

Abstract
Patients receiving lenalidomide are at an increased risk for deep venous thrombosis (DVT). Here, we prospectively investigated the DVT risk in patients with relapsed chronic lymphocytic leukemia treated with lenalidomide (n=32). Five patients developed six incidents of DVT over one year for an annual incidence of 16%. Three of these were considered drug-related. Median time to DVT was 105 days (range 56–259 days). No pulmonary embolism was detected. Hypercoagulability screen before study entry was negative in all patients who subsequently developed DVTs. Compared to normal volunteers CLL patients had increased baseline levels of D-dimer, thrombin-antithrombin, soluble vascular endothelial adhesion molecule 1 (sVCAM-1), and thrombomodulin (P<0.001). After one week on lenalidomide D-dimer, thrombomodulin, sVCAM-1, factor VIII, TNFα, and C-reactive protein were significantly increased while protein C was decreased (P<0.001). In patients with lenalidomide-related DVTs, TNFα, and sVCAM-1 were more strongly upregulated than in all other patients (P<0.05) and TNFα and sVCAM-1 levels were significantly correlated (r = 0.65, P<0.001). These data link lenalidomide associated DVTs with TNFα upregulation and endothelial cell dysfunction and suggest that aspirin may have a role for DVT prophylaxis in these patients.
Keywords: Lenalidomide, venous thrombosis, tumor necrosis factor alpha, inflammation, endothelial dysfunction
Introduction
The immunomodulating drugs (IMIDs) thalidomide and lenalidomide have shown promising activity in hematologic malignancies [1]. However, there is an increased risk of venous thromboembolism (VTE) in patients on IMIDs. The risk with single agent therapy appears low; for lenalidomide the incidence of VTE in multiple myeloma (MM) is generally <5% [2], and in patients with myelodysplastic syndrome the incidence is <1% [3]. In patients enrolled on phase II trials of single agent lenalidomide for relapsed chronic lymphocytic leukemia (CLL) pulmonary embolism has been reported in 2 of 45 patients [4] and a DVT occurred in one of 44 patients [5] suggesting that the incidence of VTE in CLL and MM may be comparable. Because of the relatively low risk VTE prophylaxis has not been routinely used in patients treated with single agent IMIDs [6]. In contrast, patients treated with IMID based combination therapies are at high risk for VTE, which in some studies affected more than 50% of patients with MM [6]. Several regimens for VTE prophylaxis have been shown to be efficacious in patients treated with IMID based combination regimens [6–8]. While the American College of Chest Physicians strongly recommends against the use of aspirin for VTE prophylaxis in high-risk cancer patients [9], two expert panels recently established guidelines for MM patients undergoing IMID based combination therapy that recommend aspirin (81–325 mg) for patients with no or one risk factor and LMWH 40 mg daily or warfarin (INR 2–3) for patients at high risk for DVT [6, 10]. There is an increased risk of bleeding during VTE prophylaxis in patients with liver or renal failure, or thrombocytopenia [6]. Thrombocytopenia in particular is a common side effect of lenalidomide in CLL [4, 5].
Recommendations for prophylaxis are complicated by the fact that the factors contributing to the increased risk of VTE during IMID therapy are not well understood [11]. In MM none of the classical coagulation parameters were associated with increased risk of VTE [12]. In MM several disease specific factors related to the presence of the monoclonal protein are thought to contribute to a hypercoagulable state including acquired protein C resistance [12] and impaired fibrinolysis [13]. Other factors described in MM that shift the balance towards activation of coagulation include high levels of von Willebrand factor antigen, plasminogen activator inhibitor-1 (PAI-1), and changes in endothelial cell function [11, 13]). Gieseler proposed the reduction of soluble TM and the development of acquired protein C resistance as the major factors inducing a prothrombotic state during the treatment of MM [11]. It has been pointed out that the absence of an increased risk of VTE in MM patients treated with single agent thalidomide or lenalidomide indicate that these drugs are not directly thrombogenic. Thus combination effects with other agents or disease specific factors appear to significantly contribute to the risk.
The mechanism of anti-tumor efficacy of the IMIDs is an area of intense study. While down-modulation of TNFα expression and anti-angiogenic properties have initially been hypothesized to play an important role, increasing evidence implicates activation of immune effector cells and modulation of cytokine levels as key mechanisms [1, 14]. Some of these effects may be disease specific. In CLL in particular, lenalidomide leads to a clinically significant inflammatory syndrome characterized by lymph node swelling in the context of a systemic cytokine release syndrome [4, 5, 15, 16]. We and others have shown that lenalidomide induces the release of many different pro-inflammatory cytokines in CLL patients including TNFα [15, 16]. These inflammatory side effects are unique to CLL and are not observed in MDS and very rarely in patients with MM, or lymphoma.
