the E2 conjugating enzyme UBC9 as well as E3 ligases. However, modification of a protein by SUMO does usually not trigger degradation, but allows non-covalent binding by proteins that contain SIMs. In vertebrates, three SUMO variants were shown to have a biological function. SUMO2 and SUMO3 are often not further distinguished, since they differ in only three amino acids. In contrast, human SUMO1 shares only 47% homology with SUMO2/3 and is functionally different.37-39 For instance, SUMO1 is mostly found in its conjugated form, whereas there seems to be a pool of free SUMO2/3, which is only conjugated upon cellular stress.39 Since PML-NBs are highly modified with all three SUMO isoforms and are enriched with sumoylated and SIM-containing proteins, they have been referred to as “sumoylation hotspots.”40 Interestingly, the distribution of the SUMO variants within a PML-NB is heterogeneous. While SUMO1 is mostly restricted to the surrounding shell, SUMO2/3 can be found at the shell and in the interior of the nuclear body (Fig. 2).22 This may reflect the differential modification of target proteins bound at different sites to the PML-NB. Notably, SUMO2/3 can also form polymeric SUMO chains, while evidence for the existence of SUMO1 chains in vivo is lacking.41 Thus, SUMO2/3 chains may protrude from the shell into the interior of the nuclear body (Fig. 2). Several reports have demonstrated a crucial role of sumoylation for the assembly and stability of PML-NBs. First, the PML protein itself is sumoylated at three lysine residues, and it has been shown that exogenously expressed mutants that lack these residues are not able to form PML-NBs in a Pml-/- background.34 Likewise, cells lacking SUMO pathway components do not form these nuclear structures.42 Second, PML can bind directly to the SUMO E2 conjugating enzyme UBC9 via its RING domain and might itself have SUMO E3 ligase activity.43,44 Third, the SIM of the PML protein is required for the formation of nuclear bodies, and the SP100 protein is sumoylated and contains a SIM.36,45,46
The above findings led to a model in which PML-NB formation is accomplished by SUMO-SIM interactions of PML proteins among themselves as well as with partner proteins like SP100.36,46,47 Consistent with this view, desumoylation of PML during mitosis is accompanied by the disassembly of the nuclear bodies resulting in the presence of unstructured mitotic accumulations of PML protein (Fig. 2).48 Moreover, the residence times of PML, SP100, and other components at the PML-NBs are influenced by their sumoylation state.35 In addition to its structural function of interlinking PML and SP100 via SUMO-SIM interactions, SUMO is also required for recruiting other protein factors to the PML-NB. In fact, most PML-NB protein components are substrates for sumoylation and/or contain a SIM.32 Examples for SUMO-SIM-mediated targeting to PML-NBs include DAXX, TDG (thymine-DNA glycosylase), BLM, CBP (CREB binding protein) or IKKε (inhibitor of nuclear factor kappa-B kinase ε), and inhibiting the SUMO interactions perturbs the sequestration of these proteins by PML-NBs and has functional consequences.33,49-51 Furthermore, as discussed in a recent review, arsenic trioxide induced sumoylation of PML and possibly other proteins in PML-NBs recruits the SUMO-targeted ubiquitin ligase RNF4 (RING finger protein 4) and leads to degradation of PML by the proteasome.52
Structure and Composition of ALT-Associated PML Nuclear Bodies
As mentioned above, PML-NBs do not contain nucleic acids in normal cells. However, in ALT-positive cells, a subset of PML-NBs co-localizes with telomeric DNA. Previously, the percentage of APB-positive cells within an asynchronous dividing ALT cell population was estimated to be 5–10% based on the detection of large co-localizations of PML and telomeres (~1 ∝m in diameter) by wide-field microscopy.23,53-55 However, confocal laser scanning microscopy can detect ~4 APBs per cell with a homogeneous size distribution in 90% of unsynchronized cells in the ALT-positive U2OS osteosarcoma cell line.56 A three-dimensional high-resolution 4Pi microscopy analysis showed that the PML protein encloses the telomeric DNA by forming a shell (Fig. 3). The thickness of the PML-formed spherical layer is similar to the one measured for normal PML-NBs and independent of the total size of the APBs, i.e., small APBs appear to form with the same PML-SP100 structural scaffold as larger ones.22 Accordingly, restricting the analysis of APBs to those of large size might underestimate their number and appearance.

Figure 3. High-resolution two-color fluorescence 4Pi-microscopy image of an APB. The PML-NB (visualized by immunofluorescence of PML protein) is shown in green and the telomeric repeat sequence, which is hybridized to a telomere-specific PNA probe, in red. The corresponding merged image (merge 1) and the 3D image reconstruction (bottom) reveal that the PML-protein forms a spherical shell that is clearly separated from the telomeric repeat sequence.22 This structure is independent from the size of the APB as can be inferred from the image of a smaller complex shown in merge 2. The scale bar is 0.5 μm.
By definition, APBs comprise PML-NB components like PML, SP100, SUMO and telomere repeat-associated proteins like TRF1, TRF2, POT1 or RAP1.23 Additionally, they contain factors that are involved in DNA damage response and repair, such as the components of the 9-1-1 complex (hRAD9, hHUS1, hRAD1), hRAD17, the phosphorylated histone variant H2A.X (γH2A.X), the RecQ-like helicase BLM, heterochromatin protein HP1 or the structural maintenance of chromosome complex (SMC5/6), which includes the SUMO E3 ligase MMS21.54,57-59 Recent studies highlight the crucial role of SUMO and MMS21 for the alternative lengthening of telomeres.59,60 In addition, proteins involved in homologous recombination localize to APBs, as for example the endonuclease MUS81, replication protein A (RPA), RAD51 and RAD52, breast cancer susceptibility protein 1 (BRCA1) or the MRN complex consisting of NBS1, MRE11 (meiotic recombination 11 protein) and RAD50.23,61,62 A detailed overview of protein factors found in APBs is given in a recent review in reference 24.
Mechanisms of APB Assembly
One mechanism for the formation of APBs could be a collision between a fully assembled PML-NB and a telomere within the cell nucleus. Several studies have addressed the mobility of PML-NBs by live-cell microscopy using fluorescently labeled constructs of PML or SP100.63-66 The majority of PML-NBs displays a restricted movement within a confined domain, a behavior that has also been observed with other nuclear bodies like Cajal bodies.64 These bodies diffuse within an accessible corral with 100–200 nm radius in the chromatin environment, and this chromatin corral again can translocate more slowly in the nucleus.64,67 Analyzing PML-NB movement with respect to a labeled telomere allows for a classification of nuclear bodies into those that are directly associated within the same chromatin region as the telomere and others that translocate in a more uncorrelated manner with respect to chromatin (Fig. 4A).65,68
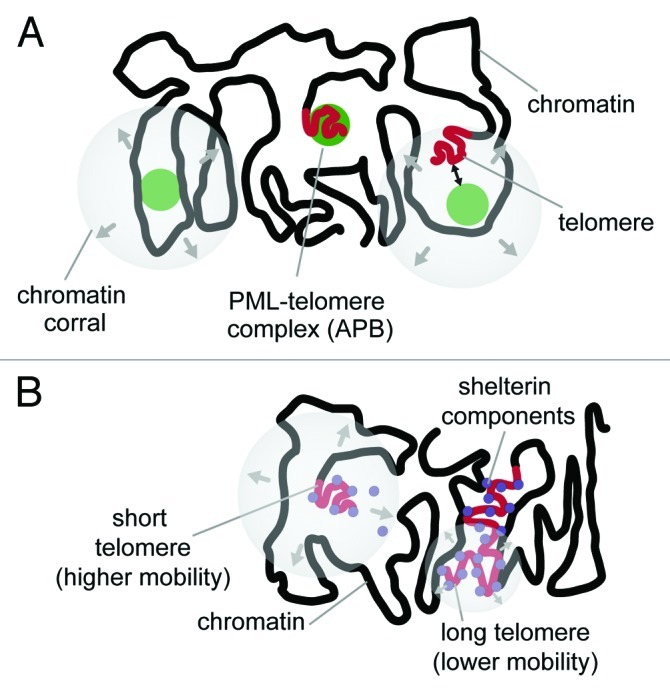
Figure 4. Dynamics of telomeres and PML-NBs within the cell nucleus. (A) PML-NBs (depicted as green spheres) can be classified into distinct groups according to their relative mobility with respect to a telomere in its proximity. One group consists of PML-NBs that display a mobility uncorrelated with that of an adjacent telomere possibly due to its location in a separated chromatin corral. Other PML-NBs translocate within the same accessible chromatin space as a telomere. These show correlated movement when PML-NBs and telomeres are detected simultaneously. In ALT-positive cells, APBs represent an additional class of PML-NBs with respect to their mobility, since these complexes are characterized by a stable association of the PML-NB to a telomere and correlated movements.65 (B) Telomeres show varying degrees of mobility depending on their repeat lengths. Long telomeres are bound by shelterin factors and move within a confined radius of 0.3–0.5 μm that is similar to that of other chromosomal loci.65,67,68 This is most likely due to an anchoring function of the associated proteins with the surrounding chromatin. In contrast, short telomeres lack sufficient binding sites, the anchoring is progressively lost, and thus, they display an increased mobility.
