

Abstract
BACKGROUND
Neuroblastoma (NBL) is a common pediatric solid tumor, and outcomes for advanced neuroblastoma remain poor despite extremely aggressive treatment. Chemotherapy resistance at relapse contributes heavily to treatment failure. The poor survival of high-risk patients urges investigation into novel treatment options and better understanding of resistance mechanisms. Based on our previous work and on data from publicly available studies, we hypothesized that Her4 contributes to resistance.
METHODS
We reduced Her4 expression with shRNA and overexpressed intracellular HER4, and tested the impact on tumor cell survival under various culture conditions. The resulting changes in gene expression after HER4 knockdown were measured by mRNA-array.
RESULTS
HER4 expression was up-regulated in tumor spheres compared with monolayer culture. With HER4 knockdown, NBL cells became less resistant to anoikis and serum starvation. Moreover, HER4 knockdown increased the chemo-sensitivity of NBL cells to cisplatin, doxorubicin, etoposide and activated ifosfamide. In mRNA-array analysis, HER4 knockdown predominately altered genes related to cell cycle regulation. In NBL spheres compared to monolayer, cell proliferation is decreased and cyclin D expression is reduced. HER4 knockdown reversed cyclin D suppression. Overexpressed intracellular HER4 slowed the cell cycle and induced chemo-resistance.
CONCLUSIONS
HER4 protects NBL cells from multiple exogenous apoptotic stimuli including anoikis, nutrient deficiency and cytotoxic chemotherapy. The intracellular fragment of HER4 is sufficient to confer this phenotype. HER4 functions as a cell cycle suppressor, maintaining resistance to cellular stress. Our finding indicates that HER4 over-expression may be associated with refractory disease, and HER4 may be an important therapeutic target.
Keywords: Neuroblastoma, HER4/ERBB4, Cellular stress, Chemoresistance, Multicellular tumor sphere
INTRODUCTION
Neuroblastoma is a common childhood cancer, most frequently diagnosed before age one.1 About half of patients are high-risk, with survival rates below 40% despite intensive multimodal therapy. Relapsed disease is rarely cured, highlighting the urgent need for new therapeutic options for high-risk neuroblastoma.
Therapies against the ERBB family of receptor tyrosine kinases (EGFR, HER2, HER3, and HER4) have improved outcomes for some common carcinomas.2 We and others have reported the relationship between ERBB receptors and neuroblastoma,3–5 suggesting ERBB signaling promotes neuroblastoma growth and survival. We reported superior in vivo growth inhibition from a pan-ERBB inhibitor compared to EGFR-specific therapy.4, 6 An immunohistochemistry study of NBL patient samples suggested that HER4 may correlate with worse outcome.5 We recently used a publicly available oncogenomic dataset to analyze the prognostic impact of HER4 expression in NBL patients.7 In four independent data sets, higher HER4 expression significantly correlated with reduced survival (Fig. 1). Notably, these DNA microarray data were generated by different probes that recognize different HER4 sequences, suggesting the observed difference is unlikely to be due to artifact. Collectively, the above evidence highly raised the importance of HER4 in NBL, which warrants further mechanistic study.
Figure 1.

HER4 forms homodimers or heterodimers with the other ERBB members to activate signaling via multiple second messengers, which is pivotal for its final biological effects.2 One means by which HER4 may contribute to malignant behavior is increased expression of Her4 in non-adherent tumor spheroids, possibly due to signaling mediated by cell-cell adhesion.8 3-D cultured tumor spheroids are known to have acquired chemo and radio resistance,9–11 which has been reported for various cancer types.9–12 Although neuroblastoma may show the same pattern of multicellular resistance13, very few studies specifically focused on elucidating the underlying mechanism.
The role of HER4 in cancer remains controversial.14–16 HER4 expression was observed to be an adverse prognostic factor in some studies,17 but a favorable factor in others, even of the same tumor type.18, 19 These disparate results may arise from expression of four alternatively spliced isoforms of HER4, which differ in subcellular localization and function.20, 21 The alternatively used α exon (JM-a/JM-d isoforms) encodes a recognition element for ADAM17-mediated proteolysis. Isoforms lacking this exon (JM-b/JM-c) cannot be cleaved by ADAM17. After two-step proteolysis by ADAM17 (required first) then γ-secretase, the cytosolic fragment of HER4 (4ICD/p80) is released from the membrane and may traffic to the nucleus.22 The CYT-1 and CYT-2 isoforms differ in their subsequent activation of downstream signaling pathways and protein degradation processes.23 Some studies failed to distinguish between membranous, cytoplasmic and nuclear expression of HER4, confounding associations between expression and outcome.
To assess the role of HER4 in neuroblastoma, we evaluated HER4 expression under different culture conditions, discovering HER4 up-regulation during cell stress. HER4 knockdown by shRNA decreased resistance to multiple adverse factors, including loss of attachment, serum starvation, and cytotoxic agents. Transduced intracellular HER4 reduced cell proliferation and protected cells from apoptosis. Our findings suggest that HER4 plays a protective role against cellular stress in neuroblastoma, indicating that HER4 expression might be an unfavorable prognostic factor, and potential therapeutic target for neuroblastoma.
MATERIALS AND METHODS
Cell culture and reagents
IMR32, CHP134, and LAI-55N, were cultured as described.4 COL, cultured as described,16 was previously thought to be an osteosarcoma. However, based on expression of CD56 and Nestin, lack of expression of other small round blue cell tumor markers, massive N-Myc amplification and in vivo growth characteristics (pseudorosette formation, metastasis to bone marrow, liver and retroorbital space in SCID mice), it is now confirmed to be neuroblastoma. All cell lines have been authenticated using STR DNA fingerprinting by Characterized Cell Line Core Facility in MDACC. For sphere culture condition, single cell suspensions mobilized from monolayers were seeded onto poly-HEMA (Sigma-Aldrich) pre-coated plates [1×105 cells/cm2]. Cisplatin (CDDP), doxorubicin (DXR) and etoposide (VP-16) were purchased from Sigma-Aldrich. 4-hydroxy-ifosfamide was purchased from Niomech.
Cell proliferation and viability assay
Cell proliferation was measured by direct nucleus counting as described,24 or by Alamar Blue assay in 96 well format.
Soft agar clonogenic assay
Soft agar clonogenesis was assessed as described.25 Cells were seeded in 0.35% top agar at 2,000 cells/well on 0.5% bottom agar in six-well plates. After 21 days, viable colonies were stained with MTT (Sigma), photographed (ChemiDoc XRS, Bio-rad), and counted by Quantitive One ® software (Bio-rad).
DNA vector design and retroviral transduction
ShRNA sequences were: human HER4 targeting shRNA (sense: 5’GATCCCCGCCAA GAAAGCGTTTGACATTCAAGAGATGTCAAACGCTTTCTTGGCTTTTTC3’; antisense: 5’TCGAGAAAAAGCCAAGAAAGCGTTTGACATCTCTTGAATGTCAAACGCTTTCTTG GCGGG3’); Scrambled control shRNA (sense: 5’GATCCCCGCGCGCTTTGTAGGATT CGTTCAAGAGACGAATCCTACAAAGCGCGCTTTTTC3’, antisense: 5’TCGAGAAAAA GCGCGCTTTGTAGGATTCGTCTCTTGAACGAATCCTACAAAGCGCGCGGG3’). The shRNA was inserted into circular pSuperior.retro.neo+gfp (OligoEngine) vector following digestion with BgLII and XhoI (New England BioLabs Inc.).