Little is known about specific risk factors for VTE in CLL in general or during lenalidomide therapy in particular. We therefore incorporated a prospective, systematic analysis of coagulation parameters into a phase II trial of single agent lenalidomide for relapsed CLL. Because of the many uncertainties related to risk of VTE in CLL with single agent lenalidomide, the risk of thrombocytopenia in our heavily pretreated patient population, and the controversies about the best agent for prophylaxis we did not use routine prophylaxis.
Methods
Patients and clinical samples
With informed consent in accordance with the Declaration of Helsinki and approval from the NHLBI institutional Review Board, 33 patients with relapsed CLL were enrolled on this investigator-initiated phase II single agent lenalidomide trial (ClinicalTrials.gov Identifier: NCT00465127) and 32 received the study drug. One patient chose not to start drug. The starting dose for the first 10 patients was 20 mg daily; the starting dose for subsequent patients was lowered to 10 mg daily because of toxicities observed in other trials using lenalidomide in CLL [17]. Lenalidomide was given for 4 cycles of 21 days on followed by 21 days off drug. Responding patients could receive additional 4 cycles. Aspirin or VTE prophylaxis was not mandated. Corticosteroids were permitted in cases with severe inflammatory side effects as reported previously [16]. A hypercoagulability screen including testing for deficiencies of proteins C, S, or antithrombin, and analysis of factor V Leiden and prothrombin gene 20210 G->A polymorphism was performed prior to therapy. Venous Doppler ultrasound studies were obtained at baseline, after completion of cycle 4, and cycle 8, and whenever a DVT was suspected. DVTs that had a temporal relationship to drug ingestion and could not be explained by immobilization or mechanical compression were considered drug-related.
Measurement of inflammatory and coagulation markers
Serum and plasma samples were collected prior to therapy, on day 8, day 22, and day 42 of cycle 1 and were stored at −80°C. Serum samples were evaluated for TNFα (R&D systems, Minneapolis, MN, detection range: 15.6 to 11218.7 pg/ml), thrombomodulin (TM, R&D systems normal range 4–67 ng/ml), soluble vascular endothelial adhesion molecule 1 (sVCAM-1, R&D systems, range: <0.6 ng/ml, normal range 349–991 μg/ml) and C-reactive protein (CRP, Siemens Healthcare, Marburg, Germany, normal range <1 mg/dl). Plasma samples were evaluated for quantitative D-dimer (normal range <0.4 μg/ml), protein C (normal range 59–144%), protein S (normal range 59–144%), factor VIII (normal range 41–184%), antithrombin (AT, normal range 57–134%) (all Diagnostica Stago, Parsippany, NJ), tissue factor (TF, Imubind tissue factor ELISA kit, American Diagnostica Stamford, CT, normal range: <10 pg/ml) and thrombin-antithrombin (TAT) complexes (normal range 1.3–4 mg/L, Enzygnost TAT micro, Siemens Healthcare Diagnostics). All tests were carried out according to manufacturer’s guidelines.
Statistical analysis
Continuous and categorical variables for different subgroups were compared using the Wilcoxon rank-sum test and the Fisher’s exact test. Change of measurements of serum and plasma markers were examined by the Wilcoxon signed-rank test for paired data. To compare the baseline coagulation markers of CLL patients with normal volunteers, the Z-test was used to determine whether the percentage of patients exceeding the normal range is significantly greater than 10%. Pearson’s correlation coefficient was used to describe correlations. All tests were two-tailed and a p-value <0.05 was used as the criterion for statistical significance. No adjustment for multiple comparisons has been made. Analyses were performed using the R statistical software (Version 2.10.1; www.r-project.org).