Visualizing the dynamics of telomeres in ALT-positive cells is somewhat more challenging than those of nuclear bodies due to the presence of ECTRs, which consist of isolated telomeric sequences bound by shelterin proteins. Thus, it is difficult to exclude that telomere labels in living cells like FISH probes or GFP-tagged telomere-binding proteins reflect to some extend also complexes with ECTRs. These extra-chromosomal particles might display a higher mobility than genuine telomeres attached to a chromosome. As an alternative approach to tag telomeres, stable integrations of lac operator (lacO) sequences next to the telomeres have been introduced.65 These lacO arrays provide high-affinity binding sites for the lac repressor LacI that can be detected as a fluorescent fusion protein in living cells.69-71 Using this system, a bimodal distribution of telomere mobility in ALT-positive U2OS cells was revealed: the main fraction of telomeres moved with lower mobility in a confined average radius of 0.36 ± 0.16 ∝m, whereas a fraction of 15% displayed an extended mobility with translocations in an average radius of 0.8 ± 0.1 ∝m in agreement with a previous report in reference 68 (Fig. 4B). Further experiments indicated an inverse correlation of telomere repeat length and telomere mobility, possibly due to the lack of interactions mediated by telomere sequence-specific binding proteins with surrounding chromatin regions. This conclusion is supported by the finding that the deletion of TRF2 results in a higher mobility.72 Thus, short telomeres display an increased mobility in the nucleus and explore a larger nuclear volume, which could facilitate interaction with a PML-NB. Such a collisional event leading to the formation of an APB-like complex was indeed observed.65 However, since both PML-NBs as well as telomeres are relatively immobile during interphase, collisions between them are rare events on the time scale of hours within a given cell.
A more efficient mechanism for the formation of APBs in ALT-positive cells appears to be the accumulation of soluble PML protein at a telomere, which was also observed in time-lapse microscopy experiments.65,68 This accumulation could be mediated by the direct interaction of the telomeric repeat binding factor TRF1 with the PML-IV isoform.73 Furthermore, several lines of evidence suggest that the assembly of PML protein at the telomere is strongly influenced by sumoylation, most likely mediated by the SUMO E3 ligase MMS21, a component of the SMC5/6 complex, that can be found in APBs.59 (1) It has been shown that MMS21 mediates sumoylation of the shelterin components TRF1, TRF2, TIN2 and RAP1.59 Moreover, mutant variants of TRF1 and TRF2 that cannot be sumoylated do not form APBs anymore. (2) It has been reported that telomeres serve as nucleation sites for the de novo assembly of PML-NBs in both non-ALT and ALT cells.74 This process involves the formation of a transient PML-telomere complex and subsequent detachment of the PML-NB from the chromosomal ends. PML-NB formation is accompanied by the presence of SMC6 at telomeres and sumoylation of PML is crucial for this process. It is tempting to speculate that in ALT cells a deregulated, increased sumoylation of telomeric proteins impairs the detachment of mature PML-NBs from telomeres by maintaining SUMO-SIM interactions. (3) Several SUMO-related proteins are capable of inducing de novo APBs in ALT-positive U2OS cells.60 This was demonstrated by recruitment experiments in U2OS cells with lacO-tagged telomeres. In these experiments, GFP-tagged proteins were bound to the telomeric lacO sites by employing a fusion construct of LacI with a high-affinity GFP-binding protein domain. The propensity of inducing APB formation was evaluated by probing the presence of PML protein at that telomere. The SUMO E3 ligase MMS21, as well as SUMO itself, were found to be highly efficient in PML recruitment to the telomeres. Interestingly, a non-conjugable SUMO1 mutant was as potent in initiating APB formation as its wild-type counterpart, in contrast to a largely reduced capability of the non-conjugable mutants of SUMO2 and SUMO3. If binding to a SIM was impeded by an additional mutation of the non-conjugable SUMO1 mutant, the ability to induce APB assembly was lost. This identified the non-covalent interactions between SUMO1 and a SIM as a major driving force for the APB nucleation event. In contrast, SUMO2 and SUMO3 appear to promote APB formation mostly via their conjugation to other proteins, which could be important for recruiting further APB components. The crucial role of SUMO-SIM interactions for formation of APBs was also apparent from the observation that various sumoylatable proteins can induce PML accumulation when highly enriched at the telomere.60
The different factors that contribute to APB assembly are depicted in the scheme in Figure 5. Various PML-NB proteins can induce a nucleation event at a telomere. From the above findings on the role of sumoylation we conclude that any process that will lead to a high local density of SUMO1 at the telomeres will initiate APB formation via providing a scaffold for SIM mediated protein accumulation. An enrichment of SUMO2/3 at the telomeres via conjugation to telomere-bound proteins would be less efficient in this respect since SUMO2/3-SIM interactions appear to play only a minor role for APB nucleation.60 In addition, it appears likely that a sumoylation feedback mechanism exists that involves MMS21 and a potential SUMO ligase activity of PML itself.43 The combination of SUMO-SIM interactions and additional sumoylation creates a structural scaffold of PML and SP100 protein, to which DNA repair and recombination factors assemble subsequently.

