

Abstract
Background
Many proteomics initiatives require a seamless bioinformatics integration of a range of analytical steps between sample collection and systems modeling immediately assessable to the participants involved in the process. Proteomics profiling by 2D gel electrophoresis to the putative identification of differentially expressed proteins by comparison of mass spectrometry results with reference databases, includes many components of sample processing, not just analysis and interpretation, are regularly revisited and updated. In order for such updates and dissemination of data, a suitable data structure is needed. However, there are no such data structures currently available for the storing of data for multiple gels generated through a single proteomic experiments in a single XML file. This paper proposes a data structure based on XML standards to fill the void that exists between data generated by proteomics experiments and storing of data.
Results
In order to address the resulting procedural fluidity we have adopted and implemented a data model centered on the concept of annotated gel (AG) as the format for delivery and management of 2D Gel electrophoresis results. An eXtensible Markup Language (XML) schema is proposed to manage, analyze and disseminate annotated 2D Gel electrophoresis results. The structure of AG objects is formally represented using XML, resulting in the definition of the AGML syntax presented here.
Conclusion
The proposed schema accommodates data on the electrophoresis results as well as the mass-spectrometry analysis of selected gel spots. A web-based software library is being developed to handle data storage, analysis and graphic representation. Computational tools described will be made available at http://bioinformatics.musc.edu/agml. Our development of AGML provides a simple data structure for storing 2D gel electrophoresis data.
Background
The dissemination of information gathered by high throughput methods is particularly challenging in the post-genomic era due to the sheer volume and diversity of experimental data. The challenge is compounded by the lack of widely accepted standards for all but the most well established methods. This is especially true in the field of proteomics [1] due to competing methodologies and the need for some measure of user intervention in data annotation.
Proteomics for a major part relies on experimental analysis to identify and elucidate proteins in the cell. One of the major experimental methods used in the identification of proteins is 2D gel electrophoresis (2DE) coupled with mass spectrometry (MS). 2DE/MS systems have the ability to identify a large number of proteins in a single sample. Studies have shown that 2DE/MS systems could identify somewhere in the region of thousands of proteins per sample [2-4]. Thus, the amount of information obtained by 2DE/MS is enormous in terms of data. In addition, the choice of 2DE "spots" to analyze through MS is often the object of human intervention. Finally, the 2DE gel matrix is characterized by heterogeneity that results in distortion in the electrophoretic migration pattern. These properties place a high demand on pre-processing, thus requiring to manually curate the gels. The experimental procedures adopted may vary depending on the researcher, but typically results in the creation of a composite representation of repeat analysis often designated as an annotated "virtual gel". The protein isolates (spots) are subsequently analyzed by mass spectrometry and referenced to the normalized position in the composite representation (i.e. the virtual gel). The procedure described outlines not only the need of a standard notation to represent the diverse data generated in the process but also the need for support-intensive manipulation by both users and computational tools specific to the analytical equipment used. Finally, the stored common representation will also need to be regularly updated by bioinformatic tools that automatically query ever-enlarging public repositories.
The eXtensible Markup Language (XML) is particularly well suited to represent biological data and methods and is presently the consensus choice in most areas [5-11]. Accordingly, XML syntax notation was used in this report to identify a suitable syntax for data collected by 2DE/MS systems, which was designated as Annotated Gel Markup Language (AGML). XML notation provides a structured document representation of 2DE/MS experimental data that can be transmitted over the Web as universally understandable, self-describing documents. The main advantage in XML is that data could be integrated in context within a single document, thereby making the data immediately meaningful to a reader, human or machine.