HER4 intracellular domain inserts 4ICD-CYT1 and 4ICD-CYT2 were amplified by PCR using described techniques22 and inserted into the multiple cloning site of the MIgR1-mKate2-C retroviral expression vector to create a 4ICD with a c-terminal mKate2 tag. The MIgR1-mKate2-C vector was generated from the Hes1-MIgR1 vector25 by replacement of Hes1 (first position) with mKate2,26 and replacement of EGFR with a puromycin resistance gene. Cells were transduced as described,25 selected for puromycin resistance, then sorted by FACS for far-red fluorescence.
Western Blot analysis
Cells were lysed and immunoblotted as described.24 Primary antibodies: [EGFR #1902-1, HER4 #1200-1 (Epitomics); PARP #9542, Cyclin D #2978 (Cell Signaling). Beta-actin (Sigma-Aldrich) served as loading control.
Cell cycle analysis
Monolayer-cultured cells were detached by cell dissociation buffer (Invitrogen). Spheres were pelleted by centrifuge, and disaggregated by incubation with cell dissociation buffer for 10 minutes in 37°C. Cell cycle was analyzed with propidium iodide (PI) and flow cytometry as described.24
mRNA microarray analysis
Col-NC and Col-SH2 cells were grown in six-well plates, four replicates each of monolayer or spheres. Total RNA was extracted using mirVana™ miRNA Isolation Kit (Ambion). Total RNA was amplified and converted into biotin-labeled cRNA using Illumina® TotalPrep™ RNA Amplification Kit. After purification, 0.75 μg cRNA was fragmented and hybridized to HumanHT-12 v4 BeadChip (Illumina). The chips were scanned with iScan system (Illumina), and the signal intensities were quantified and summarized using GenomeStudio software (Illumina). After removal of the probesets with intensities below noise, the data were quantile normalized. The significantly differentially expressed genes were identified using two-sample t-tests and a beta-uniform mixture (BUM) model to control false discovery rate (FDR) at 5%. The pathway analysis was performed using the significant genes in the Ingenuity Pathway Analysis (IPA) software.
BrdU incorporation assay
Monolayer cells were treated with BrdU for 15 minutes before harvesting, while spheres were incubated with BrdU overnight. The cells were fixed and treated with DNase followed by staining with FITC-anti-BrdU antibody (BD, 1:50), and analyzed with flow cytometry.
RESULTS
HER4 expression was density dependent and up-regulated in sphere culture
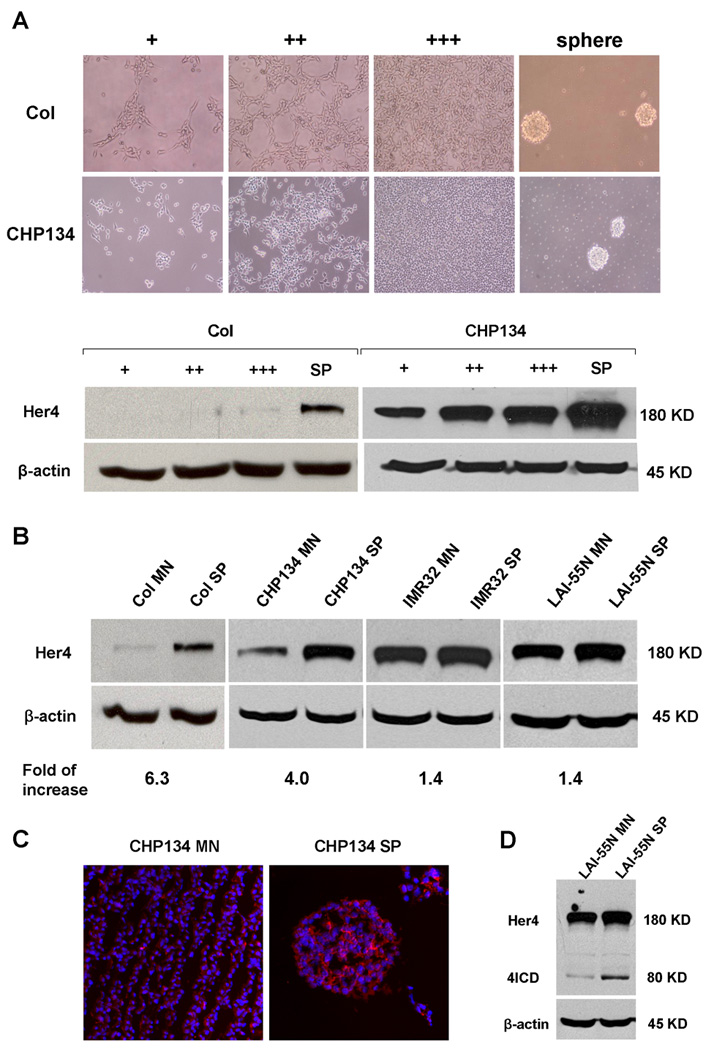
We measured expression of HER4 in neuroblastoma cell lines by western blot. In Col and CHP134 cells, HER4 expression increased with increasing cell density (Fig.1, A). When converted from monolayer to anchorage-independent culture, neuroblastoma cells tended to aggregate spontaneously and form micro tumor spheres as previously described.27 We detected a striking up-regulation of HER4 in neuroblastoma spheres; the range of up-regulation varied from 1.4 to 6.3 fold (Fig.2 B, C). Notably, cell lines with low basal levels of HER4 exhibited much higher up-regulation than cell lines with high basal levels of HER4 expression. Moreover, even in high-HER4 cells LAI-55N that showed minor up-regulation of HER4, we observed increased intracellular HER4 (Fig2. D). We also compared EGFR, HER2, HER3 expression between monolayer and sphere culture. Unlike HER4, EGFR expression did not increase in sphere culture, while basal HER2 and HER3 expression was too low to detect,4 and did not increase in sphere culture (data not shown). These data show that HER4 expression varies depending on the culture conditions, suggesting that HER4 may regulate response to different conditions.
Figure 2.