Figure 5. A model for APB assembly and ALT-specific telomere elongation. Sumoylation of telomeric proteins by the SUMO E3 ligase MMS21 triggers the accumulation of PML and SP100 at the telomere via SUMO-SIM-interactions.59 A sumoylation feedback loop leads to the complete assembly of the structural scaffold, which then recruits repair and recombination factors.60 Within these functional APBs telomeres are extended via DNA repair processes, non-replicative DNA synthesis and homologous recombination.57,60,116 It is proposed that after elongation APBs are disassembled either by dissociation of the whole PML-NB from the telomere or by complete disassembly of the PML-NB by desumoylation.74
Since multiple interactions between structural APB factors and telomeric chromatin components are involved in the assembly process, the question arises to which degree the chromatin state at the telomeres affects APB formation. Interestingly, heterochromatin appears to represent a preferential interaction surface for PML-NBs. It was shown that in fibroblasts quiescently infected with herpes simplex virus PML-NBs can accumulate viral genomes, which are associated with heterochromatin protein HP1 in their central core.75 Another example can be found in lymphocytes of patients suffering from the immunodeficiency, centromeric instability and facial dysmorphy (ICF) syndrome. These cells exhibit extremely large PML-containing bodies that form as a layered structure surrounding heterochromatic satellite DNA.76 Moreover, a direct connection between HP1 protein and APBs has been shown in RNAi-mediated knockdown experiments, which revealed the requirement of HP1α and HP1γ for APB formation.54 However, there is also evidence that in mice an open, euchromatic structure at telomeres promotes assembly of APB-like structures. In mouse cells, disruption of the normally heterochromatic structure of telomeres can result in the appearance of APBs and an ALT-like phenotype.77-79 Potentially, a less dense chromatin structure could facilitate recombination processes and binding of APB-related proteins at telomeres. However, one has to be cautious about comparing mouse and human cells with respect to their telomere biology. Mouse cells have extremely long telomeres and a less stringent regulation of telomerase activity, so that cell proliferation capability does not depend on telomere maintenance mechanisms.80,81
APB Functions
It is an ongoing question of current ALT research whether APBs are—directly or indirectly—involved in the ALT mechanism, or whether they are not associated with the telomere elongation process.54,57,82-85 Two studies that relate APB formation with senescence in ALT-positive cells argue in support of the latter view.54,82 By restoring wild-type p53 function in p53-deficient ALT cells, senescence and accumulation of large APBs was induced.54 It was proposed that upon induction of senescence the histone chaperone HIRA relocates to PML-NBs. In ALT cells this process could lead to the recruitment of HP1, telomeric DNA and possibly a histone methyltransferase.82 As a result, compaction of telomeric DNA would be promoted, which could repress recombination. As large APBs are found only in ~5% of cells within asynchronously dividing ALT cell populations, it was suggested that APBs represent a fraction of cells that have spontaneously undergone growth arrest.23,54 However, this conclusion is challenged by the finding that smaller APBs can be detected in 50–90% of proliferating ALT cells, which argues against a general association of APBs with growth arrest.56,83 Furthermore, also in non-ALT cells telomeric associations drastically increase upon induction of quiescence.86 Therefore, the primary result of senescence could be the clustering of telomeres that leads to an appearance of larger co-localizations of PML and telomeres.
On the other hand, several lines of evidence argue strongly for APBs having a functional role for ALT. First, the appearance of APBs is a robust marker for the identification of human tumors that utilize the ALT mechanism.53,87 Only in rare cases APB appearance and ALT are not linked as discussed in further detail below.26-28 Second, repression of the ALT mechanism in cell hybrids of ALT cells with telomerase-positive tumor cells is accompanied by the disappearance of APBs, whereas APBs assemble upon activation of ALT-mediated telomere maintenance.23,88 Third, experimental disruption of APBs by sequestration of the MRE11-RAD50-NBS1 complex away from the telomeres or knockdown experiments of components of the SMC5/6 complex results in simultaneous shortening of telomeres.59,89 These data can be rationalized by different mutually non-exclusive roles of APBs in alternative telomere elongation. APBs could either operate by driving ALT-relevant processes or by being required to avoid senescence or apoptosis. In support of the latter view it was proposed that linear ECTRs, which might result from telomeric recombination events, are sequestered by APBs.83 This could prevent ECTRs from being recognized as DNA double-strand breaks and from triggering cell cycle arrest. The dynamic structure of APBs and PML-NBs poses a challenge for identifying their native composition by classical purification approaches.90 Thus, an experimental demonstration that ECTRs are indeed accumulated in APBs within the cell is currently missing. Similar to their proposed function for sequestering ECTRs, APBs assembled at very short telomeres could prevent DDR induction by replacing the function of the missing shelterin proteins. However, disruption of APBs does not lead to a fast induction of senescence and apoptosis, which is in apparent contradiction to the prediction that they are needed to avoid growth arrest.59,89
Alternatively, APBs could actively promote telomere elongation by providing templates for replication- and recombination-based telomere lengthening or by recruiting proteins that facilitate these processes. Considering what we know about PML-NBs it appears likely that both PML-NBs and APBs provide catalytic surfaces for certain reactions that would proceed less efficiently in the absence of these nuclear subcompartments. For PML-NBs this is apparent from the multitude of enzymatic activities that are associated with them. Specific examples are the requirement of PML-NBs for phosphorylation-mediated p53 activation or the possible role of the PML protein as a SUMO E3 ligase in a sumoylation feedback cycle as proposed above.43,91
A role of APBs as sites that provide templates for DNA recombination or replication could involve the accumulation of ECTRs as well as the promotion of telomere clustering.15-17,68,84 Artificial enlargement of APBs using the mutated Herpes virus protein ICP0 resulted in clustering of several telomeres at the surface of these bodies.84 Also live cell imaging experiments revealed that PML bodies dynamically associate with one and more telomeres, thereby possibly facilitating intertelomeric recombination.68
Due to their ability to enrich certain proteins and/or enhance their activity, APBs can promote replication- and/or homologous recombination-mediated extension of telomeres, which was shown in several studies.57,60,84,85 Artificial enlargement of APBs resulted not only in clustering of several telomeres but also filamentous telomeric bridges were observed in metaphase spreads. The latter structures suggested that intertelomeric recombination was initiated but not resolved at the enlarged PML bodies.84 Further evidence for telomeric extension taking place in APBs comes from the detection of non-replicative DNA synthesis in these subcompartments.57,85 Additionally, a recent study revealed the induction of telomere extension through a DNA repair mechanism by artificially accumulating PML at specific telomeres in an ALT cell line. The resulting de novo formed APBs co-localized with sites of non-replicative DNA synthesis and the DSB repair marker histone γH2A.X and led to an increase of the telomere repeat length.60
In summary, strong evidence exists for a role of APBs in actively promoting alternative telomere lengthening. Dissecting their function further would require knowledge of molecular details of the ALT mechanism to test which specific reaction steps are facilitated by APBs. To date, this information is missing but several models are currently investigated and are described in a number of excellent reviews.14,18,92-94
Triggering ALT Activity by a Sequence of Deregulation Events
The existence of PML and telomere co-localizations in ALT-positive cells was discovered more than ten years ago. Their appearance was assumed to be an exclusive feature of ALT-positive immortalized cell lines and tumors.23 However, PML-telomere co-localizations have recently been reported to be present also in ALT-negative cells.55,95 In order to distinguish these colocalizations from APBs, which by definition are only present in ALT-positive cells, the term telomere-associated PML bodies (TPBs) was introduced.55 Small TPBs (<0.9 ∝m in diameter) were found in several non-neoplastic cells in the absence of any TMM.95 The highest frequency of TPBs (median 7–15% of cells) was detected in mesenchymal tissues, from which most ALT tumors arise. Interestingly, the frequency of TPBs was increased upon induction of DNA damage. This suggests that TPBs in mortal cells carry out repair processes at telomeres in a mechanism similar to that in ALT-positive tumors, but that other yet unknown factors repress ALT function. Large TPBs (> 0.9 ∝m) have also been described in 92% of low-grade astrocytomas that lack both alternative lengthened telomeres and telomerase activity.55 As ALT is activated in higher-grade astrocytomas, the emergence of an ALT phenotype was suggested to be associated with the progression from grade 1 to grade 2/3. Although direct evidence for the progression from TPB-positive cells to ALT-positive tumors is lacking, it is tempting to speculate that the appearance of TPBs is a first step toward ALT initiation, i.e., represents a pre-ALT phenotype (Fig. 6).55

Figure 6. Model for the development of ALT via multiple deregulation steps. The scheme depicts events that could lead to telomerase-independent telomere elongation. A first step toward ALT could be the deregulation of the sumoylation/desumoylation equilibrium at telomeres. A combination of additional aberrations such as the constitutive activation of the DNA damage response, reduced protection of telomeres, loss of p53 function, mutations in DAXX, ATRX and in the histone variant H3.3, as well as other yet to be identified events are likely to be required for full ALT activity. As discussed in the text, the term “ALT” represents different molecular pathways. What we refer to here as the canonical ALT mechanism is characterized by the indicated features (APBs, ECTRs, T-SCEs, heterogeneous telomere length). Besides, other telomerase-independent mechanisms for telomere maintenance exist that lack some or all of these characteristics (“non-canonical ALT”). We speculate that the deregulation of sumoylation at the telomeres could result in a cellular state termed “pre-ALT phenotype” that is still susceptible to senescence/apoptosis but displays abnormal accumulations of PML protein at telomeres, the telomere-associated PML bodies (TPBs). Upon further (epi)genetic aberrations, these structures could transform into functional APBs.