The recent proposals for a formal model to represent a proteomics workflow such as PEDRo (Proteomics Experiment Data Repository) [12] and HUPO ML http://psidev.sourceforge.net/proteomics, provides a structure for proteomics experiments. The model covers many aspects of a proteomics experiment including sample origin, separation techniques and mass spectrometry data analysis. However, it does not provide a structure for storing and dissemination of 2DE/MS data for multiple gel runs generated from a single experiment in the same XML file. PEDRo puts a much stronger emphasis on MS Analysis, while AGML is clearly 2DE centric. PEDRo and HUPO ML framework consist of many distributed entities that need software from the authors to see it. AGML however, consist of one XML file with all the relevant information on that experiment. Also, AGML is always stored as a XML file. Additionally, there are similar projects, such as SASHIMI http://sashimi.sourceforge.net/, that have been developed to provide XML markup to mass spectrometry data and GAML http://www.gaml.org that have been developed to store and archive analytical instrument data. AGML and GAML (Generalized analytical markup language) bear a structural similarity to each other, however its functions and reasons it was developed for are quite different. For example, GAML stores and archives data from analytical instruments, on the other hand AGML stores and archives data from 2DE/MS experiments.
Thus considering the diversity of the data generated through 2DE/MS experiments and a lack of a universal structure for storing and dissemination of 2DE/MS data, we propose AGML as a common representative language for storing and disseminating 2DE/MS experimental data.
Results
AGML version 1.0 was created and implemented as described in Figure 1 using Unified Modeling Language (UML). Many AGML elements are optional, although some are required to identify the data. This allows the user to use the markup even when not all the necessary information is available. The user can then enter the information as it becomes available. An AGML document describes one 2DE/MS experiment that may include one or many gels and, the conditions under which the gels were run. In addition, it also includes a virtual image, often designated as the virtual gel, generated as a composite of real gels as described under materials and methods. It should be noted that many commercial gel analysis software packages accommodate the composition of virtual gel documents. Accordingly, the virtual gel information would have to be parsed from the results file generated by the package. The following is a description of the implementation of AGML schema.
Figure 1.
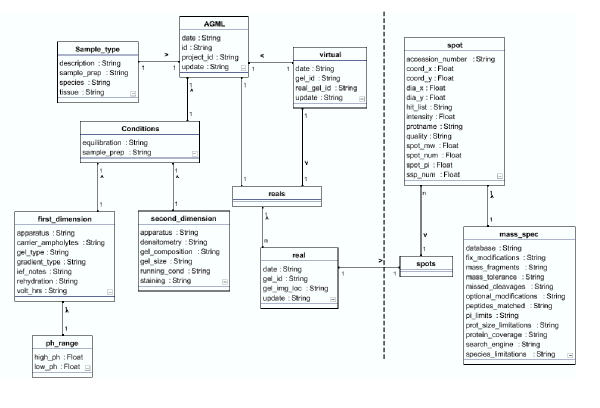
Unified Modeling Language (UML) representation of AGML data model. The dashed line separates the spot-specific from the gel-specific data described in the text as distinct AGML sections. The separation reflects an analytical distinction between the electrophoretic component from the image analysis and mass spectrometry. However, the integration in the common AGML document is seamless. It is represented here for explanatory purposes only. The UML diagram represented was generated using FUJABA http://www.fujaba.de/.
Gel information Section
This section consists of two sub-sections: <sample_type>describing the sample information and the <conditions>section describing the running conditions. The sample information includes the tissue type and the strain/species from where the sample was collected. Under the sample information the user could include as much information as deemed useful.
The next section gives a description of the running conditions under which the gels were run. It includes tags for the description of the apparatus and chemicals used in the experiment. Additionally, under <sample_prep>element the details of the sample preparation protocol or other relevant material could be included.
Under the element tag <first_dimension>, many tags are available to include different variables used during the iso electric focusing period in a 2DE experiment. For example, tags are provided to markup the gel type, gradient type, carrier ampholytes used, and the pH range employed in the iso-electric focusing. In addition, tags are also provided to markup the length of rehydration of the gel and the running conditions for the isoelectric focusing. Under the element tag <second_dimension>, information regarding the second dimension in 2DE experiment could be recorded. Tags provided in this section include the <apparatus>to identify the type of the apparatus used, <gel_composition>to describe the composition of the gel. Additional description of the tagging system is detailed for integrated documentation of individual spots and the various gels that include them. The self-descriptive nature of the XML notation greatly simplifies this task, particularly when accompanied by a UML diagram as represented in Figure 1.