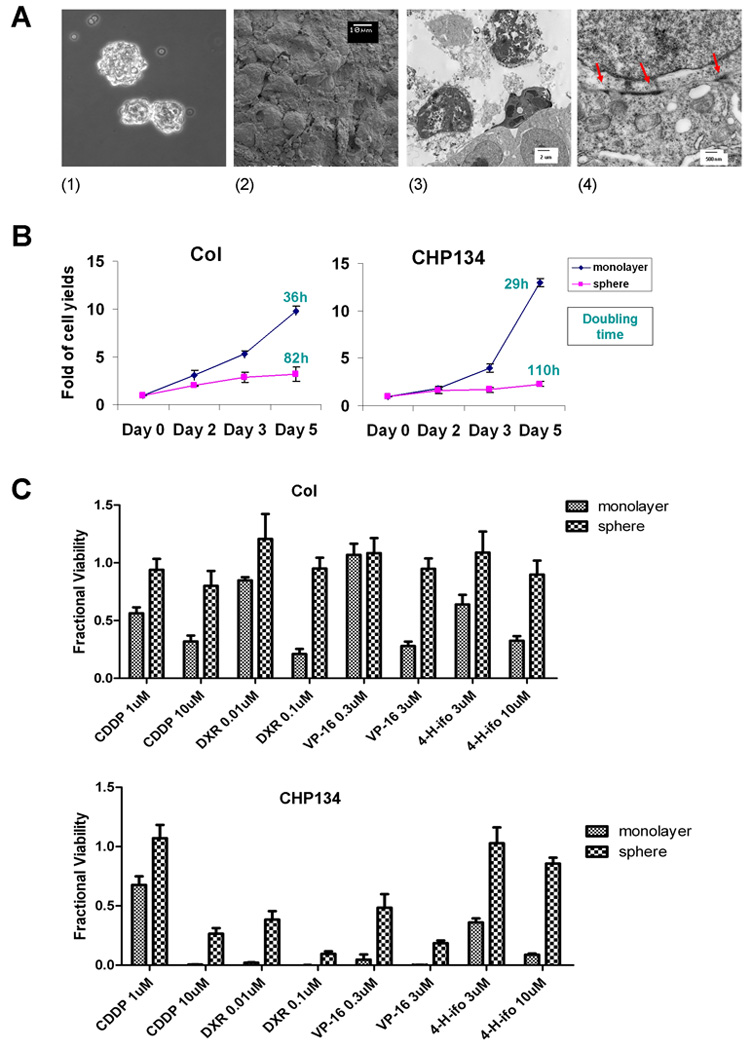
Neuroblastoma tumor spheres exhibited chemo-resistance
We seeded Col and CHP134 cells onto plates pre-coated with non-adherent substrate. After 24 hours, compact spheres were generated spontaneously (Fig. 3A, part 1). As shown by scanning electron microscopy (SEM), individual tumor cells adhered tightly with adjacent cells, such that the cell boundary was obscure (Fig. 3A, part 2). Further culture for 96 hours resulted in a mixture of healthy, necrotic and apoptotic cells in the center of the spheres (Fig. 3A, part 3). Transmission electron microscopy (TEM) revealed that tight cell-cell adhesion was well established within spheres (Fig. 3A, part 4). We compared cell proliferation in monolayer and sphere culture using Alamar Blue assay. Tumor spheres grew at a much lower rate than monolayer culture with elongated doubling time (Fig. 3B). We reasoned that since chemotherapy predominantly affects cells that are rapidly dividing, that the slower-growing cells in spheres might be more resistant to chemotherapy. To see whether the sphere culture would cause NBL cells to be less sensitive to chemotherapy, we compared cell viability between monolayer culture and spheres following exposure to drugs that are clinically used for neuroblastoma therapy: cisplatin (CDDP), doxorubicin (DXR), Etoposide (VP-16), and 4-hydroxy-ifosfamide (4-H-ifo). Col and CHP134 cells in sphere culture were more chemo-resistant than cells grown in monolayer (Fig. 3C). These results suggest that neuroblastoma cells can survive and grow in anchorage-independent conditions by forming spheres, which confers chemo-resistant phenotype, perhaps due to reduced proliferation.
Figure 3.
Knockdown of HER4 suppressed anchorage-independent survival
Since HER4 is up-regulated in sphere culture, we asked whether the up-regulation is pro-apoptotic or protective for cells. If HER4 is crucial for cells to survival, survival will decrease when HER4 is inhibited. To address this question, we used shRNA to knockdown HER4 expression in Col since it showed the highest HER4 increase when transferring from monolayer to sphere culture. We selected three clones with stable HER4 expression reduction by more than 95% (Fig. 4A). We first tested the impact of HER4 knockdown on proliferation. After 72 hours in monolayer culture, a slight decrease in cell yield was observed (Fig. 4B), possibly due to slightly increased apoptosis as shown by increased PARP cleavage in 3 shRNA clones (Fig 4A). Furthermore, we conducted a soft agar clonogenic assay to measure anchorage-independent growth. The cells with HER4 knockdown showed a significant reduction in colony formation (P<0.001) (Fig. 4C).
Figure 4.
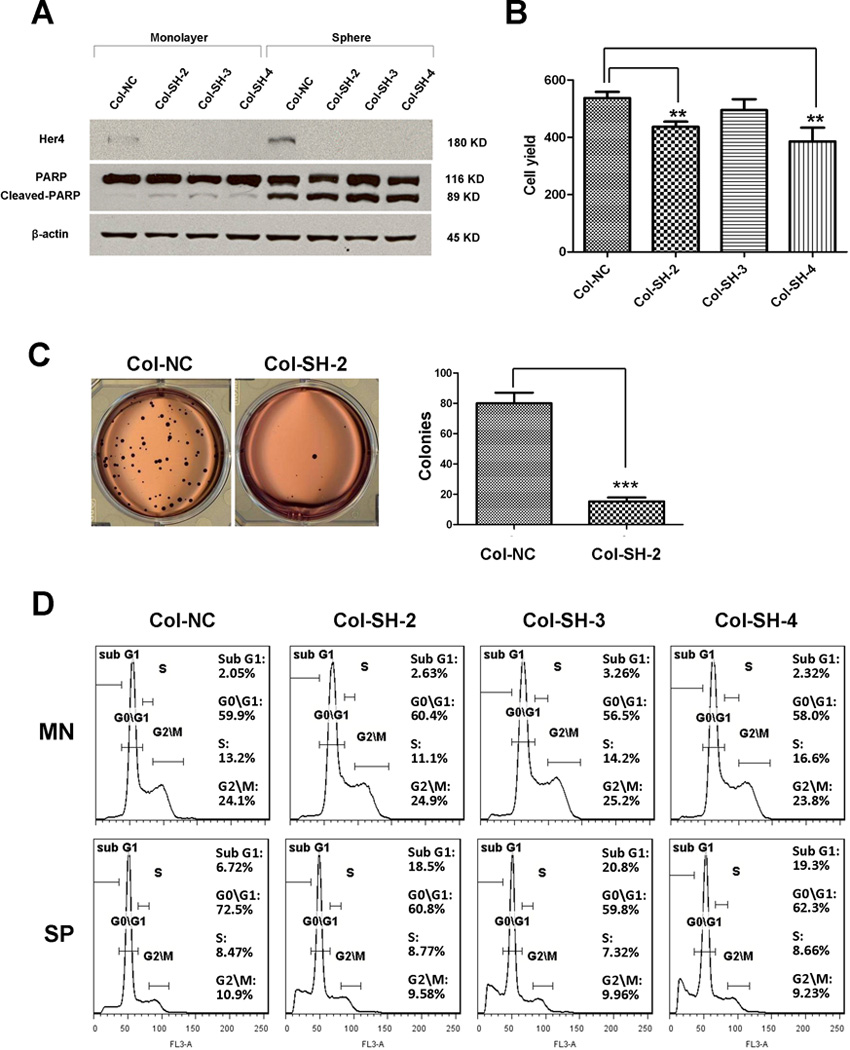
The soft agar clonogenic assay measures tumor survival as single cells in the anchorage-independent condition. Multicellular tumor spheres may be more resistant to cellular stress (see figure 3), so we wanted to determine whether HER4 knockdown could also affect the survival of tumor spheres. We conducted a cell cycle analysis based on PI staining, and quantified the sub G1 population to represent cell death. We found that cells grown in sphere culture exhibited G1 arrest compared to monolayer culture, consistent with our observation that tumor spheres are slow-growing. HER4 knockdown caused a slight increase in the sub G1 population in monolayer cells, but a larger increase in the sub G1 population when cells were grown as spheres (Fig. 4D). To test whether the sub G1 population represented apoptosis, we measured the level of PARP cleavage by immunoblotting. All 3 shRNA clones had an increased levels of cleaved PARP compared to control (Fig. 4A), indicating apoptosis. Together, these findings demonstrated that knockdown of HER4 in Col reduced survival in anchorage-independent growth by increasing apoptosis both in single cell or multi-cellular spheres. This indicates that HER4 is crucial for anoikis resistance in neuroblastoma.