Taken together, APBs are likely to emerge due to a deregulation of a normal cellular process. A likely candidate is the sumoylation pathway since de novo formation of PML-NBs was reported to take place at telomeres in a SUMO-dependent manner in both ALT and non-ALT cells.60,74 Furthermore, SUMO enrichment at telomeres is sufficient to induce APB assembly in ALT cells.60 Increased sumoylation could occur, for example via enrichment of MMS21 at the telomeres or by inhibition or downregulation of a counteracting desumoylation enzyme like one of the SENP proteins.59
Fusions of mortal cells with ALT-positive cells loose ALT activity, indicating that ALT requires loss of a normal function that represses ALT activity.96 This is also the case for hybrids between ALT and telomerase-positive cells.96,97 Although the mechanism that represses ALT-mediated DNA replication and recombination events at the telomeres in normal cells is still unknown, it appears reasonable to assume that the repressive function is not performed by a single factor. Thus, it seems likely that a series of changes is necessary to initiate ALT (Fig. 6). The above considerations imply that TPB formation might be one of these events. Yet, TPB formation alone does not seem to be sufficient to trigger ALT activity suggesting that other deregulation steps are necessary for ALT function. Likely candidates are DNA repair pathways, the telomeric chromatin state and the integrity of the telomere-bound shelterin complex. In this context p53 appears particularly interesting, since the vast majority of ALT cell lines are impaired in the p53 pathway, and there is evidence that mutations in the p53 gene are associated with ALT occurrence in glioblastoma.11,98 The p53 protein is involved in the damage response to dysfunctional telomeres and restoring functional p53 in ALT cells leads to telomere DDR-induced cell cycle arrest and senescence.54,92,99 This finding suggests that activation of ALT requires loss of normal p53 function.100,101 In line with this view, it has been proposed that reconstitution of p53 inhibits DNA synthesis in ALT cells by suppression of telomeric recombination.102 In ALT cells the DDR checkpoint kinase ATM was found to be constitutively active and ALT-positive cell lines lacking wild-type p53 show many telomeres with a DDR.99,103 We conclude that a permanently activated DDR is present in ALT cells but without triggering growth arrest due to inactivation of p53. However, the absence of functional p53 alone is not sufficient for immortalization.101
Other potential events involved in the emergence of ALT activity might be the loss of ATRX and DAXX and mutations in the histone H3 variant H3.3.104,105 ATRX and DAXX are known to interact with each other. Among other functions they are required for the non-replicative incorporation of H3.3 at telomeres.106-110 Moreover, they were suggested to facilitate heterochromatin assembly at repetitive G-rich regions, for instance at telomeres.107,109,110 Interestingly, ALT activity was found to be highly correlated with the simultaneous occurrence of mutations in the TP53, ATRX and H3F3A genes (encoding for p53, ATRX and H3.3) in a recent genome analysis of pediatric glioblastomas.105 It was shown that incorporation of mutant H3.3 results in changes in the expression profiles, which could facilitate ALT appearance.105 Furthermore, a model has been proposed, in which loss of ATRX-DAXX function inhibits the formation of heterochromatic features at the telomeres, possibly as a result of reduced incorporation of H3.3.104,105 These changes of the telomeric chromatin state could lead to increased homologous recombination associated with ALT activity. In addition, ATRX seems to be responsible for repression of the telomeric non-coding transcript TERRA, which displays elevated levels in some ALT cell lines and tumors.110-112
Disturbing the shelterin-mediated protection of telomeres might be another factor that favors ALT initiation. The shelterin complex binds telomeric DNA and participates in t-loop formation, which represses DDR and telomeric recombination.4,113 It has been demonstrated that in ALT-positive cells DDR can be partly suppressed by TRF2 overexpression.103 Furthermore, some ALT-positive cell lines have low ratios of TRF2 to telomeric DNA, i.e., a relative deficiency of TRF2 at the telomeres.103 The resulting reduced shelterin protection might favor recombination events at the telomeres. Finally, a deregulated sumoylation pathway might also decrease telomere protection. As discussed above, impaired sumoylation of the shelterin components TRF1 and TRF2 was shown to inhibit APB formation.59 As one putative sumoylation site of TRF2 is located in the TRFH domain, which mediates TRF2-dimerization, sumoylation of shelterin components could lead to their dissociation from telomeres.59,114 This supports the conclusion that deregulation of the sumoylation-desumoylation equilibrium predisposes for the emergence of an ALT phenotype.
Different Alternative Telomere Lengthening Mechanisms
As discussed above, different combinations of deregulation events are presumably able to trigger ALT activity according to the scheme depicted in Figure 6. Several findings support the hypothesis that more than one mechanism for telomerase-independent telomere elongation exists. There is the canonical ALT pathway displaying its characteristic features, namely heterogeneous telomere length, ECTRs, APBs and T-SCEs. Within this pathway two mutually non-exclusive mechanisms for telomere elongation, the unequal T-SCE and the homologous recombination dependent DNA synthesis, have been proposed.18 Furthermore, it has been demonstrated that there are different templates for recombination-mediated DNA replication of telomeres in ALT cells including the same telomere via t-loop formation, the telomere of a sister chromatid or the telomere of another chromosome.115,116 In addition, linear or circular ECTRs could serve as templates.19,23 The variety of possible templates can be related to different mechanisms for telomere elongation such as rolling circle amplification for circular forms of ECTRs or break-induced replication for recombination between telomeres.14,18,24 These recombination/repair mechanisms might operate in parallel. Thus, already within the canonical pathway there are several possible molecular routes for telomere elongation.