Results
Patient characteristics and incidence of DVTs
From May 2007 to February 2010, 32 patients were treated on this single center trial. Baseline characteristics of the study patients are summarized in Table 1: median age was 64 years (38–78), median number of prior therapies 3, and 72% had bulky lymphadenopathy. Risk factors for VTE such as obesity, indwelling catheters, cardiac disease, or renal insufficiency were present in the minority of patients and all patients had a negative Doppler ultrasound prior to starting lenalidomide. Five patients had a history of DVT dating back 1 to 45 years prior to study entry. Three of these patients were on therapeutic anticoagulation. Two patients were found to have a factor V Leiden mutation in keeping with the incidence in the general population (18). These two patients were maintained on LMWH prophylaxis during the trial. One of these patients also carried the prothrombin gene 20210 G->A polymorphism. Five patients had a low protein S level and three patients had low anti-thrombin levels. Lower extremity venous Doppler studies performed prior to the start of treatment to rule out inclusion of patients with pre-existing DVTs were negative.
Table 1.
Patient characteristics at baseline and by DVT history prior to starting therapy
| All patients n=32 | No DVT n=27 | DVT n=5 | p* | |
|---|---|---|---|---|
| Continuous variables | Median | Median | Median | |
| Age in years | 64 | 64 | 63 | n.s. |
| Prior therapies | 3 | 3 | 1 | n.s. |
| TNFα (pg/ml) | 28.2 | 26.2 | 28.2 | n.s. |
| CRP (<1 mg/dl) | 0.41 | 0.41 | 0.41 | n.s. |
| Thrombomodulin (μg/ml) | 95 | 104 | 85 | n.s. |
| VCAM (349–991 μg/ml) | 2390 | 2368 | 2562 | n.s. |
| D-dimer(<0.4μg/ml) | 0.43 | 0.42 | 0.5 | n.s. |
| Categorical variables | n | n | n | |
| Rai stage III-IV | 16 | 14 | 2 | n.s. |
| Bulky disease (LN >5 cm) | 23 | 21 | 2 | n.s. |
| Obesity (BMI>30) | 7 | 5 | 2 | n.s. |
| Venous catheter | 1 | 1 | 0 | n.s. |
| Cardiac disease | 2 | 2 | 0 | n.s. |
| Renal disease | 2 | 2 | 0 | n.s. |
| Positive lower extremity doppler | 0 | 0 | 0 | n.s. |
| Protein S low (55–134%) | 5 | 5 | 0 | n.s. |
| Protein C low (59–144%) | 0 | 0 | 0 | n.s. |
| Antithrombin low (57–137%) | 3 | 3 | 0 | n.s. |
| Factor V Leiden mutation (n=23) | 2 | 2 | 0 | n.s. |
| Prothrombine gene mutation (n=23) | 1 | 1 | 0 | n.s. |
The two-sided p-value for comparing each variable between patients with or without DVT prior to therapy.
Five patients developed six incidents of DVT over a one year observation period for an annual incidence of 16%. None of these was complicated by pulmonary embolism. The time to DVT after starting lenalidomide was variable (Figure 1). The earliest DVT occurred on day 56 in a 77 year old male with a history of provoked DVT who was maintained initially on LMWH anticoagulation and developed a DVT 2 weeks after starting oral anticoagulation. DVT on day 94 occurred in a 78 year old male who was maintained on low dose ASA. DVT at day 105 occurred in a 63 year old female with a history of unprovoked DVT who was on LMWH anticoagulation. These DVTs were classified as lenalidomide related. In contrast three other DVTs could be explained by other mechanisms. The DVT at day 158 occurred in a 53 year old patient who developed a DVT in the setting of progressive disease with massive inguinal lymphadenopathy causing venous obstruction. The DVT at day 259 occurred in a 63 year old female who had a prior history of provoked DVT and was bed-ridden due to CMV colitis. DVT at day 314 was a repeat DVT found on venous Doppler studies on routine restaging in the patient who had had a DVT at day 94. This was the only subclinical DVT observed in the study. At the time he was off lenalidomide. He had been maintained on LWMH anticoagulation for 6 months after his first DVT and resolution of the thrombus had been documented. Three patients suffered from proximal femoral vein thromboses while one patient had an iliac vein thrombosis. In the majority of cases the thromboses were extensive and 3 patients were treated with low-dose tPA delivered into the clot using a pulse-spray catheter.
Figure 1. Lenalidomide exposure and cumulative DVT incidence over 1 year.