Knockdown of HER4 reduced tolerance to serum starvation
The data above imply that in stressed conditions like loss of surface attachment, tumor cells up-regulate HER4 expression; loss of HER4 renders cells susceptible to death from stressful conditions. We wondered how generalizable this HER4 response would be to other cellular stress. Hence, we first tested whether serum starvation had any impact on HER4 expression. After culturing Col and CHP134 cells without FBS for 24, 48, and 72 hours, HER4 was up-regulated in a time dependent manner (Fig. 5A). The HER4 intracellular fragment also increased in proportion with the full length protein. To evaluate the importance of HER4 in survival during serum deprivation, Col with non-sense and HER4 specific shRNA were cultured in monolayer or as spheroids, and FBS was withdrawn 24 hours later. Cells were starved for 48 hours before measuring cell cycle. In monolayer culture, HER4 knockdown cells showed a higher sub G1 population in response to serum deprivation, whereas non-sense control cells showed few cells in the sub G1 population (Fig. 5B). In tumor spheres, the increase in the sub G1 population caused by HER4 knockdown was even more dramatic due to the co-existence of two stress factors (Fig. 5B). These data indicate that HER4 knockdown caused Col cells to become less resistant to limited nutrition. Because nutrition insufficiency is a very common event in solid tumors due to high metabolism of cancer, this finding suggests that HER4 may be critical for tumor cells to adapt to nutritional insufficiency in neuroblastoma.
Figure 5.

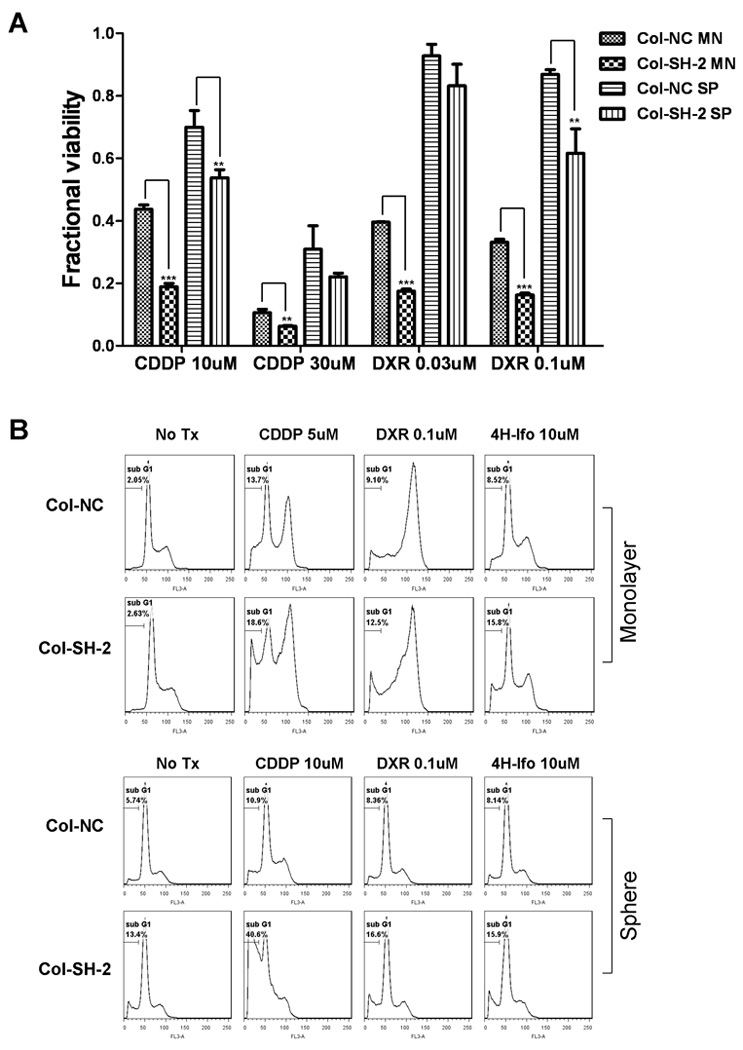
HER4 knockdown increased chemo-sensitivity
We have observed the chemo-resistance in neuroblastoma tumor spheres, and our previous data supports a role for HER4 in cellular stress response and apoptosis regulation. Therefore, we investigated whether HER4 up-regulation in tumor spheres contributes to chemo-resistance. We performed a viability assay on cell lines with knocked down HER4 after exposure to the drugs commonly used in treating neuroblastoma. After 72 hours, all HER4 knockdown cells presented lower viability upon treatment with cytotoxic drug than control cells (Fig. 6A). Furthermore, cell cycle analysis showed that HER4 knockdown clones had a dramatically higher proportion of the sub G1 population compared to non-sense control cells (Fig. 6B), indicating increased apoptosis. With HER4 knockdown, monolayer cells showed increased chemo-sensitivity and tumor spheres lost their chemo-resistance phenotype. These findings suggest that HER4 is required to provide neuroblastoma cells with a protective mechanism against cytotoxic drugs. The resistance to chemotherapy observed in neuroblastoma cells when cultured as tumor spheres is partially due to HER4 up-regulation.
Figure 6.
HER4 is involved in cell cycle regulation
To understand how HER4 contributes to stress protection and chemo-resistance, we used an mRNA microarray to identify changes in gene expression induced by HER4 knockdown. Col cells were transduced with HER4 specific or control shRNA and cultured in either monolayer or sphere condition; mRNA was isolated from four individual biological replicates under each condition. We measured the gene expression of all samples using the HumanHT-12 V4 bead-chip (Illumina). We compared control vs. knock-down samples for each condition, and differentially expressed genes were identified with 5% FDR. To categorize these changes functionally, we used Ingenuity Pathway Analysis (IPA). In monolayers, the “molecular and cellular functions” most altered by Her4 knock-down were cell cycle-associated genes. In the more stressed sphere condition, however, the most significant categorized function is “Cell Death”, as expected from the data in figures 3–6. Genes associated with “DNA Replication, Recombination, and Repair” changed with HER4 knockdown in both conditions (Fig.7A). We hypothesized that HER4 knockdown resulted in an alteration of cell cycle, which may contribute to the vulnerability of HER4 knockdown cells. To further explore how HER4 may regulate gene expression, the associations between HER4 and the identified genes with differential expression in the “Cell cycle”, “Cell Death” and “DNA Replication, Recombination, and Repair” categories mapped using the network function of IPA, centered on Her4. (Fig.7A). This network identified potential mechanisms by which HER4 may regulate cell cycle and cell death. However, more studies are required to confirm and elucidate the functional relationship of these genes.
