

Abstract
Congo red (CR) is a commonly used histological amyloid dye and a weak amyloid inhibitor. There is currently no experimentally available structure of CR bound to an amyloid fibril and the binding modes, and the mechanisms governing its inhibitory and optical properties are poorly understood. In this work, we present the first, to our knowledge, atomistically detailed picture of CR binding to protofibrils of the Alzheimer Aβ9–40 peptide. We identify three major binding modes, with the primary mode residing in the grooves formed by the β-sheets, and observe a restriction of the torsional rotation of the CR molecule upon binding. Our simulations reveal a novel, to our knowledge, electrostatic steering mechanism that plays an important role in the initial recognition and binding of CR to the positively charged surface residues of the fibril. Our simulations provide new, to our knowledge, insights into the striking spectrophotometric and inhibitory properties of CR. In particular, we show that birefringence upon CR binding is due to the anisotropic orientation of the CR dipoles resulting from the spatial ordering of these molecules in the grooves along the fibril axis. The fluorescent enhancement of the bound CR, in turn, is associated with the torsional restriction of this molecule upon binding.
Introduction
Afflicting over 20 million patients worldwide, Alzheimer’s disease (AD) is the most common neurodegenerative disease for which there is no known cure (1). It has become imperative to develop accurate diagnostic tools (2) and therapeutics (3) for this debilitating disease. A hallmark of AD (3) is the presence of amyloid plaques in the brain tissue, consisting primarily of β-sheet-rich fibrillar aggregates of the 39–42 residue long Amyloid-β (Aβ) peptide. Although the precise cause of AD is still unknown, there is ample evidence that Aβ fibrils, and to an even greater extent early aggregation intermediates such as soluble oligomers and protofibrils, are cytotoxic (3–7). Molecules that effectively interact with these Aβ species and interrupt the normal aggregation process of Aβ could serve as lead compounds for the development of novel detection and therapeutic agents for AD. The amyloidogenic dye Congo red (CR) is one such candidate. It has been used to stain amyloid fibrils in tissues for over 80 years (8–11) and has recently been reported to reduce Aβ neurotoxicity (12).
CR is a linear and amphiphilic molecule (Fig. 1): Its hydrophilic part includes two amino groups and a negatively charged sulfate group (at neutral pH), and its hydrophobic part consists of a biphenyl group at the center, along with a diazo group with two flanking naphthalene groups. CR has striking spectrophotometric properties (13–16). In aqueous solution, its absorption spectrum shows a maximal absorption at 490 nm (blue/green), leading to a red color solution, at low concentration and neutral pH. When bound to β-sheet-rich amyloid fibrils, the CR molecules adopt a specific orientation (i.e., with the long axis of the CR molecules lying parallel to the fibril axis) and become torsionally restricted. This induces a characteristic increase in absorption and a red shift in the absorption maximum from 490 to 540 nm. More importantly, this spatial ordering of CR molecules due to binding renders CR birefringent (anisotropic light transmission), diachronic (anisotropic absorption of linearly polarized light), as well as strongly fluorescent. The wavelength dependent birefringence, compounded with the anomalous dispersion around the absorption peak, leads to a characteristic apple-green birefringence when CR-stained amyloids are examined between crossed polarizer and analyzer (17). This constitutes a hallmark signature of amyloid fibrils through this traditional histological birefringence test (8–11). In addition, the enhanced fluorescence of bound CR to amyloid fibrils can serve as a second sensitive diagnostic tool for amyloidosis (15,16).
Figure 1.
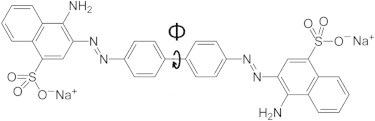
Chemical structure of CR molecule. The central torsion angle (Φ) is indicated.
CR is capable of binding to various Aβ species ranging from monomers to mature fibrils. Binding to CR has been shown to interrupt the normal aggregation of Aβ peptides, thereby reducing toxicity. For example, CR has been shown to bind to low-molecular-weight aggregates of Aβ1–40 to form 1:1 and 1:2 complexes in an NMR study (18); to solubilize toxic Aβ1–40 species such as anionic detergents (19); to bind preformed fibrils and reduce toxicity (12); and to form heterogeneous aggregates with Aβ1–40 (20).