In addition to the canonical ALT mechanism other telomerase-independent telomere maintenance mechanisms exist. In a few cases it was demonstrated that elongation of telomeres could occur in telomerase-negative cells in the absence of one or more of the canonical ALT features mentioned above.26-28 For example, one cell line was reported to lack APBs but featured all other canonical ALT characteristics.27,28 Remarkably, though lacking APBs, this telomerase-negative SV40-immortalized fibroblast cell line still showed nuclear aggregates of APB components at telomeres. These aggregates contained the SV40 large T antigen possibly formed via binding to the SV40 origin of replication sequences integrated into telomeres of this cell line.27 Due to its interaction with many proteins, the SV40 large T antigen appears to be able to replace the function of sumoylated PML in this case. Thus, although composition and structure might differ, nuclear domains comprising telomeric DNA and proteins involved in DNA processing seem to be important for telomerase-independent telomere maintenance. Moreover, the presence of ECTRs cannot always be correlated with ALT activity. Some ALT cell lines lack ECTRs, and low levels of ECTRs were detected in telomerase-positive and mortal cells.26,113,117 ECTRs were also reported to accumulate in a telomerase-positive cancer cell line upon upregulation of telomerase activity where they might result from trimming of overlengthened telomeres, most likely by resolution of the t-loop.118 Regarding the different classes of ECTRs, the so-called C-circles appear to be the most robust marker for ALT activity.19 Another example for a “non-canonical” ALT pathway is an immortalized ALT-derived human cell line that maintains its telomeres in a telomerase-independent manner in the absence of several canonical ALT features, namely APBs, heterogeneous telomere length and ECTRs.26 However, a T-SCE phenotype was observed and also C-circles were detected.19,26 These reports indicate that more than one telomerase-negative TMM exist, which is further supported by the analysis of tumor samples and a cell line that lack both detectable telomerase activity and characteristics of the ALT pathway.87,119,120
In summary, the fact that APBs and other ALT markers are not observed in some telomerase-negative tumors and cell lines does not necessarily argue against their functional role in the canonical ALT pathway. Rather it suggests that different alternative telomere lengthening mechanisms exist. It is therefore of crucial importance to decipher the precise pathways and hallmarks of these TMMs. Since canonical ALT features seem not to be universal for ALT function, a systematic reassessment of known ALT markers (and possible other TMM features) in terms of their simultaneous presence would provide valuable information in this respect. This is especially relevant for the identification of the specific TMM that is active in a particular cell line or tumor, as well as for the design of anti-cancer therapies targeting telomere maintenance.
Identification and Targeting of APBs in Tumors
In addition to being a marker for ALT activity, APBs have a yet hardly exploited potential for being used as a prognostic tumor marker. This has been demonstrated for liposarcoma and neuroblastoma where the presence of APBs is associated with poor survival.87,121-123 In addition, APBs might represent a novel therapy target. Inhibition of telomere lengthening is an attractive approach for preventing tumor growth since the shortening of the chromosomal ends during further replication rounds might finally result in induction of senescence. Moreover, the presence of a TMM is specific to cancer and stem cells. Accordingly, these drugs should have reduced side effects on healthy somatic cells. Several approaches have been developed to inhibit telomerase activity in order to treat telomerase-positive tumors.124-126 Some of them are currently tested in phase III clinical studies.127 However, therapies that employ telomerase inhibitors might lead to a selective emergence of an ALT-positive cancer cell population, even if the tumor initially was ALT-negative.128-130 Furthermore, there is evidence that both telomerase activity and ALT can coexist in human cell lines.88,131 Assuming that this also holds true for tumors, anticancer drugs targeted against telomerase will be hardly effective in these cases since ALT activity will still provide a telomere maintenance pathway. As discussed above, several lines of evidence suggest that disruption of APBs would reduce the proliferation potential of tumor cells that use the canonical ALT mechanism. It is predicted that inhibiting a factor that is able to initiate APB formation would result in telomere shortening and finally induction of senescence. One promising candidate would be the SUMO E3 ligase MMS21 that was shown to be an efficient APB-inducing factor.60 The siRNA-mediated knockdown of MMS21 disrupts APBs and induces senescence in ALT—but not in telomerase-positive cells.56,59 Thus, developing therapeutic strategies that target MMS21 or other proteins essential for APB formation and/or function could represent a currently not exploited option for the specific treatment of ALT-positive tumors.
Conclusion
Several lines of evidence indicate that APBs assemble in a SUMO-SIM-interaction-dependent manner. They represent important functional intermediates in the ALT pathway and can induce telomere elongation in the absence of active telomerase (Fig. 5). In addition to APB formation, several deregulation events that influence cellular DNA damage response, telomeric chromatin and possibly other factors are required in order to activate the ALT pathway for extending the chromosomal ends (Fig. 6). Depending on the combination of deregulating events occurring in a given cell, different ALT-phenotypes are observed. The prevalent canonical ALT pathway involves APBs, ECTRs, T-SCEs and a heterogeneous telomere length distribution. In contrast, the less frequently observed non-canonical ALT pathways lack at least one of these classical ALT hallmarks. Understanding the aberrations that trigger a specific type of ALT activity and lead to the formation of APBs, as well as further dissecting their functions will be critical to identify markers for diagnosis as well as potential drug targets for ALT-positive tumors.
Glossary
Abbreviations:
- ALT
alternative lengthening of telomeres
- APB
ALT-associated PML-NB
- ATRX
X-linked helicase II
- BLM
bloom syndrome protein
- DAXX
death-associated protein 6
- DDR
DNA damage response
- ECTRs
extrachromosomal telomeric repeats
- FISH
fluorescence in situ hybridization
- FRAP
fluorescence recovery after photobleaching
- GFP
green fluorescence protein
- γH2A.X
histone variant H2A.X with phosphorylated serine 139
- HP1
heterochromatin protein 1
- LacI
Lac repressor
- lacO
lac operator sequence
- MMS21
methyl methanesulfonate sensitivity protein 21
- NBS1
nijmegen breakage syndrome protein 1
- PML
promyelocytic leukemia
- PML-NB
PML nuclear body
- PNA
peptide nucleic acid
- POT1
protection of telomeres 1
- Rad
radiation homolog
- RAP1
repressor/activator protein 1
- SIM
SUMO interacting motif
- SMC
structural maintenance of chromosome complex
- SUMO
small ubiquitin-related modifier
- t-loop
telomeric loop
- T-SCE
telomere sister chromatid exchange
- TMM
telomere maintenance mechanism
- TRF
telomere repeat binding factor
- UBC9
ubiquitin carrier protein 9
Footnotes
Previously published online: www.landesbioscience.com/journals/nucleus/article/20326
References
- 1.Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88:657–66. doi: 10.1016/S0092-8674(00)81908-X. [DOI] [PubMed] [Google Scholar]