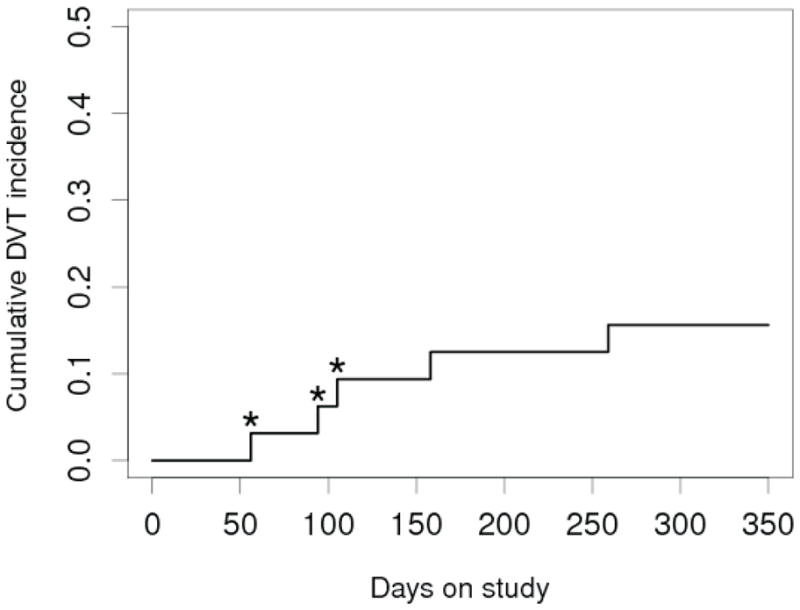
The cumulative incidence of DVT is shown. The recurrent DVT in the second patient with that was noted on day 314 on routine ultrasound is omitted from this graph. Lenalidomide related DVTs are marked with an asterisk (*).
The pretreatment hypercoagulability screen was negative in all patients who subsequently developed a DVT during the study period (Table 2). Two of these patients were fully anticoagulated due to a preceding history of DVT, and 2 had a BMI >30. Other risk factors for DVTs such as indwelling venous catheters, cardiac disease or renal insufficiency were absent. The only baseline factor significantly different between patients who developed a DVT during the study period and patients with no DVT was a history of previous DVTs (p=0.02).
Table 2.
Patient characteristics at baseline by incidence and type of a new DVT event during lenalidomide therapy
| All patients n=32 | No DVT n=27 | unrelated DVT n=2 | related DVT n=3 | |
|---|---|---|---|---|
| Continuous variables | Median | Median | Median | |
| Age in years | 64 (38–78) | 64 | 59 | 77 |
| Prior therapies | 3 (1–8) | 3 | 1 | 3 |
| Categorical variables | n | n | n | n |
| Bulky disease (LN >5 cm) | 23 | 19 | 2 | 2 |
| Obesity (BMI>30) | 7 | 5 | 1 | 1 |
| Venous catheter | 1 | 1 | 0 | 0 |
| Cardiac disease | 2 | 2 | 0 | 0 |
| Renal disease | 2 | 2 | 0 | 0 |
| History of DVT | 5 | 2 | 1 | 2 |
| Anticoagulation (Enoxa. 1mg/kg bid) | 3 | 1 | 0 | 2 |
| Enoxaparin prophylaxis (40 mg) | 2 | 2 | 0 | 0 |
| Low dose aspirin (81 mg) | 2 | 1 | 0 | 1 |
| Protein S low (55–134%) | 5 | 5 | 0 | 0 |
| Protein C low (59–144%) | 0 | 0 | 0 | 0 |
| Antithrombin low (57–137%) | 3 | 3 | 0 | 0 |
| Factor V Leiden polymorph (n=23) | 2 | 2 | 0 | 0 |
| Prothrombin gene polymorph (n=23) | 1 | 1 | 0 | 0 |
CLL patients have abnormal coagulation parameters at baseline
There is a paucity of data on the state of the coagulation system in CLL patients [18]. Here, we measured C-reactive protein (CRP), a panel of coagulation factors, and endothelial cell markers in 32 relapsed CLL patients prior to starting lenalidomide (Figure 2). Four of these parameters were significantly increased in CLL patients compared to normal controls defined as >10% of patients having levels above the normal range (P<.001). The number of patients with values exceeding the normal range was 23 (72%) for TM, 29 (91%) for sVCAM-1, 17 (53%) for D-dimer, and 10 (31%) for TAT. The levels of CRP, F VIII, Protein S and C, and AT were within the normal range for >90% of the patients. These findings suggest ongoing activation of coagulation and altered endothelial cell function at baseline in a majority of these relapsed patients with extensive disease.
Figure 2. Abnormal coagulation parameters in CLL patients prior to initiating lenalidomide therapy.