Presently, we lack detailed information at a molecular level regarding the binding of CR to the various Aβ1–40 species. This knowledge would greatly facilitate efforts toward understanding the recognition and inhibition mechanisms of CR and to optimize this compound for clinical use. There are no high-resolution structures of Aβ species in complex with CR available from experiment. Structural studies of monomers and small oligomers of Aβ are hampered by the intrinsic disorder and transient nature of these species. However, experiments (notably solid state NMR) have provided high-resolution structures of Aβ9–40 fibrillar aggregates (21) that can be used in conjunction with computational modeling to probe detailed binding interactions with amyloid dyes. In this study, we focus on the binding of CR to Aβ9–40 protofibrils with the goal not only of identifying binding modes, but more importantly of gaining insight at a molecular level into the striking optical and inhibitory properties of this molecule. CR presents much more complex optical properties than thioflavin T (ThT), the latter only exhibiting fluorescent behavior upon binding to amyloid fibrils. In addition, CR acts as a weak aggregation inhibitor, whereas ThT is not known to inhibit fibril formation. As in our work on the binding of ThT to Aβ protofibrils (22), we will use a single peptide layer to describe the fibril (as shown in Fig. 2), because this construct encompasses the maximum number of binding sites available to the dyes (indeed, in the mature fibrils with multiple peptide layers, some binding sites are buried inside the layer-to-layer interface (23,24).). Our naming protocol for the surface grooves also follows our previous study (22): we will refer to the exposed residues Y10_V12_H14_K16_V18_F20_E22_V24 for the seven grooves on the N-terminal β-sheet layer and I31_M35_V39 for the two grooves on the C-terminal β-sheet layer.
Figure 2.
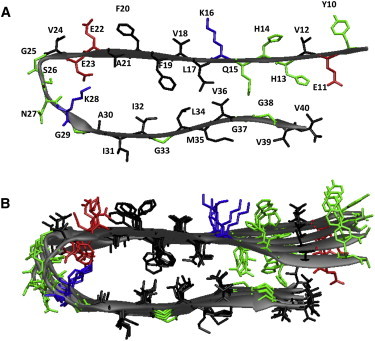
Structure of an Aβ9–40 protofibril from the solid NMR studies. (A) A single Aβ9–40 peptide in the protofibrils consists of a C-terminal β-strand (upper, residues 30–40), an N-terminal β-strand (lower, residues 10–22), and a loop. Residues 1–8 are disordered and thus omitted. (B) A protofibril, composed of six peptides. Negatively charged, positively charged, polar, and hydrophobic residues are colored in red, blue, green, and black, respectively.
Methods
System preparation
Two systems were constructed, one for the dye only and one for the protofibrils binding simulations. The dye-only system, which serves as a negative control to probe the free rotation of the dye’s central torsion, contains one CR molecule, 2 Na+ ions, and 2317 water molecules. The binding system consists of a 6-peptide protofibril, 2 CR molecules, 10 sodium ions, and 10,793 water molecules. The initial structure of the Aβ9–40 protofibril (see Fig. 2) is taken from the study by Tycko and co-workers (21), which was energy minimized and properly equilibrated for this study. Two CR molecules are initially placed ∼10 Å away from the N-/C-terminal sheet layers (see Fig. S1 in the Supporting Material). This allows simultaneous sampling of two sheet layers in a single trajectory and thus enhances sampling efficiency. 10 sodium ions (Na+) were added to neutralize the 4 negative charges carried by the 2 CR molecules and the 6 negative charges carried by the 6 peptides. The solute molecules were then placed into a truncated octahedron water box (a = b = c = ∼86 Ǻ, α = β = γ = 109.47°) containing 10,793 water molecules. The periodic water box was constructed in such a way that the solute was ∼10 Å away from the box surface and thus the minimum distance between the solute and the image was ∼20 Å.
The Duan et al. all-atom point-charge force field (25) (AMBER ff03), which shows a good balance between helix and sheet (26), was chosen to represent the peptide. The water solvent is explicitly represented by the TIP3P (27) model. The basic form of CR (pKa = 4.0) (structure shown in Fig. 1) was used in this study because it is the dominant form at neutral pH conditions. The force field parameters for CR were obtained from a previous study by one of the authors of this work (28). They are included as an appendix in the Supporting Material.
Molecular dynamics simulation
The AMBER 8 simulation package (29) was used for the molecular dynamics (MD) simulations. The system was subjected to periodic boundary conditions via both minimum image and discrete Fourier transform as part of the particle mesh Ewald method (30). After the initial energy minimization, a total of nine simulations (one for the dye only and eight for the binding system) were performed with different initial random velocities. For the binding simulations, the initial velocities were generated according to the Maxwell-Boltzmann distribution at 500 K. The simulations were started after a 10.0 ps run at 500 K to diverge the orientations and positions of the two CR molecules. A short 1.0 ns MD at 310 K in the NPT ensemble (constant pressure and temperature) was performed to adjust the system size and density, and to equilibrate the solvent. The simulations were continued at 310 K for 24.0 ns in the NVT ensemble (constant volume and temperature). For the dye-only simulation, 1.0 ns NPT plus 24.0 ns NVT simulation were conducted. SHAKE (31) was applied to constrain all bonds connecting hydrogen atoms and a time step of 2.0 fs was used. To reduce the computational cost, nonbonded forces were calculated using a two-stage RESPA approach (32) where the forces within a 10 Å radius were updated every step and those beyond 10 Å were updated every two steps. Temperature was controlled at 310 K by using Berendsen’s algorithm (33) with a coupling constant of 2.0 ps. The center of mass translation and rotation were removed every 500 steps. Studies have shown that this removes the block of ice problem (34,35). The trajectories were saved at 2.0 ps intervals for further analysis. The cumulative time of the eight binding simulations is 200 ns, ∼2 months running time of 64 Opteron CPUs (2.2 GHz).