- 2.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–14. doi: 10.1016/S0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 3.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 4.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 5.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 6.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 7.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 8.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–91. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 10.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 11.Henson JD, Reddel RR. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010;584:3800–11. doi: 10.1016/j.febslet.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JE, Broccoli D. Telomere maintenance in sarcomas. Curr Opin Oncol. 2007;19:377–82. doi: 10.1097/CCO.0b013e3281214423. [DOI] [PubMed] [Google Scholar]
- 13.Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–15. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- 15.Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 2004;24:9948–57. doi: 10.1128/MCB.24.22.9948-9957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogino H, Nakabayashi K, Suzuki M, Takahashi E, Fujii M, Suzuki T, et al. Release of telomeric DNA from chromosomes in immortal human cells lacking telomerase activity. Biochem Biophys Res Commun. 1998;248:223–7. doi: 10.1006/bbrc.1998.8875. [DOI] [PubMed] [Google Scholar]
- 17.Tokutake Y, Matsumoto T, Watanabe T, Maeda S, Tahara H, Sakamoto S, et al. Extra-chromosomal telomere repeat DNA in telomerase-negative immortalized cell lines. Biochem Biophys Res Commun. 1998;247:765–72. doi: 10.1006/bbrc.1998.8876. [DOI] [PubMed] [Google Scholar]
- 18.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 19.Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, et al. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol. 2009;27:1181–5. doi: 10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- 20.Bechter OE, Zou Y, Walker W, Wright WE, Shay JW. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004;64:3444–51. doi: 10.1158/0008-5472.CAN-04-0323. [DOI] [PubMed] [Google Scholar]
- 21.Londoño-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004;64:2324–7. doi: 10.1158/0008-5472.CAN-03-4035. [DOI] [PubMed] [Google Scholar]
- 22.Lang M, Jegou T, Chung I, Richter K, Münch S, Udvarhelyi A, et al. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J Cell Sci. 2010;123:392–400. doi: 10.1242/jcs.053496. [DOI] [PubMed] [Google Scholar]
- 23.Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999;59:4175–9. [PubMed] [Google Scholar]
- 24.Nabetani A, Ishikawa F. Alternative lengthening of telomeres pathway: recombination-mediated telomere maintenance mechanism in human cells. J Biochem. 2011;149:5–14. doi: 10.1093/jb/mvq119. [DOI] [PubMed] [Google Scholar]
- 25.Durant ST. Telomerase-independent paths to immortality in predictable cancer subtypes. J Cancer. 2012;3:67–82. doi: 10.7150/jca.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerone MA, Autexier C, Londoño-Vallejo JA, Bacchetti S. A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene. 2005;24:7893–901. doi: 10.1038/sj.onc.1208934. [DOI] [PubMed] [Google Scholar]
- 27.Fasching CL, Bower K, Reddel RR. Telomerase-independent telomere length maintenance in the absence of alternative lengthening of telomeres-associated promyelocytic leukemia bodies. Cancer Res. 2005;65:2722–9. doi: 10.1158/0008-5472.CAN-04-2881. [DOI] [PubMed] [Google Scholar]
- 28.Marciniak RA, Cavazos D, Montellano R, Chen Q, Guarente L, Johnson FB. A novel telomere structure in a human alternative lengthening of telomeres cell line. Cancer Res. 2005;65:2730–7. doi: 10.1158/0008-5472.CAN-04-2888. [DOI] [PubMed] [Google Scholar]
- 29.Koken MH, Puvion-Dutilleul F, Guillemin MC, Viron A, Linares-Cruz G, Stuurman N, et al. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J. 1994;13:1073–83. doi: 10.1002/j.1460-2075.1994.tb06356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boisvert FM, Hendzel MJ, Bazett-Jones DP. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol. 2000;148:283–92. doi: 10.1083/jcb.148.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, et al. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol. 1999;147:221–34. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong S, Müller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. Role of SUMO-1-modified PML in nuclear body formation. Blood. 2000;95:2748–52. [PubMed] [Google Scholar]
- 35.Weidtkamp-Peters S, Lenser T, Negorev D, Gerstner N, Hofmann TG, Schwanitz G, et al. Dynamics of component exchange at PML nuclear bodies. J Cell Sci. 2008;121:2731–43. doi: 10.1242/jcs.031922. [DOI] [PubMed] [Google Scholar]
- 36.Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I. Specification of SUMO1- and SUMO2-interacting motifs. J Biol Chem. 2006;281:16117–27. doi: 10.1074/jbc.M512757200. [DOI] [PubMed] [Google Scholar]
- 37.Houlard M, Berlivet S, Probst AV, Quivy JP, Héry P, Almouzni G, et al. CAF-1 is essential for heterochromatin organization in pluripotent embryonic cells. PLoS Genet. 2006;2:e181. doi: 10.1371/journal.pgen.0020181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayaydin F, Dasso M. Distinct in vivo dynamics of vertebrate SUMO paralogues. Mol Biol Cell. 2004;15:5208–18. doi: 10.1091/mbc.E04-07-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem. 2000;275:6252–8. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- 40.Van Damme E, Laukens K, Dang TH, Van Ostade X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci. 2010;6:51–67. doi: 10.7150/ijbs.6.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matic I, van Hagen M, Schimmel J, Macek B, Ogg SC, Tatham MH, et al. In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol Cell Proteomics. 2008;7:132–44. doi: 10.1074/mcp.M700173-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nacerddine K, Lehembre F, Bhaumik M, Artus J, Cohen-Tannoudji M, Babinet C, et al. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev Cell. 2005;9:769–79. doi: 10.1016/j.devcel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Quimby BB, Yong-Gonzalez V, Anan T, Strunnikov AV, Dasso M. The promyelocytic leukemia protein stimulates SUMO conjugation in yeast. Oncogene. 2006;25:2999–3005. doi: 10.1038/sj.onc.1209335. [DOI] [PubMed] [Google Scholar]
- 44.Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, et al. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J Cell Sci. 1999;112:381–93. doi: 10.1242/jcs.112.3.381. [DOI] [PubMed] [Google Scholar]
- 45.Sternsdorf T, Jensen K, Will H. Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J Cell Biol. 1997;139:1621–34. doi: 10.1083/jcb.139.7.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell. 2006;24:331–9. doi: 10.1016/j.molcel.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sternsdorf T, Jensen K, Reich B, Will H. The nuclear dot protein sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J Biol Chem. 1999;274:12555–66. doi: 10.1074/jbc.274.18.12555. [DOI] [PubMed] [Google Scholar]
- 48.Dellaire G, Eskiw CH, Dehghani H, Ching RW, Bazett-Jones DP. Mitotic accumulations of PML protein contribute to the re-establishment of PML nuclear bodies in G1. J Cell Sci. 2006;119:1034–42. doi: 10.1242/jcs.02817. [DOI] [PubMed] [Google Scholar]
- 49.Eladad S, Ye TZ, Hu P, Leversha M, Beresten S, Matunis MJ, et al. Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum Mol Genet. 2005;14:1351–65. doi: 10.1093/hmg/ddi145. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi H, Hatakeyama S, Saitoh H, Nakayama KI. Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J Biol Chem. 2005;280:5611–21. doi: 10.1074/jbc.M408130200. [DOI] [PubMed] [Google Scholar]
- 51.Renner F, Moreno R, Schmitz ML. SUMOylation-dependent localization of IKKepsilon in PML nuclear bodies is essential for protection against DNA-damage-triggered cell death. Mol Cell. 2010;37:503–15. doi: 10.1016/j.molcel.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Nagai S, Davoodi N, Gasser SM. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011;21:474–85. doi: 10.1038/cr.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217–25. [PubMed] [Google Scholar]
