

Abstract
Objective:
The effect of mHTT on human development was examined by evaluating measures of growth in children at risk for Huntington disease (HD).
Methods:
Children at risk for HD with no manifest symptoms (no juvenile HD included) were enrolled and tested for gene expansion for research purposes only. Measurements of growth (height, weight, body mass index [BMI], and head circumference) in children tested as gene-expanded (n = 20, 7–18 years of age, CAG repeats ≥39) were compared to those of a large database of healthy children (n = 152, 7–18 years of age).
Results:
Gene-expanded children had significantly lower measures of head circumference, weight, and BMI. Head circumference was abnormally low even after correcting for height, suggesting a specific deficit in brain growth, rather than a global growth abnormality.
Conclusions:
These results indicate that, compared to a control population, children who were estimated to be decades from HD diagnosis have significant differences in growth. Further, they suggest that mHTT may play a role in atypical somatic, and in particular, brain development.
Recently, the classic concept of Huntington disease (HD) as a neurodegenerative disease of the striatum has been challenged. Both studies of animal models of HD1 and preHD adults2 propose that while the primary neuropathology is neurodegeneration, abnormal brain development could contribute to the pathophysiology of the disease. Normal and mutant Huntingtin gene (HTT and mHTT) are systematically3 expressed throughout the course of development and into adulthood.4 This pattern of expression (across all stages of development as well as throughout the body) beg the question of whether there are developmental or somatic effects of mHTT.
One potential somatic feature of HD is that of weight loss,5–7 which is particularly marked in later stages of the illness. However, this symptom also appears early in the course of disease and may precede motor abnormalities.6,8,9 In addition, in a large MRI study, measures of intracranial volume (ICV) in preHD subjects were significantly smaller compared to controls, suggesting that the preHD subjects had abnormal development leading to a smaller than normal brain.2 As reduced body mass index (BMI) and ICV are both documented in preHD adults, unresolved issues include whether or not these decrements are present in childhood and what may be the developmental trajectory of these measures.
In the current study, basic anthropometric measures of height, weight, BMI, and head circumference in children at risk for HD were evaluated. Measurements of growth in gene-expanded children were compared to those of a large database of healthy children, as well as to those of gene-nonexpanded children.
METHODS
Participants and measurements.
Adults who have been clinically diagnosed with HD or tested gene-expanded through the HD Center of Excellence clinic or the HD Registry at the University of Iowa were asked to enroll their children for participation. A total of 34 participants, ages 7–18 years, were recruited. Testing for gene expansion was done using blood or saliva. Participants with a CAG repeat length equal to or greater than 39 were designated as gene-expanded (n = 20, CAG repeat range = 39–55) and the remaining 14 were tested gene-nonexpanded and therefore added into the healthy control comparisons. The results of genetic testing were not revealed to the participant, their family, or even any member of the research team. This information was deidentified and used only for research purposes.
For the gene-expanded group, estimated years to onset were calculated using a formula that accounts for age and CAG repeat length.10 The mean estimated years to onset was 31.55 years with a range of 11.63 to 54.87.
All participants were examined by an experienced clinician (either a child neurologist or psychiatrist trained in motor score ratings for HD) for neurologic signs of HD. No participant displayed any significant motor abnormalities on the Unified Huntington's Disease Rating Scale (UHDRS; no diagnostic confidence ratings above a 1).11 Healthy children were recruited from the community via local newspaper advertisements. All children in the healthy control group had no major medical, neurologic, or psychiatric illness, nor any history of learning disability as disclosed by parents during the screening process. Parental socioeconomic status of all participants was obtained using a modified Hollingshead Scale of 1 to 5, with a lower number corresponding to higher socioeconomic status.12 All participants gave informed consent prior to enrolling in the protocol, which was approved by the University of Iowa Institutional Review Board.