Spot Information Section
The spot information section consist of two sections namely <virtual>describing the virtual gel and <reals>describing the real gel characteristics. The <virtual>is a composite representation of all the annotated spots found in the distinct real gels. The main advantage in having a virtual gel is the comprehensive consensus representation of all the annotated spots, i.e. identified or characterized spots, in the entire 2DE/MS experiment.
The <reals>tag consists of the markup to identify spot information relating to a gel run in a 2DE/MS experiment. Within the <reals>, tags are provided to identify each spot within a given real gel (<real>). Since a gel has many protein spots, under the <spots>tag many <spot>elements could exist. Each <spot>tag, however, has other element that uniquely identifies that spot within the gel. These include a unique id (<ssp_num>), pH (<spot_pi>), molecular weight (spot_mw>) and additional parameters. Additionally, if this spot was subjected to MS and identified, tags are provided to include the data generated through this process. These include among other tags <database>, <search_engine>and, <species_limitations>(see Fig. 1 and web link).
When all the data specific to real gels are completed, the virtual gel can be automatically composed within the AGML document. The virtual gel consists of all the spot information as <reals>, but with an additional tag, <real_gel_id>, to indicate from which real gel the spot information comes from. The software to generate a virtual gel and real gels was written in PHP and Java® (see availability). The tag specifications are constantly being updated; as such please see the AGML 1.0 web site for the latest tag descriptions (see availability).
Data Input and Data Display
The method employed was generated for processing and display of data generated by PDQUEST® software (Bio-RAD, Hercules, CA, USA). However, the proposed XML schema can be used to annotate any type of 2DE gel data. The AGML data is displayed by way of a Java applet through a web interface (Fig. 2). The current implementation displays a web page that includes the virtual gel and sample information. Links are provided to access the real gels and spot information. Text file generated from PDQUEST can easily be uploaded, XML generated and viewed. The spots that are in the virtual gel are highlighted in the real gels with a different color to indicate where each spot came from.
Figure 2.
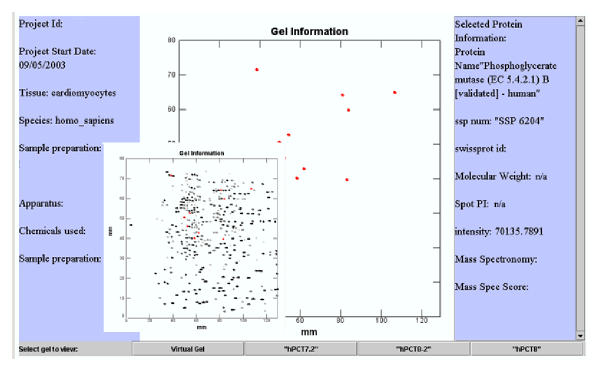
A Sample screenshot of the AGML Visualizer in action. AGML Visualizer software is capable of reading AGML documents and display a visual representation of virtual and real gels described in the AGML document instance. In this representative figure, the real gel is shown as the small figure atop the virtual gel. Left side of the gel depicts all the pertinent information regarding the gel (gel information as described in methods). The right side of the gel displays the information on a particular spot (spot information as described in methods). The AGML Visualizer is based on the AGML schema proposed in this paper.
Discussion
The aim of this paper is to propose standard XML syntax for data exchange and visualization of 2DE/MS experimental data that is designated as the annotated gel markup language (AGML). However, this does not limit adapting AGML, a XML application, for data storage [13]. The proposed AGML syntax captures the essence of a 2D gel experiment and its pertinent MS data, and conveys enough information to analyze and replicate the results. The need to go beyond a format for data storage in the development of the AGML syntax is justified by the diverse set of methods involved and, the enduring obstacles to full automation. The need for a common format to manipulate as well as to store the data is captured by the concept of annotated "virtual gel". This practical solution was reflected in the identification of the data model and ultimately mimicked by the AGML schema.