Clustering analysis
We grouped the stable dye-fibril complexes (≥20 atom contacts between dye and protein) from the eight simulations into different structural families based on the root mean-square deviation (RMSD) of the dye molecule (cutoff 3.5 Å) after alignment with the protofibrils, using the Daura clustering protocol (36). In detail, the aligned complexes are clustered into different structural families using a recursive algorithm in which neighboring structures for each structure are identified using the RMSD cutoff, the largest structural family is then identified and removed to form a new structure family, and the process is continued for the remaining structures until all structures have been included into a structural family. The centroid of each cluster is shown in the Supporting Material. The clusters are then merged further into seven binding types based on the binding sites (see Fig. S5).
Rotation relaxation time
The autocorrelation function of the central torsion angle was fit to an exponential decay function ().
Results
Binding of CR to Aβ9–40 protofibrils
We constructed a binding system (see Fig. S1) consisting of an Aβ9–40 protofibril and two free CR molecules, one placed above and one below the protofibril. Over the course of all eight independent binding simulations of 25 ns, the structure of the protofibril was stable at 310 K except for an initial relaxation within the first ns (see Cα RMSD over time plots in Fig. S2). The overall convergence of the binding simulations is confirmed by the steady number of contacts between CR and the protofibrils (Fig. S3). We found that the two copies of CR bound to opposite sides of the protofibrils in six out of eight simulations, (see Fig. S4, 1–6). Two exceptions include a), one CR binding to the edges of the two layers and another binding to the C-terminal sheet layer surface; b), two CR monomers forming a dimer and then binding to the C-terminal sheet layer surface (Fig. S4, 7 and 8). The dimer binding is consistent with our previous binding simulations (28) and the experimental observation (37) by Król and co-workers that CR might bind to amyloid fibrils in a supramolecular form. In general, though, the two CR molecules (seven of out of the eight simulations) are noninteracting, which allows us to separate the eight simulations into 16 binding trajectories of one CR molecule for further analysis.
Charge-charge interaction plays a role in recognition of the N-terminal sheet layer: an electrostatic steering mechanism
Fig. 3 shows the initial and final contacts between the two CR molecules (denoted by CR-1 and CR-2) and the protofibril in a typical binding simulation (see Fig S4 for all eight simulations). The initial contacting modes differ: Whereas only one side of the CR molecule (CR-1) touches down onto the N-terminal sheet layer, both sides of the other CR molecule (CR-2) form initial contacts with the C-terminal sheet layer. Whereas the favorable charge-charge interaction between the positively charged K16 of the N-terminal sheet layer and one of the two negatively charged sulfate groups of the CR-1 molecule appears to be responsible for the initial steering of the dye to the N-terminal sheet layer, the nonspecific hydrophobic interactions appear to be responsible for the initial nonspecific contacts of the dye to the C-terminal sheet layer. In the final snapshot of this trajectory, both sides of the CR-1 molecule are in contact with the protofibril in its final bound state (Fig. 3, bottom): one side forms two salt bridges with the K16 residues and the other side is in contact with hydrophobic residue V18 and aromatic residue F20, leading to an ordered binding mode. CR-2, on the other hand, forms contacts with hydrophobic residues I31 and M35 of the fibril, leading to a disordered binding mode. Table S1 lists a summary of the initial contacts and the final binding mode of the CR molecule to the N-/C-terminal sheet layer in the seven binding trajectories in which there is no CR dimer formation. Clearly, whereas the charge-charge interaction between CR-1 and K16 of the N-terminal sheet leads to the initial salt-bridge formation and the final ordered binding modes in six out of the seven trajectories, the nonspecific hydrophobic interactions between CR-2 and the C-terminal sheet layer leads to the initial nonspecific contacts and the final disordered binding modes in five out of the seven trajectories. Hence, the specific charge-charge interactions lead to preferential orientations in the binding process of the dye to the N-terminal sheet layer.
Figure 3.
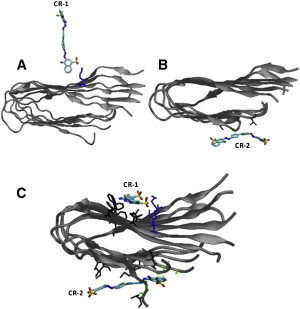
Snapshots showing the initial (A and B) and the last (C) contacts between each CR molecule and the protofibril in a typical binding trajectory having two CR molecules.