- 54.Jiang WQ, Zhong ZH, Nguyen A, Henson JD, Toouli CD, Braithwaite AW, et al. Induction of alternative lengthening of telomeres-associated PML bodies by p53/p21 requires HP1 proteins. J Cell Biol. 2009;185:797–810. doi: 10.1083/jcb.200810084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slatter T, Gifford-Garner J, Wiles A, Tan X, Chen YJ, MacFarlane M, et al. Pilocytic astrocytomas have telomere-associated promyelocytic leukemia bodies without alternatively lengthened telomeres. Am J Pathol. 2010;177:2694–700. doi: 10.2353/ajpath.2010.100468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osterwald S, Wörz S, Reymann J, Sieckmann F, Rohr K, Erfle H, et al. A three-dimensional colocalization RNA interference screening platform to elucidate the alternative lengthening of telomeres pathway. Biotechnol J. 2012;7:103–16. doi: 10.1002/biot.201000474. [DOI] [PubMed] [Google Scholar]
- 57.Nabetani A, Yokoyama O, Ishikawa F. Localization of hRad9, hHus1, hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengthening of telomeres-associated promyelocytic leukemia body. J Biol Chem. 2004;279:25849–57. doi: 10.1074/jbc.M312652200. [DOI] [PubMed] [Google Scholar]
- 58.Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, et al. The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet. 2002;11:3135–44. doi: 10.1093/hmg/11.25.3135. [DOI] [PubMed] [Google Scholar]
- 59.Potts PR, Yu H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 2007;14:581–90. doi: 10.1038/nsmb1259. [DOI] [PubMed] [Google Scholar]
- 60.Chung I, Leonhardt H, Rippe K. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J Cell Sci. 2011;124:3603–18. doi: 10.1242/jcs.084681. [DOI] [PubMed] [Google Scholar]
- 61.Zeng S, Xiang T, Pandita TK, Gonzalez-Suarez I, Gonzalo S, Harris CC, et al. Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol. 2009;11:616–23. doi: 10.1038/ncb1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu G, Jiang X, Lee WH, Chen PL. Assembly of functional ALT-associated promyelocytic leukemia bodies requires Nijmegen Breakage Syndrome 1. Cancer Res. 2003;63:2589–95. [PubMed] [Google Scholar]
- 63.Chen YC, Kappel C, Beaudouin J, Eils R, Spector DL. Live cell dynamics of promyelocytic leukemia nuclear bodies upon entry into and exit from mitosis. Mol Biol Cell. 2008;19:3147–62. doi: 10.1091/mbc.E08-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Görisch SM, Wachsmuth M, Ittrich C, Bacher CP, Rippe K, Lichter P. Nuclear body movement is determined by chromatin accessibility and dynamics. Proc Natl Acad Sci U S A. 2004;101:13221–6. doi: 10.1073/pnas.0402958101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jegou T, Chung I, Heuvelman G, Wachsmuth M, Görisch SM, Greulich-Bode KM, et al. Dynamics of telomeres and promyelocytic leukemia nuclear bodies in a telomerase-negative human cell line. Mol Biol Cell. 2009;20:2070–82. doi: 10.1091/mbc.E08-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muratani M, Gerlich D, Janicki SM, Gebhard M, Eils R, Spector DL. Metabolic-energy-dependent movement of PML bodies within the mammalian cell nucleus. Nat Cell Biol. 2002;4:106–10. doi: 10.1038/ncb740. [DOI] [PubMed] [Google Scholar]
- 67.Wachsmuth M, Caudron-Herger M, Rippe K. Genome organization: balancing stability and plasticity. Biochim Biophys Acta. 2008;1783:2061–79. doi: 10.1016/j.bbamcr.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 68.Molenaar C, Wiesmeijer K, Verwoerd NP, Khazen S, Eils R, Tanke HJ, et al. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 2003;22:6631–41. doi: 10.1093/emboj/cdg633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belmont AS, Li G, Sudlow G, Robinett C. Visualization of large-scale chromatin structure and dynamics using the lac operator/lac repressor reporter system. Methods Cell Biol. 1999;58:203–22. doi: 10.1016/S0091-679X(08)61957-3. [DOI] [PubMed] [Google Scholar]
- 70.Belmont AS, Straight AF. In vivo visualization of chromosomes using lac operator-repressor binding. Trends Cell Biol. 1998;8:121–4. doi: 10.1016/S0962-8924(97)01211-7. [DOI] [PubMed] [Google Scholar]
- 71.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, et al. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–8. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krajewski WA, Nakamura T, Mazo A, Canaani E. A motif within SET-domain proteins binds single-stranded nucleic acids and transcribed and supercoiled DNAs and can interfere with assembly of nucleosomes. Mol Cell Biol. 2005;25:1891–9. doi: 10.1128/MCB.25.5.1891-1899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brouwer AK, Schimmel J, Wiegant JC, Vertegaal AC, Tanke HJ, Dirks RW. Telomeric DNA mediates de novo PML body formation. Mol Biol Cell. 2009;20:4804–15. doi: 10.1091/mbc.E09-04-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Everett RD, Chelbi-Alix MK. PML and PML nuclear bodies: implications in antiviral defence. Biochimie. 2007;89:819–30. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 76.Luciani JJ, Depetris D, Usson Y, Metzler-Guillemain C, Mignon-Ravix C, Mitchell MJ, et al. PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J Cell Sci. 2006;119:2518–31. doi: 10.1242/jcs.02965. [DOI] [PubMed] [Google Scholar]
- 77.Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T, et al. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol. 2007;178:925–36. doi: 10.1083/jcb.200703081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, et al. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol. 2006;8:416–24. doi: 10.1038/ncb1386. [DOI] [PubMed] [Google Scholar]
- 79.Benetti R, García-Cao M, Blasco MA. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat Genet. 2007;39:243–50. doi: 10.1038/ng1952. [DOI] [PubMed] [Google Scholar]
- 80.Prowse KR, Greider CW. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci U S A. 1995;92:4818–22. doi: 10.1073/pnas.92.11.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene. 1998;16:1723–30. doi: 10.1038/sj.onc.1201933. [DOI] [PubMed] [Google Scholar]
- 82.Jiang WQ, Nguyen A, Cao Y, Chang AC, Reddel RR. HP1-mediated formation of alternative lengthening of telomeres-associated PML bodies requires HIRA but not ASF1a. PLoS One. 2011;6:e17036. doi: 10.1371/journal.pone.0017036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fasching CL, Neumann AA, Muntoni A, Yeager TR, Reddel RR. DNA damage induces alternative lengthening of telomeres (ALT) associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res. 2007;67:7072–7. doi: 10.1158/0008-5472.CAN-07-1556. [DOI] [PubMed] [Google Scholar]
- 84.Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londoño-Vallejo A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A. 2009;106:15726–31. doi: 10.1073/pnas.0907689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu G, Lee WH, Chen PL. NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G2 phases in immortalized telomerase-negative cells. Implication of NBS1 in alternative lengthening of telomeres. J Biol Chem. 2000;275:30618–22. doi: 10.1074/jbc.C000390200. [DOI] [PubMed] [Google Scholar]
- 86.Nagele RG, Velasco AQ, Anderson WJ, McMahon DJ, Thomson Z, Fazekas J, et al. Telomere associations in interphase nuclei: possible role in maintenance of interphase chromosome topology. J Cell Sci. 2001;114:377–88. doi: 10.1242/jcs.114.2.377. [DOI] [PubMed] [Google Scholar]
- 87.Costa A, Daidone MG, Daprai L, Villa R, Cantù S, Pilotti S, et al. Telomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progression. Cancer Res. 2006;66:8918–24. doi: 10.1158/0008-5472.CAN-06-0273. [DOI] [PubMed] [Google Scholar]
- 88.Perrem K, Colgin LM, Neumann AA, Yeager TR, Reddel RR. Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol Cell Biol. 2001;21:3862–75. doi: 10.1128/MCB.21.12.3862-3875.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang WQ, Zhong ZH, Henson JD, Neumann AA, Chang AC, Reddel RR. Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol Cell Biol. 2005;25:2708–21. doi: 10.1128/MCB.25.7.2708-2721.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hancock R. Internal organisation of the nucleus: assembly of compartments by macromolecular crowding and the nuclear matrix model. Biol Cell. 2004;96:595–601. doi: 10.1016/j.biolcel.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 91.Möller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Dröge W, et al. PML is required for homeodomain-interacting protein kinase 2 (HIPK2)-mediated p53 phosphorylation and cell cycle arrest but is dispensable for the formation of HIPK domains. Cancer Res. 2003;63:4310–4. [PubMed] [Google Scholar]