Figure 7.
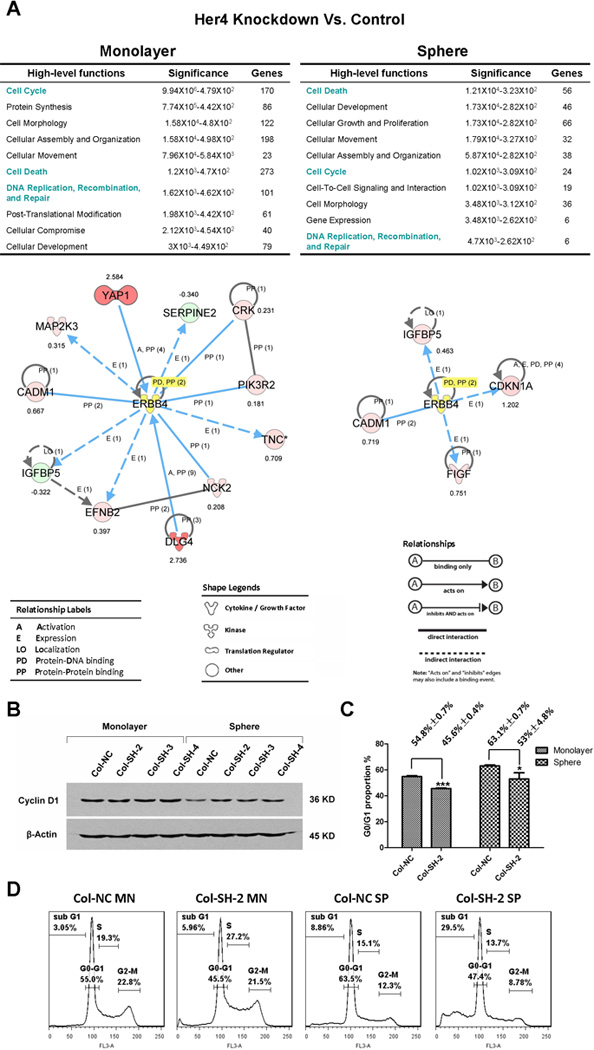
To follow up on the potential role of HER4 in cell cycle that we observed in microarray, we measured cell cycle-related molecules in HER4 knockdown and control cells by western blot. Similar to findings previously reported by others 27, we found that cyclin D was suppressed in spheres compared to monolayer (Fig.7B), which was reflected in reduced proliferation and G1 arrest (54.8%±0.7% vs. 63.1%±0.7%, P<0.001) (Fig.7C and 7D). However, HER4 knockdown partially rescued the suppression of cyclin D caused by sphere culture. In parallel, cell cycle analysis revealed a significantly smaller proportion of G1 phase in HER4 knockdown cells (Fig.7C). Together, these data suggest that cells would respond protectively to stress by arresting cell cycle in G1, allowing more time for DNA repair. However, HER4 knockdown impairs this mechanism and drives cell cycle progression, leading to increased apoptosis.
The HER4 intracellular domain suppressed the cell cycle and conferred chemo-resistance
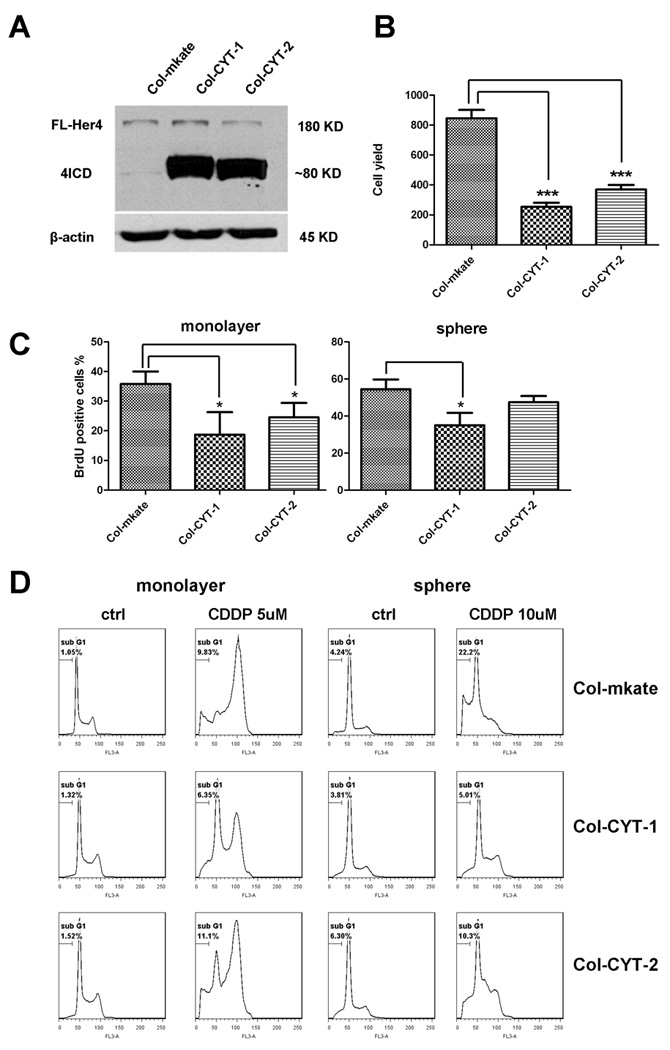
Since HER4 is a receptor tyrosine kinase (RTK), we wanted to address whether the activity of the tyrosine kinase is responsible for the stress-protective role of HER4. Because there is no HER4 specific RTK inhibitor, we used a CI-1033, a pan-ERBB inhibitor, to inhibit HER4 activation. Meanwhile, an antibody (Thermo, clone H4.72.8) recognizing the ectodomain of HER428 was also used to inhibit the binding of heregulin, a ligand of HER4, which blocked the activation of the HER4 tyrosine kinase. However, neither CI-1033 nor the HER4 blocking antibody reproduced the impact of HER4 knockdown on the cells (data not shown). Since the intracellular fragment of HER4 can function as a transcription factor,18, 29, 30 we asked whether intracellular HER4 is sufficient to confer stress-protection. To study how intracellular HER4 would impact the cells, we transduced Col with the intracellular HER4 isoforms CYT-1 and CYT-2. Both HER4 isoforms reduced cell yield after 3 days of growth (Fig 8A and 8B). To identify whether the loss of cell yield is due to reduced proliferation, we performed a BrdU incorporation assay to measure S phase cells. Consistent with the proliferation assay, Col-CYT-1 and Col-CYT-2 had a lower percentage of dividing cells than control cells as represented by the BrdU positive proportion (Fig. 8C). We further tested whether this suppressed cell cycle would confer chemoresistance. While both isoforms conferred resistance to cisplatin, overexpression of the CYT-1 HER4 isoform produced a more resistant phenotype (Fig. 8D). These results support the notion that the HER4 intracellular fragment regulates the cell cycle. Together with previous data, our findings indicate that neuroblastoma cells adapt to cell stress by up-regulating HER4 to acquire a relatively slow-growing phenotype and to protect themselves from apoptosis.