Three major binding modes of CR to the protofibril
To identify the binding modes, stable complexes (atom contacts ≥20) from all trajectories were clustered into different structural groups as described in the Methods section. Representative structures of the abundant structural families (>1% of the population) are shown in the Supporting Material (Fig. S5). The top 16 structural families can be further merged into seven binding types based on their binding sites (Fig. 4): type I), binding in the H14_V12 groove of the N-terminal sheet layer; type II), binding in the F20_V18 groove of the N-terminal sheet layer; type III), binding in the G29_I31 groove of the C-terminal sheet layer; type IV), binding in the I31_M35 groove of the C-terminal sheet layer; type V), binding parallel to the β-strands of the N-terminal sheet layer; type VI), binding parallel to the β-strands of the C-terminal sheet layer; and type VII), binding at the edge of the two sheet layers. These seven binding types can be further grouped into three binding modes, summarized in Table 1 along with their abundance: A), binding in the grooves perpendicular to the β-strand (58%) and along the fibril axis; B), binding parallel to the β-strand (21%); C), binding to an edge of the two sheet layers (12%). In mode A, binding to the N-terminal sheet layer (50%) is more abundant than binding to the C-terminal sheet layer (8%). This is likely due to favorable charge-charge interaction between K16 and the sulfate groups of CR.
Figure 4.
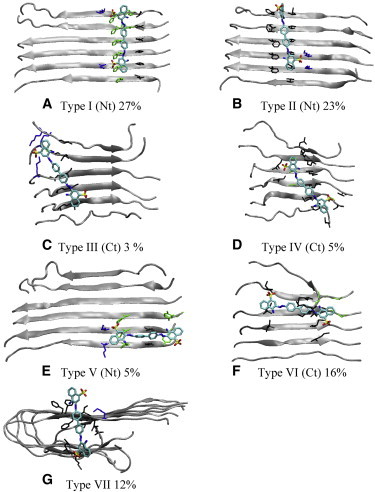
Seven binding sites (A–G) observed from the binding simulations. Abundance is noted. Nt: N-terminal sheet layer; Ct: C-terminal sheet layer. For clarity, the unbound sheet layer is not shown.
Table 1.
Features of the binding modes (Fig. 4)
| Binding modes | A | B | C | |
|---|---|---|---|---|
| Binding type | I–II (Nt sheet layer) | III–IV(Ct sheet layer) | V–VI | VII |
| Feature | Perpendicular to β-stand | Parallel to β-stand | Sheet edges | |
| Abundance | 58% (50% + 8%) | 21% | 12% |
Rotational restriction of the central torsion angle of CR
Much like ThT, binding of CR to amyloid fibrils leads to fluorescence. It has been suggested that the observed fluorescence is associated with rotational restriction of these molecules. To examine how rotation of the two rings of the CR molecule is affected by binding, we used the protocol developed in our earlier study on ThT (22) to characterize the preferential orientation of the central torsion angle of CR and its rotation relaxation time under different environments. To investigate torsional preference, we calculated the probability distributions of the central torsion angle (Fig. 5 A). Because the dye molecule roughly has C2 rotation symmetry, the torsion angle ranges between 0° and 90°. The distribution for CR in free and bound form is very similar and the most probable angle is ∼30°, which is consistent with steric hindrance between the neighboring hydrogen atoms on the two central rings (Fig. 5 B). For the rotation relaxation time, we calculated the autocorrelation of each dye molecule in each binding trajectory (Fig. S8). The summary of these results is listed in Table 2. Clearly, the rotational relaxation time of CR increases from 0.2 ps in bulk water to ∼4.0 ps on the surface of the protofibril. The 20-fold increase of the rotational relaxation time is due to larger friction forces/viscosity resulting from stronger rotational restrictions imposed by the more rigid environment (surface residues) on the dye molecules.
Figure 5.
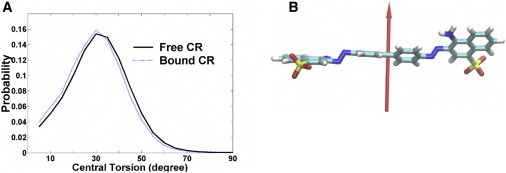
Central torsion angle distributions of dye molecules at free and bound states (A) and dipole moment vector of dye molecule at the bound state (B).
Table 2.