Table 1 displays the demographic information for the 2 at-risk groups (gene-expanded [n = 20] and gene-nonexpanded [n = 14]), and the healthy control (HC) group (n = 138). Analysis of variance was used to compare demographic variables across groups. There were no significant differences in age or sex distribution among the 3 groups. There was a significant difference in socioeconomic status measure among the 3 groups. Post hoc analysis revealed that the parental socioeconomic status of gene-nonexpanded group was significantly lower than that of the HC group [t(2,169) = 0.51, p = 0.007].
Table 1.
Demographic information of gene-nonexpanded, gene-expanded, healthy control, and combined control group
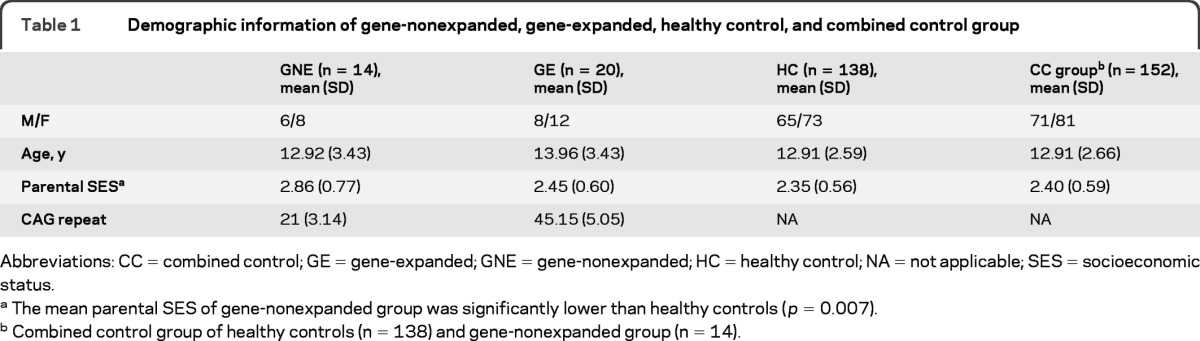
Abbreviations: CC = combined control; GE = gene-expanded; GNE = gene-nonexpanded; HC = healthy control; NA = not applicable; SES = socioeconomic status.
The mean parental SES of gene-nonexpanded group was significantly lower than healthy controls (p = 0.007).
Combined control group of healthy controls (n = 138) and gene-nonexpanded group (n = 14).
For the initial analysis, the gene-nonexpanded group (n = 14) and the healthy control group (n = 138) were merged, comprising a total control sample of 152. Table 1 shows the demographics for this combined control (CC) group. t-Test results showed no significant differences in age, sex distribution, or parental socioeconomic status between the gene-expanded group and the CC group.
Measurements.
Basic anthropometric measures of height (cm), weight (kg), and head circumference (cm) were obtained by trained research nurses in the University of Iowa's Institute for Clinical and Translational Sciences. All participants were measured on the same equipment using the same measurement protocols.
Statistical analysis.
All analyses were performed using the statistical software package SAS.13 The first analysis compared gene-expanded subjects to the CC group (table 2). General Linear Model procedure was used to compare all anthropometric measures (height, weight, head circumference, BMI = weight kg/height m2) between groups with fixed variable of CAG expansion status (gene-expanded vs combined control), adjusting for age, sex, and parental socioeconomic status. As head size is related to overall stature, the analysis of head circumference also contained height as a covariate. All possible interaction terms among age, sex, and group were entered into the models. We also added the quadratic term for age (age2) to investigate potential nonlinear interactions between age and growth measurements. Interaction terms were deleted from the model if they were not significant. An α level of 0.05 was used for significance tests.
Table 2.