AGML syntax could easily be adapted by other applications to present the data in XML format. In this specific application the 2DE experimental data was generated using PDQUEST coupled with a MicroMass MALDI-TOF instrument using MassLynx and Micromass global server software for protein identification (MicroMass, Manchester, U.K.). The data generated from a 2DE/MS experiment using the above instrumentation is stored using the manufacturer specific formatting as tab-delimited files. This text file is then converted to AGML syntax through a web interface using software written in PHP (see availability). The conversion of the tab-delimited file to AGML syntax and, if requested, web-based graphical representation, is fully automated. The latter application illustrates the advantage of using AGML as a common format as the graphical displaying is in effect a web-based service available for any dataset represented in our proposed AGML syntax notation. Registered users can then decide whether to deposit the AGML to the database. Since AGML conforms to the XML rules, it's highly flexible and simple to modify [5]. This adaptability of the syntax, also known as content scalability, helps in defining new elements when new information is acquired through 2DE/MS experiments. This is a great asset in an emerging field like proteomics where new information is discovered at a rapid pace, which requires a constant adaptation of the prevailing data model.
In the field of bioinformatics experimental data needs to be analyzed, stored, updated and exchanged often by researchers [5]. To this effect, a bioinformatics infrastructure built around AGML will fulfill all these aspects for 2DE/MS experimental data. The ultimate goal of developing the AGML syntax, is to enable proteomics research to move into the 'browsing mode' of searching through existing information databases along similar lines as proposed by Aebersold [14,15].
The proposed AGML document contains the experimental procedure, the experimental results and the composite virtual gel. It is useful to compare the proposed standards with related work in transcriptomics. For example, MAGE-ML [16] has the representation of DNA array data in XML format as the sole purpose. A similar focus on representation of data is found in ProML [17], which only includes the protein sequence information while not making allowances for the description of the methodological procedure followed. The advantage in incorporating both the experimental procedure and the results, as we have proposed in AGML, is that the data could be understood in the context of the experiment. The methodological detail facilitates repeating the experiment documented in AGML by another researcher. Arguably, the need to include methodological detail in AGML reflects unresolved methodological challenges in proteomic profiling based on 2D gel electrophoresis, a lesser problem in sequencing or transcriptomics projects.
AGML can be incorporated to be used with large descriptors of proteomics information. Using XML namespace rules, AGML markup can easily be incorporated into any other schema. Specifically, the PEDRo model proposed by Taylor et al. explicitly accommodates the representation of 2DE/MS data [12] where AGML could be particularly useful. AGML by no way replaces the structure envisioned by PEDRo; instead proposes an XML format for handling 2DE/MS data that can be incorporated into the existing large schemas such as PEDRo. In order to transmit subsets of information from this repository, the PEDRo model has to employ other methods [12]. Accordingly, the PEDRo model could use AGML syntax to transmit the description of individual 2DE/MS experiments. For that purpose, the AGML could greatly benefit from general-purpose XML translation languages (e.g. XSLT). Additionally, with the wide use of resource description framework (RDF, http://www.w3.org/RDF), AGML can easily be incorporated into other proposed model frameworks that have been written in XML. Thus incorporating the strengths of AGML, such as storing multiple gel runs per experiment in the same file, with the strengths of other proposed models such as PEDRo and HUPO ML.
Conclusions
AGML notation provides users with a compact representation scheme for 2DE/MS experimental data that can be delivered over the World Wide Web as universally understandable self-defining documents. In addition to the advantages inherent to representing information as alphanumeric strings that can be easily stored, transmitted between machines and between applications using emerging XML technologies such as SOAP, AGML is particular suitable for usage in the development of proteomics web-services. The emphasis of AGML to accommodate not only the data but also methodological detail reflects unresolved methodological challenges in proteomics profiling based on 2D Gel Electrophoresis. The proposed AGML syntax was developed for an integrative bioinformatics infrastructure encompassing a facility for high throughput 2DE/MS, a computational biology group analyzing the data and, finally, researchers and clinicians collecting the samples in a concerted effort to identify proteomic profile patterns indicative of various degrees of active disease or predisposition thereof. As such AGML syntax is ideal for incorporation into complex proteomics schemas such as PEDRo, as well as storing information as a standalone applications, due to XML's self-describing nature, for future reference.