Rotational relaxation times of the central torsion angle of CR under different environments
| Environment τ(ps) | Cong red |
|---|---|
| Free in water | 0.2 |
| Binding to Aβ9–40 | 4.0 ± 1.0 |
Discussion and Conclusion
When CR-stained amyloid deposits are microscopically examined between cross polarizer and analyzer, one can observe an apple-green birefringence, which is a hallmark of amyloids. This histological hallmark (8–11) is believed to be caused by the spatial ordering of CR molecules upon binding to amyloids. The importance of the spatial ordering of CR molecules in producing the birefringence is confirmed by the fact that the spectrophotometric properties of the fibril-bound CR molecules can be replicated in nonfibrillar systems, provided that the CR molecules become ordered. For example, Howie et al. (17) have shown that the smearing of CR drops with a single stroke in one direction on a glass slide can produce ordering of CR molecules in streaks (i.e., the long axis of CR molecules is parallel to the smearing direction/the streak axis), which leads to the same birefringence as in amyloid systems. Because amyloid fibrils are rich in β-sheet content (more specifically with a cross-β-structure), as established from x-ray powder diffraction (38), solid state NMR (39) and computational studies (23,24,40–42), and because CR can bind to a number of amyloid fibrils (suggesting a common recognition motif), it has been proposed that CR might bind in the characteristic grooves present on the β-sheet surface of amyloids (43). The simulations presented in this work provide the first atomistically detailed picture of CR binding to amyloid fibrils of the Aβ40 peptide and reveal that the primary binding mode is indeed located in the grooves, with the long axis of the CR molecules residing parallel to the long axis of the protofibril axis. The detailed atomistic information gleaned from simulation enables us to rationalize both the spectrophotometric properties and the inhibitory properties of this molecule.
Our simulations reveal that the CR molecules adopt a specific ordering upon binding to the amyloid fibril. By examining the ordering of the CR molecules in the main binding site, we can obtain new molecular insights into how spatial ordering affects the photo-matter interaction in light transmission. The refractive index of an organic compound in bulk is determined by several molecular descriptors, including the polarizability of the molecule, the charge distribution in the molecule, and the hydrogen bonding capability in the bulk (44). Here, we focus on the molecular charge distribution to provide a qualitative explanation of the birefringence of fibril-bound CR. Recall that the interaction of an electric field of linearly polarized light wave and a dipole of a charged molecule is given by the dot product of the dipole moment and the electric field vector. When the two vectors are parallel, the interaction is strongest, leading to a bigger change of light speed. Conversely, when they are perpendicular, the interaction is weakest, leading to a smaller change of light speed. Hence, this angle-dependent interaction will modulate the transmission speed of light. In our simulations, the preferred conformation of CR molecules in the major binding mode is planar (see Fig. 4, I–VI, and Fig. 5), and parallel to the local β-strand surface. Our previous quantum mechanical calculations show that monopole and dipole of the CR molecule in this planar conformation are −2 charge and 13.0 Debye (28) (Fig. 5 B). Of particular importance is the fact that the dipole direction is perpendicular to the plane of the CR molecule (i.e., perpendicular to the local β-strand surface). Due to the twist angle (∼15°) between two neighboring β-strands in a β-sheet (Fig. 6 A), the β-strands in the β-sheets adopt a long-range helical rotation (∼24 strands) along the fibril axis (i.e., the β-sheets are not planar but rotated along the fibril axis as shown in Fig. S6) (38,45). With these two basic facts (i.e., the CR dipole is perpendicular to the local β-strand plane and the β-strand plane is rotated along the fibril axis), we can schematically represent the dipole orientation of the CR molecules in the major binding mode from different viewpoints (Fig. 6, B and C). When viewed along the fibrils axis (Fig. 6 B), the projection of the termini of the β-strand in one β-sheet layer is roughly a circle due to the helical rotation, with the dipole of the CR molecules tangent to the circle as a result of the dipole being perpendicular to the β-strand plane. When the linearly polarized light trace is along the fibril axis, the interaction between the electric field of the light wave and the dipoles is isotropic, regardless of the oscillating plane of the electric field. Therefore, the transmission velocity of any linearly polarized light is the same and the fibril axis becomes the optical axis for this birefringent material. In contrast, if viewed perpendicular to the optical/fibril axis (i.e., along the β-strand direction), the projection of the termini of the β-strands in one β-sheet layer can be represented by a wave line as a result once again of the rotation of the β-strands (Fig. 6 C). The projection of the CR dipole in a plane (e.g., the paper plane in Fig. 6 C) passing the fibril axis will also be modulated by the rotation of the β-strands due to the orthogonal relation between the dipole direction and β-strand direction (i.e., it is maximal when the β-strand is perpendicular to the paper plane and minimal when the β-strand is parallel to the paper plane). The dipole direction of the CR molecules is always perpendicular to the optical axis and there is no dipole along the fibril axis. When the linearly polarized trace is perpendicular to the optical axis (i.e., the paper plane is the polarization plane), the transmission velocity of the light wave will be anisotropic and depend on the angle between the CR dipole direction and the direction of the oscillating electric field. The biggest velocity difference (this difference could be negative or positive depending on the wavelength, leading to negative and positive birefringence) appears when the angle is either 0 or 180 degrees (0/180) for one light wave (ordinary light) and 90/270 degrees for another light wave (extraordinary light). To get the brightest birefringence interference for CR-strained amyloid between crossed polarizer and analyzer, the angle should be 45/225 degrees. Negative birefringence leads to transmission of the color blue, positive yellow, and the mixture appears as apple-green (13,17).
Figure 6.