Figure 8.
DISCUSSION
Resisting cell death is one of the hallmarks of cancer.31 Under less than ideal conditions such as serum starvation, loss of attachment, hypoxia, etc., cancer cells usually show higher resistance to death than normal cells. Improved understanding of the mechanisms providing this stress resistance may clarify the reasons for treatment failure in cancer patients. In this study, we observed altered expression of HER4 in neuroblastoma cells in response to changes in the culture environment. In particular, there was up-regulation of HER4 when cells were exposed to diverse stressors. This led us to consider two opposing roles for the function of HER4 inside the cell. First, HER4 could mediate pro-apoptotic signaling to trigger cell death. Second, HER4 could mediate up-regulation or down-regulation of diverse genes required for cells to survive under stress. Both models have been proposed by other investigators.8, 18 With this study, we wished to determine whether HER4 acts to promote cell survival or cell death in NBL cells, and the mechanism by which it acts. Identifying how HER4 contributes to malignant behavior could provide new therapeutic opportunities.
We first observed density-dependent HER4 expression: HER4 was up-regulated when in vitro-cultured cells became more confluent. A high level of confluency causes increased cell-cell adhesion due to the close proximity of cells to one another. Cell-cell adhesion mediates important biological functions, including transduction of proliferative signals8. Low confluency cell culture is considered a poor experimental model because it lacks cell-cell adhesion and less resembles a solid tumor. Our findings show that cells in higher confluency express more HER4 than cells in lower confluency, which suggests cell-cell adhesion may trigger downstream signals to increase HER4 expression. Cancer cells are widely known typically to have no contact inhibition, and this feature is considered a manifestation of malignancy.31 Here we show that HER4 is up-regulated upon reaching confluence. This up-regulation could be one of the mechanisms that regulate the suppression of contact inhibition in cancer cells. Furthermore, over-confluence is stressful due to a lack of sufficient nutrients, space, etc., which would cause an increase in reactive oxygen species and induce senescence.32 Thus, the up-regulation of HER4 when over-confluent could possibly be protecting cells from stress. A similar finding has been observed earlier with regards to HSP27, a well-known cell stress protector.33
We also found that when the neuroblastoma cells were grown on non-adherent plates, they aggregate spontaneously to form compact spheroids, which has been observed in different cancer types.9, 34, 35 The multicellular tumor sphere is usually considered as a model that holds an intermediate complexity between monolayer cell culture and an in vivo model.9 It could be a useful tool to mimic avascular tumor regions and micrometastases in the pre-vascularized stage. It is regarded as a better model than monolayer cell culture because it resembles solid tumors more closely in several respects, including tight cell-cell adhesion,27, 36 moderate proliferation rate,27 hypoxic microenvironment,37 and chemo/radiation resistance.10, 12, 27 One important factor that might cause therapy ineffectiveness is the intrinsic resistance to the adverse environment that comes from the three-dimensional structure within the bulk of tumors--termed as multicellular resistance (MCR).10 Our data showed that HER4 up-regulation is one of the events that occur during the acquisition of multicellular resistance. We assume HER4 may be one important factor that contributes to multicellular resistance, because HER4 knockdown causes a more vulnerable phenotype to the stressors used in this experiment. This would strengthen the point that it could serve as a therapeutic target.
Acquisition of anoikis resistance is seen as one of the prominent features of tumorigenesis31 and is associated with metastasis. Some groups used the multicellular tumor sphere model to study anoikis resistance.27, 38, 39 Although tumor cells in this model are not fully “homeless” due to the multicellular structure of the sphere, tumor cells do suffer from loss of attachment in early stages when cells are still in suspension as single cells. For tumor spheres as a whole, the anchorage-independent culture condition is still stressful, and they showed decreased proliferation. A proteomic analysis of neuroblastoma tumor spheres revealed changes in proteins that regulate the cell stress response.35 Thus, the stress that cells undergo in tumor spheres could cause a reactive up-regulation of HER4 if HER4 is indeed a cell stress regulator. Our results showed that knockdown of HER4 made neuroblastoma cells more susceptible to death from loss of attachment, which indicates that HER4 protects cells from anoikis by regulating response to cellular stress. Since anoikis resistance is associated with metastasis, it is possible to use HER4 as a target to reduce tumor metastasis. The effect of reducing tumor metastasis may be increased by combining HER4 targeted therapy with established regimens for treating malignant tumors with chemotherapeutics or radiation therapy.
Serum starvation is a well-established approach for induction of a broad range of cellular stress. It induces oxidative stress and causes an accumulation of intracellular reactive oxygen species (ROS).40 In this study, we reported the up-regulation of HER4 upon serum starvation among neuroblastoma cell lines and 293T cells (data not shown). We also showed that HER4 knockdown rendered the cells less tolerant to serum deprivation. HER4 signaling has been found to protect neural-origin tumor cells from serum starvation41 and oxidative stress.42 Another group has found that ROS activates NRG-1β/HER4 paracrine signaling in the heart, suggesting HER4 is involved in cardiac adaptation to oxidative stress.43 Together with our findings, these data support that stress-induced HER4 expression may function protectively to maintain homeostasis and prevent apoptosis.
The role of HER4 in cancer has been controversial.14, 18, 44–47 One possible explanation for the conflicting observations may be that different isoforms and the subcellular localizations of HER4 differ in function.15, 20 HER4 intracellular domain (4ICD) has been proven to function as a transcription factor in the nucleus.29, 48, 49 In this study, our experimental model using neuroblastoma proposes another idea to explain the contradiction. Our results showed that HER4 sufficiently protects cancer cells from different kinds of cellular stressors which can contribute to the malignancy. In contrast, insertion and overexpression of 4ICD suppresses cell cycle and proliferation, which makes HER4 take on an “inhibitory” role. One recent study suggested that high HER4 expression correlates with low mitosis karyorrhexis index (MKI), an indicator of mitosis activity. However, they also found that high HER4 expression was associated with clinical high risk groups, metastasis, and poorer survival,5 which is consistent with our laboratory findings. The regulation of tumor dormancy affects metastasis and therapeutic response.50, 51 In essence, these observations point to the idea that tumors growing more slowly may be harder to treat. Together with our findings, it’s suggested that although HER4 may reduce proliferation, its overexpression renders a refractory phenotype, which may be highly dependent on microenvironment. Although the prognostic relevance of HER4 in neuroblastoma needs to be confirmed with larger scale clinical-pathological analysis, levels of HER4 expression should at least be considered as one important factor that influences outcome.
In conclusion, HER4 expression can be induced upon various cellular stresses in neuroblastoma and functions to protect cells from apoptosis. HER4 knockdown increases the vulnerability of neuroblastoma cells to chemotherapy. Intracellular HER4 may provide protection from stress by slowing proliferation and conferring increased chemo-resistance. Further investigation is required to understand the consequences of HER4 intracellular signaling and whether signaling at the membrane is required in conjunction with the transcriptional regulation provided by the soluble HER4 fragment to provide protection from stress. Thus, HER4 has been suggested as a potential therapeutic target in neuroblastoma and Ewing’s sarcoma8, which may raise interest in developing new strategies against HER4 in pediatric solid tumors.