- 92.Cesare AJ, Reddel RR. Telomere uncapping and alternative lengthening of telomeres. Mech Ageing Dev. 2008;129:99–108. doi: 10.1016/j.mad.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 93.Royle NJ, Foxon J, Jeyapalan JN, Mendez-Bermudez A, Novo CL, Williams J, et al. Telomere length maintenance--an ALTernative mechanism. Cytogenet Genome Res. 2008;122:281–91. doi: 10.1159/000167814. [DOI] [PubMed] [Google Scholar]
- 94.Tarsounas M, West SC. Recombination at mammalian telomeres: an alternative mechanism for telomere protection and elongation. Cell Cycle. 2005;4:672–4. doi: 10.4161/cc.4.5.1689. [DOI] [PubMed] [Google Scholar]
- 95.Slatter TL, Tan X, Yuen YC, Gunningham S, Ma SS, Daly E, et al. The alternative lengthening of telomeres pathway may operate in non-neoplastic human cells. J Pathol. 2012;226:509–18. doi: 10.1002/path.2981. [DOI] [PubMed] [Google Scholar]
- 96.Perrem K, Bryan TM, Englezou A, Hackl T, Moy EL, Reddel RR. Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids. Oncogene. 1999;18:3383–90. doi: 10.1038/sj.onc.1202752. [DOI] [PubMed] [Google Scholar]
- 97.Ishii Y, Tsuyama N, Maeda S, Tahara H, Ide T. Telomerase activity in hybrids between telomerase-negative and telomerase-positive immortal human cells is repressed in the different complementation groups but not in the same complementation group of immortality. Mech Ageing Dev. 1999;110:175–93. doi: 10.1016/S0047-6374(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 98.Chen YJ, Hakin-Smith V, Teo M, Xinarianos GE, Jellinek DA, Carroll T, et al. Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res. 2006;66:6473–6. doi: 10.1158/0008-5472.CAN-06-0910. [DOI] [PubMed] [Google Scholar]
- 99.Stagno D’Alcontres M, Mendez-Bermudez A, Foxon JL, Royle NJ, Salomoni P. Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J Cell Biol. 2007;179:855–67. doi: 10.1083/jcb.200703020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mekeel KL, Tang W, Kachnic LA, Luo CM, DeFrank JS, Powell SN. Inactivation of p53 results in high rates of homologous recombination. Oncogene. 1997;14:1847–57. doi: 10.1038/sj.onc.1201143. [DOI] [PubMed] [Google Scholar]
- 101.Rogan EM, Bryan TM, Hukku B, Maclean K, Chang AC, Moy EL, et al. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol Cell Biol. 1995;15:4745–53. doi: 10.1128/mcb.15.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Razak ZR, Varkonyi RJ, Kulp-McEliece M, Caslini C, Testa JR, Murphy ME, et al. p53 differentially inhibits cell growth depending on the mechanism of telomere maintenance. Mol Cell Biol. 2004;24:5967–77. doi: 10.1128/MCB.24.13.5967-5977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, et al. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 2009;16:1244–51. doi: 10.1038/nsmb.1725. [DOI] [PubMed] [Google Scholar]
- 104.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 106.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A. 2003;100:10635–40. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Elsaesser SJ, Allis CD. HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harb Symp Quant Biol. 2010;75:27–34. doi: 10.1101/sqb.2010.75.008. [DOI] [PubMed] [Google Scholar]
- 108.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–80. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Law MJ, Lower KM, Voon HPJ, Hughes JR, Garrick D, Viprakasit V, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010;143:367–78. doi: 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 110.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–91. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ng LJ, Cropley JE, Pickett HA, Reddel RR, Suter CM. Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res. 2009;37:1152–9. doi: 10.1093/nar/gkn1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sampl S, Pramhas S, Stern C, Preusser M, Marosi C, Holzmann K. Expression of Telomeres in Astrocytoma WHO Grade 2 to 4: TERRA Level Correlates with Telomere Length, Telomerase Activity, and Advanced Clinical Grade. Transl Oncol. 2012;5:56–65. doi: 10.1593/tlo.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang RC, Smogorzewska A, de Lange T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004;119:355–68. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 114.Fairall L, Chapman L, Moss H, de Lange T, Rhodes D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol Cell. 2001;8:351–61. doi: 10.1016/S1097-2765(01)00321-5. [DOI] [PubMed] [Google Scholar]
- 115.Muntoni A, Neumann AA, Hills M, Reddel RR. Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum Mol Genet. 2009;18:1017–27. doi: 10.1093/hmg/ddn436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet. 2000;26:447–50. doi: 10.1038/82586. [DOI] [PubMed] [Google Scholar]
- 117.Pickett HA, Henson JD, Au AY, Neumann AA, Reddel RR. Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum Mol Genet. 2011;20:4684–92. doi: 10.1093/hmg/ddr402. [DOI] [PubMed] [Google Scholar]
- 118.Pickett HA, Cesare AJ, Johnston RL, Neumann AA, Reddel RR. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009;28:799–809. doi: 10.1038/emboj.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hakin-Smith V, Jellinek DA, Levy D, Carroll T, Teo M, Timperley WR, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet. 2003;361:836–8. doi: 10.1016/S0140-6736(03)12681-5. [DOI] [PubMed] [Google Scholar]
- 120.Brachner A, Sasgary S, Pirker C, Rodgarkia C, Mikula M, Mikulits W, et al. Telomerase- and alternative telomere lengthening-independent telomere stabilization in a metastasis-derived human non-small cell lung cancer cell line: effect of ectopic hTERT. Cancer Res. 2006;66:3584–92. doi: 10.1158/0008-5472.CAN-05-2839. [DOI] [PubMed] [Google Scholar]
- 121.Cairney CJ, Hoare SF, Daidone MG, Zaffaroni N, Keith WN. High level of telomerase RNA gene expression is associated with chromatin modification, the ALT phenotype and poor prognosis in liposarcoma. Br J Cancer. 2008;98:1467–74. doi: 10.1038/sj.bjc.6604328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Venturini L, Motta R, Gronchi A, Daidone M, Zaffaroni N. Prognostic relevance of ALT-associated markers in liposarcoma: a comparative analysis. BMC Cancer. 2010;10:254. doi: 10.1186/1471-2407-10-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Onitake Y, Hiyama E, Kamei N, Yamaoka H, Sueda T, Hiyama K. Telomere biology in neuroblastoma: telomere binding proteins and alternative strengthening of telomeres. J Pediatr Surg. 2009;44:2258–66. doi: 10.1016/j.jpedsurg.2009.07.046. [DOI] [PubMed] [Google Scholar]
- 124.Hashizume R, Ozawa T, Gryaznov SM, Bollen AW, Lamborn KR, Frey WH, 2nd, et al. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol. 2008;10:112–20. doi: 10.1215/15228517-2007-052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, et al. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70:9494–504. doi: 10.1158/0008-5472.CAN-10-0233. [DOI] [PubMed] [Google Scholar]
- 126.Corey DR. Telomerase inhibition, oligonucleotides, and clinical trials. Oncogene. 2002;21:631–7. doi: 10.1038/sj.onc.1205063. [DOI] [PubMed] [Google Scholar]
- 127.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–79. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 128.Villa R, Folini M, Lualdi S, Veronese S, Daidone MG, Zaffaroni N. Inhibition of telomerase activity by a cell-penetrating peptide nucleic acid construct in human melanoma cells. FEBS Lett. 2000;473:241–8. doi: 10.1016/S0014-5793(00)01540-4. [DOI] [PubMed] [Google Scholar]
- 129.Kelland LR. Overcoming the immortality of tumour cells by telomere and telomerase based cancer therapeutics--current status and future prospects. Eur J Cancer. 2005;41:971–9. doi: 10.1016/j.ejca.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 130.Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148:651–63. doi: 10.1016/j.cell.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cerone MA, Londono-Vallejo JA, Bacchetti S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum Mol Genet. 2001;10:1945–52. doi: 10.1093/hmg/10.18.1945. [DOI] [PubMed] [Google Scholar]