Parameters of inflammation, coagulation, and endothelial cell function at baseline (n=32) are normalized to the corresponding upper limit of normal that is set as 100%. The Whisker plot shows the 25th, 50th, and 75th percentile as part of the box, the dashed line indicates the range.
Lenalidomide induced inflammation, TNFα upregulation, and activation of coagulation
Lenalidomide induces a prominent inflammatory state in CLL, especially during the first cycles [16]. CRP, as a sensitive marker of inflammation, significantly increased on lenalidomide therapy in all patients with a median upregulation of 4.5-fold on day 8 and regressed to pretreatment levels by the end of the 3 week off drug period in the majority of patients (Figure 3A). TNFα, a key inflammatory cytokine,[19] was also up-regulated a median 3-fold by day 8 (Figure 3B) and regressed to almost baseline levels during the off drug period. Inflammation and in particular TNFα have been reported to cause endothelial dysfunction [20]. To estimate lenalidomide’s effects on endothelial cells in vivo, we measured sVCAM-1 and sTM, whose serum levels increase in states of endothelial dysfunction [21]. Both markers were significantly increased in patients while on lenalidomide therapy (Figure 3C and D). D-dimer levels also increased by day 8 of lenalidomide therapy indicating increased fibrin turnover and returned to pre treatment values thereafter (Figure 3E). Protein C levels rapidly decreased in the initial phase of lenalidomide therapy and returned to pre-treatment values when the drug was stopped (Figure 3F). Factor VIII levels increased on lenalidomide therapy and remained increased at day 42 (figure 3G). On day 8 of lenalidomide levels of TAT, Protein S, and TF were unchanged (data not shown). Effects continued through 3 weeks of drug therapy as indicated by persistent elevation of CRP (P<0.01, n=24), TNFα (P<0.01, n=25), sVCAM1 (P<0.01, n=25), sTM (P<0.05, n=20) and factor VIII (P<0.01, n=21) on day 22. In summary, lenalidomide induces an inflammatory state, leads to endothelial dysfunction, and activates the coagulation system.
Figure 3. Effects of lenalidomide on markers of inflammation, and endothelial dysfunction and on coagulation factors.

Measurements were obtained prior to therapy (pre), on day 8 of lenalidomide and on day 42 of cycle 1, 3 weeks off lenalidomide (post). Only patients with all 3 measurements are shown. The 0, 25, 50, 75 and 100th percentiles are shown in box charts. (A) CRP (n=32) (B) TNFα (n= 31) (C) sVCAM1 (n=30) (D) sTM (n=27) (E) D-dimer(n= 31) (F) Protein C (n= 29) (G) FVIII (n=24).
TNFα and endothelial dysfunction as mediators of lenalidomide-related VTE
To explore whether lenalidomide induced changes in the coagulation system contribute to an increased incidence of VTE we compared patients with lenalidomide related DVTs (n=3) to all other patients (n=29). Up-regulation of TNFα, TF and sVCAM1 was significantly higher in patients with lenalidomide-related DVTs compared to patients with no DVT or DVTs unrelated to lenalidomide (Figures 4A, B). sTM was also relatively more increased in patients with lenalidomide related DVTs but this difference did not quite reach statistical significance (Figure 4C). Next, we examined correlations between TNFα and markers of endothelial dysfunction. TNFα blood levels on day 8 correlated highly with both sVCAM-1 and sTM (Figure 4E and F).
Figure 4. Correlation between TNFα, endothelial dysfunction and lenalidomide-related DVTs.

A–D. Change of expression levels in patients without DVTs, DVTs unrelated to lenalidomide (Unrel. DVT) and related to lenalidomide (Rel. DVT), with exact p-value for comparison between patients with lenalidomide-related DVT and all other patients using the Wilcoxon rank-sum test: A. TNFα B. TF C. sVCAM1 and D. sTM. E–F. Pearson’s correlation between TNFα on day 8 of lenalidomide and E. sVCAM1 (n=30) and F. sTM (n=32).