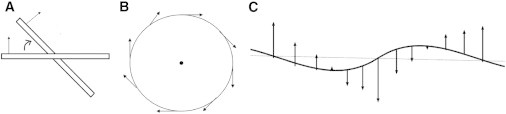
Scheme for showing dipole orientation of CR molecules bound along the fibril axis (e.g., type I–IV of Fig. 4) on a β-sheet layer with helical rotation. (A) Twisting between two β-stands in a β-sheet layer. (B) Viewpoint along the fibril axis. (C) Viewpoint along the β-strand direction, which is perpendicular to the fibril axis. Arrow represents CR dipole vector direction.
The dipole model presented previously, which is based on the orientation of the CR molecules in the major binding site identified from simulation, rationalizes the experimentally observed birefringence. This interaction analysis between the electronic field of light and the dipole of the bound CR molecules in the major binding mode can be extended to explain the experimentally observed dichromatic feature (i.e., anisotropic light absorption) of the spatially ordered CR molecules. The only difference between the two cases is that the transition dipole involving both electronically excited states and the ground state should be used instead of the dipole of the ground state (as was used in the previous transmission analysis).
Another interesting optical feature of CR is enhanced fluorescence upon binding to amyloid fibrils. This is a common feature shared by other amyloid dyes, such as ThT. The enhancement has been extensively studied experimentally and theoretically in the case of ThT (46–48). Torsional rotation restriction between two rings of ThT in the electronically excited state has been suggested as the key factor for reducing nonradioactive relaxation and thereby improving quantum yield. Indeed, our previous analysis based on binding simulations of ThT to the same Aβ9–40 profibril have shown that the rotation relaxation of ThT increases from 0.4 to 0.7 ps in bulk water to 6.1–10.5 ps s on the profibril surface in the ground state (22). In this study, the same analysis shows a similar trend with the relaxation time increasing from 0.2 to ∼4.0 ps for the central torsion angle of CR. We suggest based on these simulations that rotational restriction should also be an important component of CR fluorescence in the bound state. This fluorescence feature could serve as an additional sensitive diagnosis tool for amyloids.
In addition to shedding light into the optical properties of bound CR, our simulations also provide insights into the chemical specificity of the binding between CR and Aβ fibrils. In earlier work, one of the authors of this work examined the binding of CR to protofibrils of the model amyloidogenic fragment GNNQQNY of the yeast prion Sup35 (28) and identified two major binding sites, the first in the grooves formed by residues GNN, with binding of CR parallel to the fibril axis (73%), and the second site with CR binding parallel to the β-strands (11%). In the case of the Aβ9–40 profibril, which has a more complex supramolecular structure (strand-loop-strand, versus simple β-sheet), we identify seven binding sites (Fig. 4, A–G), which can be grouped into three main binding modes: groove binding (58%), β-strand binding (21%, parallel to β-strand), and edge binding (12%, perpendicular to β-strand). In earlier work, we also examined the binding of the ThT dye to the same Aβ9–40 profibril (22) and in that case, we identified two major binding modes: groove binding (52%) and loop binding (48%). The nature of the most abundant binding mode (located in the grooves) is the same for CR binding to protofibrils of both the Sup35 fragment and the Aβ9–40 peptide, highlighting the fact that CR recognize a common structural motif. Interestingly, our earlier work on ThT binding to the same Aβ9–40 profibril (22) as well as to other fragments (49,50) also revealed that the main binding mode of ThT is located in the grooves, implying that CR and ThT share the same primary recognition motif. Nonetheless, it is important to point out that the chemical differences between CR and ThT, and between the peptides making up the different fibrils, lead to variations of the secondary binding modes. Hence, CR and ThT are not perfect competitive binders, as their minor binding modes are located at different sites (e.g., β-strand and edge of the Aβ9–40 profibril in the case of CR and loop of the Aβ9–40 profibril in the case of ThT).
Our simulations also uncover a novel, to our knowledge, electrostatic steering mechanism for the binding of CR to the charged N-terminal sheet layer of the Aβ9–40 profibril. Indeed, we see that charge-charge interactions between K16 of the fibril and CR play an important role in directing the binding of CR to the charged fibril surface. Favorable charge interaction between K16 and the sulfate group lead to a steering effect in the initial binding contact stage (Fig. 4). Binding with these salt bridges (70% of Fig. 4, I–III, V, and VII), are more abundant than the binding without the salt bridges (21% of Fig. 4, IV and VI). This electrostatic steering was not observed for either the binding of CR to the GNNQQNY protofibrils or the binding of ThT to the Aβ9–40 profibril. The lack of any positively charged residue in the GNNQQNY peptide can explain the former, whereas the deeper burial of the positively charge ThT (with its methyl group) provides an explanation for the latter.