ACKNOWLEDGEMENT
EM was conducted by Kenneth Dunner, Jr. in the High Resolution Electron Microscopy Facility (www.mdanderson.org/HREMF) funded by institutional core grant #CA16672. Microarray was done with the help of Qiuyu Wu and Kim Seung Wook from Dr. Fidler’s lab, MDACC.
Funding support: This work was supported by grants 1R01-CA149501 (Hughes), 1K08CA118730, the Physician-Scientist Program of MD Anderson Cancer Center (Hughes); the Jori Zemel Children's Bone Tumor Foundation (Hua).
Footnotes
Financial disclosure: No disclosure.
REFERENCES
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. The Lancet. 2007;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 3.Ho R, Minturn JE, Hishiki T, et al. Proliferation of Human Neuroblastomas Mediated by the Epidermal Growth Factor Receptor. Cancer Res. 2005 Nov;65(21):9868–9875. doi: 10.1158/0008-5472.CAN-04-2426. 2005. [DOI] [PubMed] [Google Scholar]
- 4.Richards KN, Zweidler-McKay PA, Van Roy N, et al. Signaling of ERBB receptor tyrosine kinases promotes neuroblastoma growth in vitro and in vivo. Cancer. 2010 Jul 1;116(13):3233–3243. doi: 10.1002/cncr.25073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Izycka-Swieszewska E, Wozniak A, Drozynska E, et al. Expression and significance of HER family receptors in neuroblastic tumors. Clinical and Experimental Metastasis. 2011:1–12. doi: 10.1007/s10585-010-9369-1. [DOI] [PubMed] [Google Scholar]
- 6.Hughes DP, Thomas DG, Giordano TJ, McDonagh KT, Baker LH. Essential erbB family phosphorylation in osteosarcoma as a target for CI-1033 inhibition. Pediatr Blood Cancer. 2006 May 1;46(5):614–623. doi: 10.1002/pbc.20454. [DOI] [PubMed] [Google Scholar]
- 7.Oncogenomics. [[accessed Nov 18th, 2011]]; Available from URL: http://home.ccr.cancer.gov/oncology/oncogenomics/
- 8.Kang H-G, Jenabi JM, Zhang J, et al. E-Cadherin Cell-Cell Adhesion in Ewing Tumor Cells Mediates Suppression of Anoikis through Activation of the ErbB4 Tyrosine Kinase. Cancer Research. 2007 Apr 1;67(7):3094–3105. doi: 10.1158/0008-5472.CAN-06-3259. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santini MT, Rainaldi G. Three-dimensional spheroid model in tumor biology. Pathobiology. 1999 May-Jun;67(3):148–157. doi: 10.1159/000028065. [DOI] [PubMed] [Google Scholar]
- 10.Desoize B, Jardillier J. Multicellular resistance: a paradigm for clinical resistance? Crit Rev Oncol Hematol. 2000 Nov-Dec;36(2–3):193–207. doi: 10.1016/s1040-8428(00)00086-x. [DOI] [PubMed] [Google Scholar]
- 11.Olive PL, Durand RE. Drug and radiation resistance in spheroids: cell contact and kinetics. Cancer and Metastasis Reviews. 1994;13(2):121–138. doi: 10.1007/BF00689632. [DOI] [PubMed] [Google Scholar]
- 12.Durand RE, Olive PL. Resistance of tumor cells to chemo- and radiotherapy modulated by the three-dimensional architecture of solid tumors and spheroids. Methods Cell Biol. 2001;64:211–233. doi: 10.1016/s0091-679x(01)64015-9. [DOI] [PubMed] [Google Scholar]
- 13.Bilir A, Erguven M, Yazihan N, Aktas E, Oktem G, Sabanci A. Enhancement of vinorelbine-induced cytotoxicity and apoptosis by clomipramine and lithium chloride in human neuroblastoma cancer cell line SH-SY5Y. Journal of Neuro-Oncology. 2010;100(3):385–395. doi: 10.1007/s11060-010-0209-6. [DOI] [PubMed] [Google Scholar]
- 14.Gullick WJ. c-erbB-4/HER4: friend or foe? J Pathol. 2003 Jul;200(3):279–281. doi: 10.1002/path.1335. [DOI] [PubMed] [Google Scholar]
- 15.Mill CP, Gettinger KL, Riese Ii DJ. Ligand stimulation of ErbB4 and a constitutively-active ErbB4 mutant result in different biological responses in human pancreatic tumor cell lines. Exp Cell Res. 2011;317(4):392–404. doi: 10.1016/j.yexcr.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes DP, Thomas DG, Giordano TJ, Baker LH, McDonagh KT. Cell surface expression of epidermal growth factor receptor and Her-2 with nuclear expression of Her-4 in primary osteosarcoma. Cancer Res. 2004 Mar 15;64(6):2047–2053. doi: 10.1158/0008-5472.can-03-3096. [DOI] [PubMed] [Google Scholar]
- 17.Hollmen M, Maatta JA, Bald L, Sliwkowski MX, Elenius K. Suppression of breast cancer cell growth by a monoclonal antibody targeting cleavable ErbB4 isoforms. Oncogene. 2009 Mar 12;28(10):1309–1319. doi: 10.1038/onc.2008.481. [DOI] [PubMed] [Google Scholar]
- 18.Naresh A, Long W, Vidal GA, et al. The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 2006 Jun 15;66(12):6412–6420. doi: 10.1158/0008-5472.CAN-05-2368. [DOI] [PubMed] [Google Scholar]
- 19.Vidal GA, Clark DE, Marrero L, Jones FE. A constitutively active ERBB4/HER4 allele with enhanced transcriptional coactivation and cell-killing activities. Oncogene. 2007 Jan 18;26(3):462–466. doi: 10.1038/sj.onc.1209794. [DOI] [PubMed] [Google Scholar]
- 20.Sundvall M, Veikkolainen V, Kurppa K, et al. Cell Death or Survival Promoted by Alternative Isoforms of ErbB4. Mol Biol Cell. 2010;21(23):4275–4286. doi: 10.1091/mbc.E10-04-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muraoka-Cook RS, Sandahl MA, Strunk KE, et al. ErbB4 splice variants Cyt1 and Cyt2 differ by 16 amino acids and exert opposing effects on the mammary epithelium in vivo. Mol Cell Biol. 2009 Sep;29(18):4935–4948. doi: 10.1128/MCB.01705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elenius K, Corfas G, Paul S, et al. A novel juxtamembrane domain isoform of HER4/ErbB4. Isoform-specific tissue distribution and differential processing in response to phorbol ester. J Biol Chem. 1997 Oct 17;272(42):26761–26768. doi: 10.1074/jbc.272.42.26761. [DOI] [PubMed] [Google Scholar]
- 23.Kainulainen V, Sundvall M, Maatta JA, Santiestevan E, Klagsbrun M, Elenius K. A natural ErbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J Biol Chem. 2000 Mar 24;275(12):8641–8649. doi: 10.1074/jbc.275.12.8641. [DOI] [PubMed] [Google Scholar]