Methods
Design principles
For the purpose of AGML, we attempted to include all essential information that is required to identify a spot generated in a 2D-gel electrophoresis experiment. In order to accomplish this we used meaningful syntax familiar to investigators in the field to markup the data and kept the markup to the sufficient minimum. The latter is important to reduce the size of the document, which is important in storage and transmission. Also, the syntax used does not constrain the meaning of the data it holds. The syntax was designed merely to provide a placeholder in context for the data. An AGML document can represent one 2DE/MS experiment consisting of the sample information, running conditions and spot data of several gels (known as real gels) that were run per experiment (Fig. 1). In addition, the AGML document should also contain a composite representation of the set of real gels, known as the virtual gel, that represents the annotated spots of the real gels. The virtual gel concept developed in AGML provides a representation of all the gels per experiment that have their spots identified and annotated represented in one virtual gel. This is somewhat different from other uses of the virtual gel concept in proteomics where it is meant to represent a reconstructed gel, whose molecular weight information is acquired by mass spectrometry rather than by gel electrophoresis [18]. In keeping to these guidelines we propose the following structure for the XML application AGML. An implementation decision was taken to follow a recommendation that all tags be elements with no attributes [19]. This is because the elements in AGML schema represent the logical units of information and further clarification, in our view, was not required.
Structure of AGML documents
The AGML documents consist of two main sections: the gel information section, and the spot information section (Fig. 1). The gel information section describes the information about the sample and the conditions under which the sample was run. The spot information section consists of the spot information in each gel. Two subsections make up the spot information section, one that holds information about the 2DE gels (known as real gels) and the other the reconstructed virtual gel. The virtual gel represents the annotated spots in the real gels. In addition, under the root element, elements are included to identify a specific AGML document. Thus, the main features of the structure of an AGML document are as follows:
Gel information section
1) Sample section
Describes the pertinent information about the sample.
2) Conditions sections
Describes 2DE-running conditions. In addition, it also contains the instruments used, protocols and reagents.
Spot information section
3) Virtual gel section
Describes the virtual representation of all the annotated spots of real gel data.
4) Real gel section
Stores the data from the 2DE experiment that were run per experiment. It can contain any number of 2DE gels that describes the specific experiment. It can also include additional information from the mass spectrum of the protein spot that was used for the protein identification.
XML schema for the proposed AGML structure and the elements defined in the schema are available on the AGML web site http://bioinformatics.musc.edu/agml.
Authors' contributions
RS and JSA devised the schema and wrote the manuscript. RS wrote the PHP scripts to handle data and to generate the AGML documents. LHJ made the web interface and maintains the AGML Visualizer. JA and MS provided experimental expertise. JA contributed in writing the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We would like to thank Shuyuan Wu for the critical reading of the manuscript and Jennifer Franklin for initial writing of the software for AGML Visualizer. This work was supported by the NHLBI Proteomics Initiative through contract N01-HV-28181.
Contributor Information
Romesh Stanislaus, Email: stanisrc@musc.edu.
Liu Hong Jiang, Email: jiangliu@musc.edu.
Martha Swartz, Email: swartzm@musc.edu.
John Arthur, Email: arthurj@musc.edu.
Jonas S Almeida, Email: almeidaj@musc.edu.