Results of comparison in growth measures between gene-expanded group and combined control group
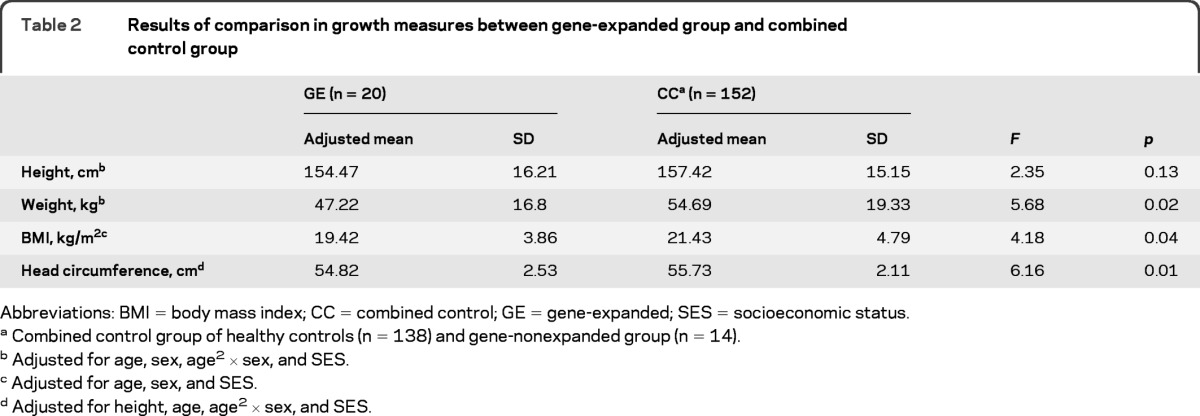
Abbreviations: BMI = body mass index; CC = combined control; GE = gene-expanded; SES = socioeconomic status.
Combined control group of healthy controls (n = 138) and gene-nonexpanded group (n = 14).
Adjusted for age, sex, age2 × sex, and SES.
Adjusted for age, sex, and SES.
Adjusted for height, age, age2 × sex, and SES.
This analysis was repeated comparing the 3 groups of at-risk gene-expanded, at-risk gene-nonexpanded, and HC (table 3). The relationship between anthropometric measures and CAG length was tested (within the gene-expanded group), employing a nonparametric spearman correlation, controlling for age and parental socioeconomic status.
Table 3.
Results of comparison in growth measures between gene-nonexpanded, gene-expanded, and healthy control group
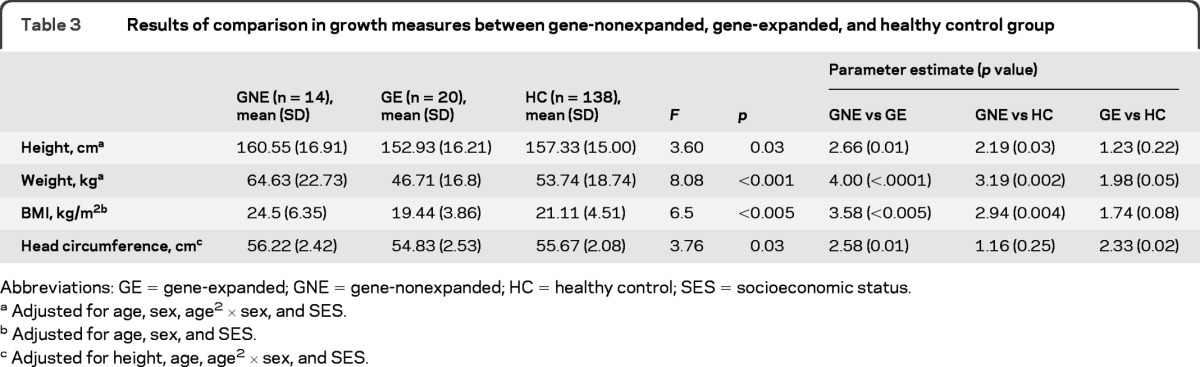
Abbreviations: GE = gene-expanded; GNE = gene-nonexpanded; HC = healthy control; SES = socioeconomic status.
Adjusted for age, sex, age2 × sex, and SES.
Adjusted for age, sex, and SES.
Adjusted for height, age, age2 × sex, and SES.