Discussion
VTE is a frequent complication of cancer and patients with hematologic malignancies are at highest risk. One study reported a 28-fold increase in the incidence of VTE in patients with hematologic malignancies as compared to controls [22]. The specific incidence of VTE in CLL patients is not well defined and little is known about disease specific predisposing factors. In our cohort with a maximum observation period of one year, five of 32 CLL patients (16%) developed six incidents of DVT while receiving lenalidomide. This is a higher rate of VTE then has been reported for single agent lenalidomide treatment in MM [2], MDS [3], or in other phase II studies of lenalidomide in CLL where it was <5% [4, 5]. While five patients in our study had a history of prior VTE, this is also considerably higher than the expected annual recurrence rate of 5–6% in patients with prior proximal DVT or PE [23]. Thus, additional disease specific and treatment related factors appear to have contributed to a hypercoagulable state. Consistent with disease related activation of coagulation and endothelial cell dysfunction, a majority of our patients showed elevations of TAT complexes, D-dimer, sTM, and sVCAM1 prior to starting lenalidomide (Figure 2), a finding that has not been previously reported. However, these baseline characteristics failed to identify patients with subsequent DVTs. In two patients DVTs could be attributed to immobility and local compression, respectively. In three patients DVTs were related to lenalidomide and all of these occurred in the first 3 months of therapy during a period of inflammation.
Immune activation is thought to be a major contributor to the anti-tumor effects of lenalidomide [1, 14]. We and others have shown that lenalidomide induced immune activation frequently causes significant systemic symptoms, lymph node swelling, increased CRP, and cytokine release [4, 5, 15, 16]. While the association between chronic inflammation and cardiovascular disease has been firmly established [24] the relationship with VTE remains less clear [25]. Supporting such a link is the observation that acute infections in a community setting have been associated with a 2-fold increase in VTE rates in the subsequent weeks to months [26]. Increased levels of TNFα, IL-6, IL-8, and MCP-1 have been found in patients with a history of VTE. In these studies blood collections took place after the thrombotic event was diagnosed. Thus, it is possible that the increase in these cytokines is reactive. In contrast, we collected plasma samples on all study participants prospectively and found lenalidomide induced upregulation of CRP, TNFα, and FVIII consistent with an acute phase reaction. sVCAM1 and sTM were significantly elevated indicating worsening endothelial dysfunction on lenalidomide (Figure 3). In addition, protein C, a crucial anticoagulant, was significantly decreased (Figure 3). Non-specific suppression of protein synthesis in the liver is unlikely to explain the effect on protein C given that protein S, AT, and albumin showed no significant changes. While serum markers were measured in the first cycle, drug related DVTs manifested clinically over a period of 3 months from the start of treatment. It is noteworthy that patients with pronounced inflammatory reactions typically also had more pronounced reactions in subsequent cycles. It thus appears that changes we observed in cycle one do not to immediately cause DVTs, but may primarily contribute an additional risk factor.
Several factors could contribute to inflammation induced VTE. However, in our series only lenalidomide induced upregulation of TNFα, sVCAM1, and TF were significantly associated with subsequent thrombosis (Figure 4). To study the relative predictive value of a prior history of DVT and increases in TNFα and sVCAM1 we compared a number of logistic regression models with lenalidomide associated DVTs as the primary outcome. In these models either the degree of increase in TNFα or sVCAM was more predictive than DVT history, and the best prediction for the development of a drug related DVT was the combination of history of DVT with increase in TNFα (p=0.0013; data not shown). Whether lenalidomide-induced upregulation of TNFα and sVCAM1 could be useful to identify patients at highest risk for treatment related VTE awaits confirmation in a larger cohort of patients.
Our in vivo observations are consistent with in vitro findings that culturing HUVEC cells in the presence of IMIDs and TNFα lead to increased TF activity [27]. Thus, our data indicate a possible mechanism for lenalidomide related VTE that centers on drug induced upregulation of TNFα with consequent endothelial dysfunction and increased activation of TF. Endothelial cell damage has also been invoked to explain the increased incidence of DVT when anti-angiogenesis drugs were combined with chemotherapy agents such as cisplatin or gemcitabine [28, 29].
Effective strategies for DVT prevention in lenalidomide treated patients may therefore profitably focus on protecting the endothelium by reducing the inflammatory syndrome and in particular TNFα up-regulation. While aspirin is not considered an effective DVT prophylaxis in typical medical or surgical settings, it has proven to be effective in MM patients and some ongoing trials of lenalidomide in CLL also incorporate aspirin prophylaxis. Interestingly, in vitro studies also indicated a mild inhibitory effect of aspirin on TF [27]. There has been no apparent dose related difference between 81 mg and 325 mg aspirin for VTE prophlaxis in MM. In CLL, the pronounced inflammatory reaction to lenalidomide raises the question whether aspirin at a dose with an increased anti-inflammatory effect could be more effective. However, the dose of aspirin that would be optimal for down-regulation of TNFα is not clear, and its effectiveness may also be influenced by regulation of other inflammatory cytokines[30]. Thus the optimal prophylaxis for lenalidomide related DVTs remains to be defined.