In addition, the three binding modes observed in our simulations provide insights into the inhibition mechanism of CR to Aβ fibrils formation. Groove binding and β-strand binding on the β-sheet surface may play a role in interrupting sheet-to-sheet stacking, whereas edge binding, a mode that has also been observed in the binding of two nonsteroidal antiinflammatory drugs (51,52) to the concave edges of the Aβ fibril, may play a role in blocking strand-to-sheet extension. Edge binding may be an important player in inhibition: this binding mode was not observed in binding simulations of ThT (which does not inhibit aggregation) to the Aβ9–40 profibril. Additionally, the exposed charged and polar groups of CR should improve the solubility of the bound Aβ species (19), which may prevent further oligomerization (note that whereas CR has a −2 charge, the noninhibiting ThT has a +1 charge). Future work will involve the simulation of CR to monomers and oligomers of Aβ to further elucidate the role of CR in inhibiting the aggregation process.
Acknowledgments
This project is funded by the David and Lucile Packard Foundation and the National Science Foundation (MCB 0642086 and 1158577) and the National Institutes of Health (AG027818). The computer time was provided by the Lonestar and Ranger clusters at the Texas Advanced Computing Center (XSEDE grant MCA 05S027).
Supporting Material
References
- 1.Goedert M., Spillantini M.G. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Drzezga A. Amyloid-plaque imaging in early and differential diagnosis of dementia. Ann. Nucl. Med. 2010;24:55–66. doi: 10.1007/s12149-009-0330-9. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto M., Rockenstein E., Masliah E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med. 2003;4:21–36. doi: 10.1385/NMM:4:1-2:21. [DOI] [PubMed] [Google Scholar]
- 5.Lorenzo A., Razzaboni B., Yankner B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 6.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 7.Bucciantini M., Calloni G., Stefani M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004;279:31374–31382. doi: 10.1074/jbc.M400348200. [DOI] [PubMed] [Google Scholar]
- 8.Benhold H. Specific staining of amyloid by Congo red. Muenchen. Med. Wochenschr. 1922:1537–1538. [Google Scholar]
- 9.Divry P. Histochemical study of senile plaques. J. Neurol. Psychiatry. 1927;27:643–657. [Google Scholar]
- 10.Klunk W.E., Jacob R.F., Mason R.P. Quantifying amyloid β-peptide (Abeta) aggregation using the Congo red-Abeta (CR-abeta) spectrophotometric assay. Anal. Biochem. 1999;266:66–76. doi: 10.1006/abio.1998.2933. [DOI] [PubMed] [Google Scholar]
- 11.Klunk W.E., Pettegrew J.W., Abraham D.J. Quantitative evaluation of Congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J. Histochem. Cytochem. 1989;37:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 12.Lorenzo A., Yankner B.A. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc. Natl. Acad. Sci. USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howie A.J., Brewer D.B. Optical properties of amyloid stained by Congo red: history and mechanisms. Micron. 2009;40:285–301. doi: 10.1016/j.micron.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Jin L.W., Claborn K.A., Kahr B. Imaging linear birefringence and dichroism in cerebral amyloid pathologies. Proc. Natl. Acad. Sci. USA. 2003;100:15294–15298. doi: 10.1073/pnas.2534647100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linke R.P. Highly sensitive diagnosis of amyloid and various amyloid syndromes using Congo red fluorescence. Virchows Arch. 2000;436:439–448. doi: 10.1007/s004280050471. [DOI] [PubMed] [Google Scholar]
- 16.Sen S., Başdemir G. Diagnosis of renal amyloidosis using Congo red fluorescence. Pathol. Int. 2003;53:534–538. doi: 10.1046/j.1440-1827.2003.01513.x. [DOI] [PubMed] [Google Scholar]
- 17.Howie A.J., Brewer D.B., Jones A.P. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab. Invest. 2008;88:232–242. doi: 10.1038/labinvest.3700714. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen M.O., Mikkelsen K., Nielsen N.C. NMR reveals two-step association of Congo red to amyloid β in low-molecular-weight aggregates. J. Phys. Chem. B. 2010;114:16003–16010. doi: 10.1021/jp108035y. [DOI] [PubMed] [Google Scholar]
- 19.Lendel C., Bolognesi B., Gräslund A. Detergent-like interaction of Congo red with the amyloid β peptide. Biochemistry. 2010;49:1358–1360. doi: 10.1021/bi902005t. [DOI] [PubMed] [Google Scholar]