To assess the relationship between measures of growth and age or growth trajectory, we plotted each measure across the age range for each group and calculated the relationship between age and that measure. This analysis was cross-sectional with each measure representing 1 individual as there were no intrasubject longitudinal data.
RESULTS
Gene-expanded compared to CC group.
Height.
The results showed a significant main effect of age, sex, and an age2 by sex interaction on height. The main effects of age and sex were as expected with higher age being associated with higher height values, and males being taller than females. Further evaluation of the sex by age2 interaction showed that males have a significantly different growth trajectory compared to females. The growth trajectories of our sample (figure 1) matched those produced by the Centers for Disease Control14 and used as standard growth curves for most health care providers.15 In these charts, the height of females reaches a peak and plateaus around age 16 while the males do not plateau through this age range, peaking their height after the age of 18 years. The trajectories in our sample match those of the standardized trajectories supporting the use of our healthy sample as a valid control sample.
Figure 1. Relationship between height and age.

This scatterplot demonstrates the distribution of height by age for each participant including those from both gene-expanded group and combined controls. Both male and female growth trajectories of our current sample closely match the standardized trajectories produced by the Centers for Disease Control and Prevention.
After controlling for all covariates and interactions, the mean height of the gene-expanded group was lower than the mean for the CC group; however, this difference did not reach statistical significance (F6,165 = 2.35, p = 0.13).
Weight.
There was a significant main effect of age, sex, and an age2 by sex interaction on weight. Similar to height, the main effects were as expected with higher weight being associated with higher age, and males having greater weight than females. In parallel to height, the weight of females peaked near age 17, yet the males did not plateau in this age range.
The model did produce a significant effect of group (F6,165 = 5.68, p = 0.02) with children in the gene-expanded group having significantly lower mean weight than the children in the CC group.
Body mass index.
There was a main effect of age on BMI, but no main effect of sex nor sex by age (or age2) interaction. There was a main effect of group (F4,167 = 4.18, p = 0.04): mean BMI was significantly lower in children in the gene-expanded group when compared to that of the CC group.
Head circumference.
The analysis showed a significant effect of height, age, and sex on head circumference. Main effects of sex and height showed that being male and higher measures of height were associated with larger head circumference. The gene-expanded group showed a significantly smaller mean head circumference than the CC group (F5,166 = 5.65, p = 0.018).
There was no sex by group interaction for any measure indicating that both males and females in the gene-expanded group showed the same pattern of deficits compared to the CC group. However, given the small sample size (only 8 males in the gene-expanded group), larger samples will be needed to evaluate possible sex differences more thoroughly.
Since the gene-nonexpanded group was quite small and to investigate the possibility that outliers in that group were driving the findings, we repeated all above analysis using nonparametric methods (ranking the variables). The findings remained the same with no change in significance.
Figure 2 shows the trajectories of the 2 groups for each growth measurement. There was no group by age (or age2) interaction, nor any triple interaction between sex, group, and age (or age2). This suggests that the 2 groups (and the sexes within both groups) had equivalent trajectories of growth. As there was no significant interaction between sex, age, and group, the growth trajectory for each group is accurately represented by combining the sexes in each group.
Figure 2. Growth trajectory and Z score of growth measurements of CAG gene-expanded and gene-nonexpanded group.

Growth trajectory in (A) height, (B) weight, (C) body mass index (BMI), and (D) head circumference of children with and without CAG gene expansion is depicted. The dashed lines indicate the 95% confidence limits for the fitted trend lines. (E) Findings in growth measurements of children at risk for Huntington disease in comparison with healthy controls. Z score of 0 represents healthy controls.