Acknowledgments
This work was supported by the NIH Intramural Research program. This work was presented in part at the annual American Society of Hematology meeting December 6th, 2009 in New Orleans, LA.
We are most indebted to our patients whose participation made this research possible. We thank Khanh Nghiem and Alison Cooper for performing the coagulation studies. Dr. Richard Chang conducted the thrombolysis procedure.
Footnotes
Authorship contribution:
GA, JNL, AW designed research, GA, JNL, AMC, LS, PMC, AW performed research, GA, JNL, AW contributed vital reagents, GA, JNL, SS, AW contributed to clinical monitoring and collected data, GA, JNL, AW analyzed and interpreted data, XT performed statistical analysis, and GA, JNL, AW wrote the manuscript.
Disclosure:
The authors declare no competing financial interests.
The publisher or recipient acknowledges right of the US government to retain a nonexclusive, royalty-free license to any copyright covering the article.
Reference List
- 1.Chanan-Khan AA, Cheson BD. Lenalidomide for the treatment of B-cell malignancies. J Clin Oncol. 2008;26:1544–1552. doi: 10.1200/JCO.2007.14.5367. [DOI] [PubMed] [Google Scholar]
- 2.Bennett CL, Angelotta C, Yarnold PR, et al. Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer. JAMA. 2006;296:2558–2560. doi: 10.1001/jama.296.21.2558-c. [DOI] [PubMed] [Google Scholar]
- 3.Yang X, Brandenburg NA, Freeman J, et al. Venous thromboembolism in myelodysplastic syndrome patients receiving lenalidomide: results from postmarketing surveillance and data mining techniques. Clin Drug Investig. 2009;29:161–171. doi: 10.2165/00044011-200929030-00003. [DOI] [PubMed] [Google Scholar]
- 4.Chanan-Khan A, Miller KC, Musial L, et al. Clinical efficacy of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia: results of a phase II study. J Clin Oncol. 2006;24:5343–5349. doi: 10.1200/JCO.2005.05.0401. [DOI] [PubMed] [Google Scholar]
- 5.Ferrajoli A, Lee BN, Schlette EJ, et al. Lenalidomide induces complete and partial remissions in patients with relapsed and refractory chronic lymphocytic leukemia. Blood. 2008;111:5291–5297. doi: 10.1182/blood-2007-12-130120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palumbo A, Rajkumar SV, Dimopoulos MA, et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia. 2008;22:414–423. doi: 10.1038/sj.leu.2405062. [DOI] [PubMed] [Google Scholar]
- 7.Baz R, Li L, Kottke-Marchant K, et al. The role of aspirin in the prevention of thrombotic complications of thalidomide and anthracycline-based chemotherapy for multiple myeloma. Mayo Clin Proc. 2005;80:1568–1574. doi: 10.4065/80.12.1568. [DOI] [PubMed] [Google Scholar]
- 8.Klein U, Kosely F, Hillengass J, et al. Effective prophylaxis of thromboembolic complications with low molecular weight heparin in relapsed multiple myeloma patients treated with lenalidomide and dexamethasone. Ann Hematol. 2009;88:67–71. doi: 10.1007/s00277-008-0561-1. [DOI] [PubMed] [Google Scholar]
- 9.Geerts WH, Pineo GF, Heit JA, et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338S–400S. doi: 10.1378/chest.126.3_suppl.338S. [DOI] [PubMed] [Google Scholar]
- 10.Palumbo A, Davies F, Kropff M, et al. Consensus guidelines for the optimal management of adverse events in newly diagnosed, transplant-ineligible patients receiving melphalan and prednisone in combination with thalidomide (MPT) for the treatment of multiple myeloma. Ann Hematol. 2010;89:803–811. doi: 10.1007/s00277-010-0925-1. [DOI] [PubMed] [Google Scholar]
- 11.Gieseler F. Pathophysiological considerations to thrombophilia in the treatment of multiple myeloma with thalidomide and derivates. Thromb Haemost. 2008;99:1001–1007. doi: 10.1160/TH08-01-0009. [DOI] [PubMed] [Google Scholar]
- 12.Elice F, Fink L, Tricot G, et al. Acquired resistance to activated protein C (aAPCR) in multiple myeloma is a transitory abnormality associated with an increased risk of venous thromboembolism. Br J Haematol. 2006;134:399–405. doi: 10.1111/j.1365-2141.2006.06208.x. [DOI] [PubMed] [Google Scholar]