- 20.Bose P.P., Chatterjee U., Arvidsson P.I. Effects of Congo red on Aβ(1–40) fibril formation process and morphology. ACS Chem. Neuros. 2010;1:315–324. doi: 10.1021/cn900041x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petkova A.T., Yau W.M., Tycko R. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C., Bowers M.T., Shea J.E. On the origin of the stronger binding of PIB over thioflavin T to protofibrils of the Alzheimer amyloid-β peptide: a molecular dynamics study. Biophys. J. 2011;100:1316–1324. doi: 10.1016/j.bpj.2011.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buchete N.V., Hummer G. Structure and dynamics of parallel β-sheets, hydrophobic core, and loops in Alzheimer’s Aβ fibrils. Biophys. J. 2007;92:3032–3039. doi: 10.1529/biophysj.106.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng J., Ma B.Y., Nussinov R. Molecular dynamics simulations of Alzheimer Abeta40 elongation and lateral association. Front. Biosci. 2008;13:3919–3930. doi: 10.2741/2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan Y., Wu C., Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 26.Hornak V., Abel R., Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparisons of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 28.Wu C., Wang Z.X., Duan Y. Dual binding modes of Congo red to amyloid protofibril surface observed in molecular dynamics simulations. J. Am. Chem. Soc. 2007;129:1225–1232. doi: 10.1021/ja0662772. [DOI] [PubMed] [Google Scholar]
- 29.Wang J.M., Wolf R.M., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 30.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 31.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 32.Procacci P., Berne B.J. Multiple time-scale methods for constant-pressure molecular-dynamics simulations of molecular-systems. Mol. Phys. 1994;83:255–272. [Google Scholar]
- 33.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Comput. Phys. 1984;81:3684–3690. [Google Scholar]
- 34.Chiu S.W., Clark M., Jakobsson E. Collective motion artifacts arising in long-duration molecular dynamics simulations. J. Comput. Chem. 2000;21:121–131. [Google Scholar]
- 35.Harvey S.C., Tan R.K.Z., Cheatham T.E. The flying ice cube: velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998;19:726–740. [Google Scholar]
- 36.Daura X., Gademann K., Mark A.E. Peptide folding: when simulation meets experiment. Angew. Chem. Int. Ed. 1999;38:236–240. [Google Scholar]
- 37.Stopa B., Piekarska B., Król M. The structure and protein binding of amyloid-specific dye reagents. Acta Biochim. Pol. 2003;50:1213–1227. [PubMed] [Google Scholar]
- 38.Sunde M., Serpell L.C., Blake C.C. Common core structure of amyloid fibrils by synchrotron x-ray diffraction. J. Mol. Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 39.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 2006;39:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 40.Ma B.Y., Nussinov R. Stabilities and conformations of Alzheimer’s β -amyloid peptide oligomers (Abeta 16–22, Abeta 16–35, and Abeta 10–35): sequence effects. Proc. Natl. Acad. Sci. USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchete N.V., Tycko R., Hummer G. Molecular dynamics simulations of Alzheimer’s β-amyloid protofilaments. J. Mol. Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 42.Zheng J., Jang H., Nussinov R. Modeling the Alzheimer Abeta17–42 fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys. J. 2007;93:3046–3057. doi: 10.1529/biophysj.107.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krebs M.R.H., Bromley E.H.C., Donald A.M. The binding of thioflavin-T to amyloid fibrils: localization and implications. J. Struct. Biol. 2005;149:30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Katritzky A.R., Sild S., Karelson M. Correlation and prediction of the refractive indices of polymers by QSPR. J. Chem. Inf. Comput. Sci. 1998;38:1171–1176. [Google Scholar]
- 45.Kajava A.V., Baxa U., Steven A.C. β arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolfe L.S., Calabrese M.F., Xiong Y. Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc. Natl. Acad. Sci. USA. 2010;107:16863–16868. doi: 10.1073/pnas.1002867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stsiapura V.I., Maskevich A.A., Buganov O.V. Charge transfer process determines ultrafast excited state deactivation of thioflavin T in low-viscosity solvents. J. Phys. Chem. A. 2010;114:8345–8350. doi: 10.1021/jp105186z. [DOI] [PubMed] [Google Scholar]
- 48.Stsiapura V.I., Maskevich A.A., Kuznetsova I.M. Computational study of thioflavin T torsional relaxation in the excited state. J. Phys. Chem. A. 2007;111:4829–4835. doi: 10.1021/jp070590o. [DOI] [PubMed] [Google Scholar]
- 49.Wu C., Wang Z.X., Shea J.E. The binding of thioflavin T and its neutral analog BTA-1 to protofibrils of the Alzheimer’s disease Abeta(16–22) peptide probed by molecular dynamics simulations. J. Mol. Biol. 2008;384:718–729. doi: 10.1016/j.jmb.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu C., Biancalana M., Shea J.E. Binding modes of thioflavin-T to the single-layer β-sheet of the peptide self-assembly mimics. J. Mol. Biol. 2009;394:627–633. doi: 10.1016/j.jmb.2009.09.056. [DOI] [PubMed] [Google Scholar]
- 51.Raman E.P., Takeda T., Klimov D.K. Molecular dynamics simulations of Ibuprofen binding to Abeta peptides. Biophys. J. 2009;97:2070–2079. doi: 10.1016/j.bpj.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takeda T., Chang W.L.E., Klimov D.K. Binding of nonsteroidal anti-inflammatory drugs to Aβ fibril. Proteins. 2010;78:2849–2860. doi: 10.1002/prot.22804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.