Figure 2C depicts the BMI trajectory for both groups. Although the group by age interaction was not significant, there did appear (by viewing the figure) to be a lower slope of growth over time for the gene-expanded group. To evaluate further, a post hoc analysis evaluated extreme groups with gene-expanded participants of age 7–12 years (n = 7) compared to controls in the same age range (n = 88) and gene-expanded participants ages 13 years and above (n = 13) compared to controls in the same age range (n = 64). The gene-expanded participants of ages 7–12 had no significant difference in BMI when compared to healthy peers (F4,95 = 0.06, p = 0.809). On the contrary, the gene-expanded participants ages 13 and above had significantly lower BMI (mean [SD] = 20.69 [1.58]) compared to control subjects in the same age range (mean [SD] = 23.79 [1.58]; F4,65 = 4.03, p = 0.048).
Gene-expanded vs gene-nonexpanded vs HC group.
For all measures, the pattern of significant main effects and interactions were the same as for the 2-group analysis (gene-expanded compared to CC group). We report post hoc analyses of interest to compare the findings across the 3 groups.
Height.
Gene-expanded children were not significantly shorter than the other 2 comparison groups. However, the gene-nonexpanded group was significantly taller than both the HC group and the gene-expanded group.
Weight.
Analysis revealed a stair-step pattern in which gene-nonexpanded children were the heaviest and significantly so compared to the HC group, and the HC group was significantly heavier than the gene-expanded group.
Body mass index.
Similar to weight, the group with the highest BMI was the gene-nonexpanded group, which was significantly different compared to the HC group. The gene-expanded group was lower in BMI compared to the HC group; however, this was not statistically significant.
Head circumference.
Head circumference was not significantly different between the gene-nonexpanded group and the HC group. However, the gene-expanded group had significantly smaller head circumference compared to both the HC group and the gene-nonexpanded group.
Figure 2E illustrates the z scores of the growth measurement results of children at risk for HD relative to the HC group. In sum, when all 3 groups were compared to each other, both at-risk groups were clearly atypical. That is, the gene-nonexpanded group, compared to the HC group, was significantly taller, weighed more, and therefore had a greater BMI. Conversely, the gene-expanded children were found to weigh less and to have smaller head circumferences compared to healthy controls.
Length of CAG repeat and growth measurements.
Results of a Spearman nonparametric test revealed a significant correlation between longer CAG repeats and smaller head circumference in the gene-expanded group (rs = −0.58, p = 0.01). The correlation results between the length of CAG repeat and height and BMI was essentially zero (height: rs = −0.08, p = 0.77; BMI: rs = −0.10, p = 0.69). The relation between the length of CAG repeat and weight was negative indicating higher CAG repeats were associated with lower measures; however, they did not reach significance (weight: rs = −0.21, p = 0.41).
DISCUSSION
In the current study, the children who were tested as gene-expanded were, on average, greater than an estimated 3 decades from the onset of disease. The results indicated that these preHD children have subtle abnormalities in anthropometric measures of growth—namely, lower weight, BMI, and head circumference—compared to controls.
The findings of lower weight and BMI in preHD children extend the results shown in preHD adults and HD patients. Marked weight loss or reduced body mass is observed in both early and, in particular, later phase of the disease.5,16,17 Although constant caloric burn due to involuntary choreic movements is one possible explanation for the weight loss in manifest HD, lower body weight in spite of higher daily caloric intake and unaltered physical activity level has been seen in preHD adults.6,18 Given that the preHD children were free of involuntary movements further supports the idea of a primary abnormality in metabolic rate underlying this issue in HD.18–21
Defects in energy metabolism that may or may not be related to mitochondrial function have not only long been the center of studies of the pathogenesis,22–24 but also been the target of therapeutic approaches of HD in humans and animal models.25,26 Mitochondrial dysfunction influenced by mHTT is present in the brain27,28 and in non-neuronal tissue.29 Findings from the current study suggest that abnormalities in energy metabolism may manifest first as abnormalities in growth.