References
- Tyers M, Mann M. From genomics to proteomics. Nature. 2003;422:193–197. doi: 10.1038/nature01510. [DOI] [PubMed] [Google Scholar]
- Gorg A, Obermaier C, Boguth G, Harder A, Scheibe B, Wildgruber R, Weiss W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 2000;21:1037–1053. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1037::AID-ELPS1037>3.3.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Fey SJ, Larsen PM. 2D or not 2D. Two-dimensional gel electrophoresis. Curr Opin Chem Biol. 2001;5:26–33. doi: 10.1016/S1367-5931(00)00167-8. [DOI] [PubMed] [Google Scholar]
- Harry JL, Wilkins MR, Herbert BR, Packer NH, Gooley AA, Williams KL. Proteomics: capacity versus utility. Electrophoresis. 2000;21:1071–1081. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1071::AID-ELPS1071>3.3.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Achard F, Vaysseix G, Barillot E. XML, bioinformatics and data integration. Bioinformatics. 2001;17:115–125. doi: 10.1093/bioinformatics/17.2.115. [DOI] [PubMed] [Google Scholar]
- Freier A, Hofestadt R, Lange M, Scholz U, Stephanik A. BioDataServer: a SQL-based service for the online integration of life science data. In Silico Biol. 2002;2:37–57. [PubMed] [Google Scholar]
- Kitano H. Standards for modeling. Nat Biotechnol. 2002;20:337. doi: 10.1038/nbt0402-337a. [DOI] [PubMed] [Google Scholar]
- Lacroix Z. Biological data integration: wrapping data and tools. IEEE Trans Inf Technol Biomed. 2002;6:123–128. doi: 10.1109/TITB.2002.1006299. [DOI] [PubMed] [Google Scholar]
- Martin AC. Can we integrate bioinformatics data on the Internet? Trends Biotechnol. 2001;19:327–328. doi: 10.1016/S0167-7799(01)01733-4. [DOI] [PubMed] [Google Scholar]
- Matsuno H, Doi A, Hirata Y, Miyano S. XML documentation of biopathways and their simulations in Genomic Object Net. Genome Inform Ser Workshop Genome Inform. 2001;12:54–62. [PubMed] [Google Scholar]
- Juty NS, Spence HD, Hotz HR, Tang H, Goryanin I, Hodgman TC. Simultaneous modelling of metabolic, genetic and product-interaction networks. Brief Bioinform. 2001;2:223–232. doi: 10.1093/bib/2.3.223. [DOI] [PubMed] [Google Scholar]
- Taylor CF, Paton NW, Garwood KL, Kirby PD, Stead DA, Yin Z, Deutsch EW, Selway L, Walker J, Riba-Garcia I, et al. A systematic approach to modeling, capturing, and disseminating proteomics experimental data. Nat Biotechnol. 2003;21:247–254. doi: 10.1038/nbt0303-247. [DOI] [PubMed] [Google Scholar]
- Bos B. XML representation of relational database. 1997. http://www.w3.org/XML/RDB.html
- Aebersold R. Constellations in a cellular universe. Nature. 2003;422:115–116. doi: 10.1038/422115a. [DOI] [PubMed] [Google Scholar]
- Marte B. proteomics. Nature. 2003;422:191. doi: 10.1038/422191a. [DOI] [Google Scholar]
- Spellman PT, Miller M, Stewart J, Troup C, Sarkans U, Chervitz S, Bernhart D, Sherlock G, Ball C, Lepage M, et al. Design and implementation of microarray gene expression markup language (MAGE-ML) Genome Biol. 2002;3:RESEARCH0046. doi: 10.1186/gb-2002-3-9-research0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch D, Zimmer R, Lengauer T. ProML – the protein markup language for specification of protein sequences, structures and families. In Silico Biol. 2002;2:313–324. [PubMed] [Google Scholar]
- Walker AK, Rymar G, Andrews PC. Mass spectrometric imaging of immobilized pH gradient gels and creation of "virtual" two-dimensional gels. Electrophoresis. 2001;22:933–945. doi: 10.1002/1522-2683()22:5<933::AID-ELPS933>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Cover R. SGML/XML Elements versus Attributes: When should I use Elements, and when should I use Attributes? 2000. http://www.oasis-open.org/cover/elementsAndAttrs.html