- 24.Geryk-Hall M, Yang Y, Hughes DP. Driven to death: inhibition of farnesylation increases Ras activity in osteosarcoma and promotes growth arrest and cell death. Mol Cancer Ther. 2010 May;9(5):1111–1119. doi: 10.1158/1535-7163.MCT-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang P, Yang Y, Zweidler-McKay PA, Hughes DP. Critical role of notch signaling in osteosarcoma invasion and metastasis. Clin Cancer Res. 2008 May 15;14(10):2962–2969. doi: 10.1158/1078-0432.CCR-07-1992. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Pletnev S, Shcherbo D, Chudakov DM, et al. A Crystallographic Study of Bright Far-Red Fluorescent Protein mKate Reveals pH-induced cis-trans Isomerization of the Chromophore. Journal of Biological Chemistry. 2008 Oct 24;283(43):28980–28987. doi: 10.1074/jbc.M800599200. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawlor ER, Scheel C, Irving J, Sorensen PH. Anchorage-independent multi-cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene. 2002 Jan 10;21(2):307–318. doi: 10.1038/sj.onc.1205053. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Levkowitz G, Tzahar E, et al. An immunological approach reveals biological differences between the two NDF/heregulin receptors, ErbB-3 and ErbB-4. J Biol Chem. 1996 Mar 29;271(13):7620–7629. [PubMed] [Google Scholar]
- 29.Linggi B, Carpenter G. ErbB-4 s80 intracellular domain abrogates ETO2-dependent transcriptional repression. J Biol Chem. 2006 Sep 1;281(35):25373–25380. doi: 10.1074/jbc.M603998200. [DOI] [PubMed] [Google Scholar]
- 30.Jones FE. HER4 intracellular domain (4ICD) activity in the developing mammary gland and breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(2):247–258. doi: 10.1007/s10911-008-9076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Ho JH, Chen YF, Ma WH, Tseng TC, Chen MH, Lee OK. Cell Contact Accelerates Replicative Senescence of Human Mesenchymal Stem Cells Independent of Telomere Shortening and p53 Activation: Roles of Ras and Oxidative Stress. Cell Transplant. 2010 Dec 22; doi: 10.3727/096368910X546562. [DOI] [PubMed] [Google Scholar]
- 33.Garrido C, Ottavi P, Fromentin A, et al. HSP27 as a Mediator of Confluence-dependent Resistance to Cell Death Induced by Anticancer Drugs. Cancer Research. 1997 Jul 1;57(13):2661–2667. 1997. [PubMed] [Google Scholar]
- 34.Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol. 2010 Jul 1;148(1):3–15. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Kumar HR, Zhong X, Hoelz DJ, et al. Three-dimensional neuroblastoma cell culture: proteomic analysis between monolayer and multicellular tumor spheroids. Pediatr Surg Int. 2008 Nov;24(11):1229–1234. doi: 10.1007/s00383-008-2245-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green SK, Francia G, Isidoro C, Kerbel RS. Antiadhesive antibodies targeting E-cadherin sensitize multicellular tumor spheroids to chemotherapy in vitro. Mol Cancer Ther. 2004 Feb;3(2):149–159. [PubMed] [Google Scholar]
- 37.Valcarcel M, Arteta B, Jaureguibeitia A, et al. Three-dimensional growth as multicellular spheroid activates the proangiogenic phenotype of colorectal carcinoma cells via LFA-1-dependent VEGF: implications on hepatic micrometastasis. J Transl Med. 2008;6:57. doi: 10.1186/1479-5876-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Díaz-Montero CM, McIntyre BW. Acquisition of anoikis resistance in human osteosarcoma cells. European Journal of Cancer. 2003;39(16):2395–2402. doi: 10.1016/s0959-8049(03)00575-6. [DOI] [PubMed] [Google Scholar]
- 39.Douma S, van Laar T, Zevenhoven J, Meuwissen R, van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430(7003):1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- 40.Guo D, Han J, Adam B-L, et al. Proteomic analysis of SUMO4 substrates in HEK293 cells under serum starvation-induced stress. Biochemical and Biophysical Research Communications. 2005;337(4):1308–1318. doi: 10.1016/j.bbrc.2005.09.191. [DOI] [PubMed] [Google Scholar]
- 41.Erlich S, Goldshmit Y, Lupowitz Z, Pinkas-Kramarski R. ErbB-4 activation inhibits apoptosis in PC12 cells. Neuroscience. 2001;107(2):353–362. doi: 10.1016/s0306-4522(01)00350-5. [DOI] [PubMed] [Google Scholar]
- 42.Goldshmit Y, Erlich S, Pinkas-Kramarski R. Neuregulin rescues PC12-ErbB4 cells from cell death induced by H(2)O(2). Regulation of reactive oxygen species levels by phosphatidylinositol 3-kinase. J Biol Chem. 2001 Dec 7;276(49):46379–46385. doi: 10.1074/jbc.M105637200. [DOI] [PubMed] [Google Scholar]
- 43.Kuramochi Y, Cote GM, Guo X, et al. Cardiac Endothelial Cells Regulate Reactive Oxygen Species-induced Cardiomyocyte Apoptosis through Neuregulin-1β/erbB4 Signaling. Journal of Biological Chemistry. 2004 Dec 3;279(49):51141–51147. doi: 10.1074/jbc.M408662200. 2004. [DOI] [PubMed] [Google Scholar]
- 44.Hollmen M, Elenius K. Potential of ErbB4 antibodies for cancer therapy. Future Oncol. 2010 Jan;6(1):37–53. doi: 10.2217/fon.09.144. [DOI] [PubMed] [Google Scholar]
- 45.Sartor CI, Zhou H, Kozlowska E, et al. Her4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cells. Mol Cell Biol. 2001 Jul;21(13):4265–4275. doi: 10.1128/MCB.21.13.4265-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sundvall M, Iljin K, Kilpinen S, Sara H, Kallioniemi OP, Elenius K. Role of ErbB4 in breast cancer. J Mammary Gland Biol Neoplasia. 2008 Jun;13(2):259–268. doi: 10.1007/s10911-008-9079-3. [DOI] [PubMed] [Google Scholar]
- 47.Thor AD, Edgerton SM, Jones FE. Subcellular localization of the HER4 intracellular domain, 4ICD, identifies distinct prognostic outcomes for breast cancer patients. Am J Pathol. 2009 Nov;175(5):1802–1809. doi: 10.2353/ajpath.2009.090204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilmore-Hebert M, Ramabhadran R, Stern DF. Interactions of ErbB4 and Kap1 Connect the Growth Factor and DNA Damage Response Pathways. Mol Cancer Res. 2010 Sep 21; doi: 10.1158/1541-7786.MCR-10-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Komuro A, Nagai M, Navin NE, Sudol M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem. 2003 Aug 29;278(35):33334–33341. doi: 10.1074/jbc.M305597200. [DOI] [PubMed] [Google Scholar]
- 50.Sleeman JP, Nazarenko I, Thiele W. Do all roads lead to Rome? Routes to metastasis development. International Journal of Cancer. 2011;128(11):2511–2526. doi: 10.1002/ijc.26027. [DOI] [PubMed] [Google Scholar]
- 51.Ranganathan AC, Adam AP, Aguirre-Ghiso JA. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle. 2006 Aug;5(16):1799–1807. doi: 10.4161/cc.5.16.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]