In contrast to the at-risk gene-nonexpanded children, gene-expanded children were found to have lower weight and BMI. All at-risk children come from homes in which a parent or grandparent has HD. Especially for those children whose parents are affected, there may well be significant disruption in the family dealing with such a disabling disease. Given the multifactorial nature of childhood development, we cannot entirely exclude the contribution of environmental factors on growth in the gene-expanded children. However, growth deficit in the gene-expanded group compared to gene-nonexpanded group points to the genetic influence on these decrements, as the 2 at-risk groups both come from potentially chaotic homes but differ only in the gene expansion status. Although none of the current sample of at-risk children exhibited clinically significant psychiatric symptoms such as depression, future studies clarifying any possible factors affecting the child's nutritional intake would be of benefit.
The higher measures of height, weight, and BMI in the gene-nonexpanded group in comparison to the gene-expanded group can potentially be attributed to environmental factors. The children in the gene-nonexpanded group had significantly lower parental socioeconomic status compared to healthy controls. Whereas height shows a high genetic influence with a heritability close to 0.8, weight is largely driven by environmental factors,30–32 with greater incidence of childhood obesity associated with lower parental education and social class.33,34 Moreover, weight gain to obesity early in infancy and childhood has been shown to be accompanying greater height and growth measures during development.35–37 Therefore, the increase in stature seen in our gene-nonexpanded children is likely due to the metabolic effects of being overweight rather than a primary effect on growth hormone.
The significantly smaller head circumference observed in preHD children also mirrors the findings of a previous study showing substantially lower ICV in preHD adults than controls.2 Head circumference is a significant predictor of brain volume and is highly correlated with ICV,38,39 which is an indicator of maximal brain growth,40 as the enlargement of the cranium is a passive translation of the expansion of the enclosed neural mass.41 Especially in light of the normal achievement of height in this group, the small head circumference suggests a specific brain growth abnormality rather than an overall or general growth abnormality. That is, given a relatively normal height, the lower head circumference indicates that the head is disproportionately small, and therefore showing an abnormality of brain growth. Furthermore, the significant association between longer CAG repeat length and smaller head circumference highlights the direct genetic impact of expanded CAG repeat on this measure. These results suggest that the CAG repeats above the threshold of 39 repeats not only have a negative influence on development but also exert its influence in a dosage-dependent style.
PreHD children did not show any significant alterations in growth trajectories compared to controls. These results may be linked to the limited power in our sample. Though our healthy control sample was large and comparable to the standards used by health care providers, our gene-expanded group was small. Although the difference did not reach statistical significance, the BMI trajectory of preHD children appeared to have a lower slope compared to that of the controls. Perhaps a larger sample would provide the power needed for statistical significance, as further post hoc analysis confirmed a significant difference in BMI between preHD children and age-matched controls to emerge at a postpubescent age range (13–18 years).
It is important to highlight that the abnormalities in growth measures are statistically significant, yet clinically subtle. While the findings do not support clinically relevant abnormalities, the results may be essential to understand both the roots as well as the scope of HD.
Finally, the present study is limited by small sample size. Though large enough to detect significant differences, we were not able to accurately assess subtle effects of potential sex by group interactions or growth trajectories. The current study is ongoing and additional accumulation of both cross-sectional and longitudinal data will further our understanding of the effect of CAG expansion on developmental processes in HD.