- 13.Uaprasert N, Voorhees PM, Mackman N, et al. Venous thromboembolism in multiple myeloma: current perspectives in pathogenesis. Eur J Cancer. 2010;46:1790–1799. doi: 10.1016/j.ejca.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Ramsay AG, Gribben JG. Immune dysfunction in chronic lymphocytic leukemia T cells and lenalidomide as an immunomodulatory drug. Haematologica. 2009;94:1198–1202. doi: 10.3324/haematol.2009.009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andritsos LA, Johnson AJ, Lozanski G, et al. Higher doses of lenalidomide are associated with unacceptable toxicity including life-threatening tumor flare in patients with chronic lymphocytic leukemia. J Clin Oncol. 2008;26:2519–2525. doi: 10.1200/JCO.2007.13.9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aue G, Njuguna N, Tian X, et al. Lenalidomide-induced upregulation of CD80 on tumor cells correlates with T-cell activation, the rapid onset of a cytokine release syndrome and leukemic cell clearance in chronic lymphocytic leukemia. Haematologica. 2009;94:1266–1273. doi: 10.3324/haematol.2009.005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol. 2007;25:5047. doi: 10.1200/JCO.2007.14.2141. [DOI] [PubMed] [Google Scholar]
- 18.Negaard HF, Iversen PO, Ostenstad B, et al. Hypercoagulability in patients with haematological neoplasia: no apparent initiation by tissue factor. Thromb Haemost. 2008;99:1040–1048. doi: 10.1160/TH07-09-0541. [DOI] [PubMed] [Google Scholar]
- 19.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–497. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Park Y, Wu J, et al. Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond) 2009;116:219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elice F, Jacoub J, Rickles FR, et al. Hemostatic complications of angiogenesis inhibitors in cancer patients. Am J Hematol. 2008;83:862–870. doi: 10.1002/ajh.21277. [DOI] [PubMed] [Google Scholar]
- 22.Blom JW, Doggen CJ, Osanto S, et al. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715–722. doi: 10.1001/jama.293.6.715. [DOI] [PubMed] [Google Scholar]
- 23.Baglin T, Douketis J, Tosetto A, et al. Does the clinical presentation and extent of venous thrombosis predict likelihood and type of recurrence? A patient level meta-analysis. J Thromb Haemost. 2010;8:2436–2442. doi: 10.1111/j.1538-7836.2010.04022.x. [DOI] [PubMed] [Google Scholar]
- 24.Ridker PM, Cushman M, Stampfer MJ, et al. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 25.Fox EA, Kahn SR. The relationship between inflammation and venous thrombosis. A systematic review of clinical studies. Thromb Haemost. 2005;94:362–365. doi: 10.1160/TH05-04-0266. [DOI] [PubMed] [Google Scholar]
- 26.Smeeth L, Cook C, Thomas S, et al. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367:1075–1079. doi: 10.1016/S0140-6736(06)68474-2. [DOI] [PubMed] [Google Scholar]
- 27.Valsami S, Ruf W, Leikauf MS, et al. Immunomodulatory drugs increase endothelial tissue factor expression in vitro. Thromb Res. 2011;127:264–271. doi: 10.1016/j.thromres.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 28.Kuenen BC, Rosen L, Smit EF, et al. Dose-finding and pharmacokinetic study of cisplatin, gemcitabine, and SU5416 in patients with solid tumors. J Clin Oncol. 2002;20:1657–1667. doi: 10.1200/JCO.2002.20.6.1657. [DOI] [PubMed] [Google Scholar]
- 29.Zangari M, Siegel E, Barlogie B, et al. Thrombogenic activity of doxorubicin in myeloma patients receiving thalidomide: implications for therapy. Blood. 2002;100:1168–1171. doi: 10.1182/blood-2002-01-0335. [DOI] [PubMed] [Google Scholar]
- 30.Kast RE. Aspirin, TNF-alpha, NFkB, and survival in multiple myeloma: the importance of measuring TNF-alpha. Inflammopharmacology. 2006;14:256–259. doi: 10.1007/s10787-006-1532-6. [DOI] [PubMed] [Google Scholar]