Supplementary Material
GLOSSARY
- BMI
body mass index
- CC
combined control
- HD
Huntington disease
- ICV
intracranial volume
- preHD
prediagnostic stage of HD
Footnotes
Editorial, page 621
AUTHOR CONTRIBUTIONS
Jessica K. Lee: drafting of the manuscript, analysis and interpretation of the data, critical revision of the manuscript. Kathy Mathews: critical revision of the manuscript. Bradley Schlaggar: critical revision of the manuscript. Joel Perlmutter: critical revision of the manuscript. Jane S. Paulsen: critical revision of the manuscript. Eric Epping: critical revision of the manuscript. Leon Burmeister: statistical analysis. Peg Nopoulos: obtaining funding, study concept, analysis and interpretation of the data, drafting of the manuscript, critical revision of the manuscript, final approval of the version to be published.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Molero AE, Gokhan S, Gonzalez S, Feig JL, Alexandre LC, Mehler MF. Impairment of developmental stem cell-mediated striatal neurogenesis and pluripotency genes in a knock-in model of Huntington's disease. Proc Natl Acad Sci USA 2009; 106: 21900– 21905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nopoulos PC, Aylward EH, Ross CA, et al. Smaller intracranial volume in prodromal Huntington's disease: evidence for abnormal neurodevelopment. Brain 2011; 134: 137– 142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Orth M, Cooper JM, Bates GP, Schapira AH. Inclusion formation in Huntington's disease R6/2 mouse muscle cultures. J Neurochem 2003; 87: 1– 6 [DOI] [PubMed] [Google Scholar]
- 4. Bhide PG, Day M, Sapp E, et al. Expression of normal and mutant huntingtin in the developing brain. J Neurosci 1996; 16: 5523– 5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farrer LA, Yu PL. Anthropometric discrimination among affected, at-risk, and not-at-risk individuals in families with Huntington disease. Am J Med Genet 1985; 21: 307– 316 [DOI] [PubMed] [Google Scholar]
- 6. Marder K, Zhao H, Eberly S, Tanner CM, Oakes D, Shoulson I. Dietary intake in adults at risk for Huntington disease: analysis of PHAROS research participants. Neurology 2009; 73: 385– 392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trejo A, Tarrats RM, Alonso ME, Boll MC, Ochoa A, Velasquez L. Assessment of the nutrition status of patients with Huntington's disease. Nutrition 2004; 20: 192– 196 [DOI] [PubMed] [Google Scholar]
- 8. Farrer LA, Meaney FJ. An anthropometric assessment of Huntington's disease patients and families. Am J Phys Anthropol 1985; 67: 185– 194 [DOI] [PubMed] [Google Scholar]
- 9. Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, Roos RA. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008; 71: 1506– 1513 [DOI] [PubMed] [Google Scholar]
- 10. Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet 2004; 65: 267– 277 [DOI] [PubMed] [Google Scholar]
- 11. Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord 1996; 11: 136– 142 [DOI] [PubMed] [Google Scholar]
- 12. Hollingshead A, Redlich F. Social Class and Mental Illness: A Community Study. New York: John Wiley; 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. SAS Institute SAS/STAT User's Guide. Cary, NC: SAS Institute; 2003. [Google Scholar]
- 14. Centers for Disease Control and Prevention CDC Growth Charts: United States. Available at: http://www.cdc.gov/growthcharts/ Accessed June 2011
- 15. Kuczmarski RJ, Ogden CL, Guo SS, et al. CDC Growth Charts for the United States: methods and development. Vital Health Stat 11 2000; 2002: 1– 190 [PubMed] [Google Scholar]
- 16. Djousse L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH. Weight loss in early stage of Huntington's disease. Neurology 2002; 59: 1325– 1330 [DOI] [PubMed] [Google Scholar]
- 17. Robbins AO, Ho AK, Barker RA. Weight changes in Huntington's disease. Eur J Neurol 2006; 13: e7 [DOI] [PubMed] [Google Scholar]
- 18. Mochel F, Charles P, Seguin F, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One 2007; 2: e647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goodman AO, Murgatroyd PR, Medina-Gomez G, et al. The metabolic profile of early Huntington's disease–a combined human and transgenic mouse study. Exp Neurol 2008; 210: 691– 698 [DOI] [PubMed] [Google Scholar]
- 20. Underwood BR, Broadhurst D, Dunn WB, et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 2006; 129: 877– 886 [DOI] [PubMed] [Google Scholar]
- 21. van der Burg JM, Bacos K, Wood NI, et al. Increased metabolism in the R6/2 mouse model of Huntington's disease. Neurobiol Dis 2008; 29: 41– 51 [DOI] [PubMed] [Google Scholar]
- 22. Damiano M, Galvan L, Deglon N, Brouillet E. Mitochondria in Huntington's disease. Biochim Biophys Acta 2010; 1802: 52– 61 [DOI] [PubMed] [Google Scholar]
- 23. Oliveira JM. Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J Neurochem 2010; 114: 1– 12 [DOI] [PubMed] [Google Scholar]
- 24. Powers WJ, Videen TO, Markham J, et al. Selective defect of in vivo glycolysis in early Huntington's disease striatum. Proc Natl Acad Sci USA 2007; 104: 2945– 2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mochel F, Haller RG. Energy deficit in Huntington disease: why it matters. J Clin Invest 2011; 121: 493– 499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosenstock TR, Duarte AI, Rego AC. Mitochondrial-associated metabolic changes and neurodegeneration in Huntington's disease: from clinical features to the bench. Curr Drug Targets 2010; 11: 1218– 1236 [DOI] [PubMed] [Google Scholar]
- 27. Orr AL, Li S, Wang CE, et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 2008; 28: 2783– 2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis 2006; 22: 388– 400 [DOI] [PubMed] [Google Scholar]
- 29. Turner C, Cooper JM, Schapira AH. Clinical correlates of mitochondrial function in Huntington's disease muscle. Mov Disord 2007; 22: 1715– 1721 [DOI] [PubMed] [Google Scholar]
- 30. Singh GK. Childhood Obesity in the United States, 1976–2008: Trends and Current Racial/Ethnic, Socioeconomic, and Geographic Disparities: A 75th Anniversary Publication: Maternal and Child Health Bureau. Rockville, MD: US Department of Health and Human Services; 2010. [Google Scholar]
- 31. Silventoinen K, Sammalisto S, Perola M, et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res 2003; 6: 399– 408 [DOI] [PubMed] [Google Scholar]
- 32. Visscher PM, Medland SE, Ferreira MA, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2006; 2: e41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gerald LB, Anderson A, Johnson GD, Hoff C, Trimm RF. Social class, social support and obesity risk in children. Child Care Health Dev 1994; 20: 145– 163 [DOI] [PubMed] [Google Scholar]
- 34. Lamerz A, Kuepper-Nybelen J, Wehle C, et al. Social class, parental education, and obesity prevalence in a study of six-year-old children in Germany. Int J Obes 2005; 29: 373– 380 [DOI] [PubMed] [Google Scholar]
- 35. Dunger DB, Ahmed ML, Ong KK. Early and late weight gain and the timing of puberty. Mol Cell Endocrinol 2006; 254–255: 140– 145 [DOI] [PubMed] [Google Scholar]
- 36. Buchan IE, Bundred PE, Kitchiner DJ, Cole TJ. Body mass index has risen more steeply in tall than in short 3-year olds: serial cross-sectional surveys 1988–2003. Int J Obes 2007; 31: 23– 29 [DOI] [PubMed] [Google Scholar]
- 37. Murasko JE. Socioeconomic status, height, and obesity in children. Econ Hum Biol 2009; 7: 376– 386 [DOI] [PubMed] [Google Scholar]
- 38. Bartholomeusz HH, Courchesne E, Karns CM. Relationship between head circumference and brain volume in healthy normal toddlers, children, and adults. Neuropediatrics 2002; 33: 239– 241 [DOI] [PubMed] [Google Scholar]
- 39. Bray PF, Shields WD, Wolcott GJ, Madsen JA. Occipitofrontal head circumference–an accurate measure of intracranial volume. J Pediatr 1969; 75: 303– 305 [DOI] [PubMed] [Google Scholar]
- 40. Courchesne E, Chisum HJ, Townsend J, et al. Normal brain development and aging: quantitative analysis at in vivo MR imaging in healthy volunteers. Radiology 2000; 216: 672– 682 [DOI] [PubMed] [Google Scholar]
- 41. Moss ML, Salentijn L. The primary role of functional matrices in facial growth. Am J Orthod 1969; 55: 566– 577 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.