

Abstract
Intramolecular chaperones (IMCs), which are specific domains/segments encoded in the primary structure of proteins, exhibit chaperone-like activity against the aggregation of the other domains in the same molecule. In this research, we found that the truncation of the linker greatly promoted the thermal aggregation of the isolated C-terminal domain (CTD) of rabbit muscle creatine kinase (RMCK). Either the existence of the linker covalently linked to CTD or the supply of the synthetic linker peptide additionally could successfully protect the CTD of RMCK against aggregation in a concentration-dependent manner. Truncated fragments of the linker also behaved as a chaperone-like effect with lower efficiency, revealing the importance of its C-terminal half in the IMC function of the linker. The aggregation sites in the CTD of RMCK were identified by molecular dynamics simulations. Mutational analysis of the three key hydrophobic residues resulted in opposing effects on the thermal aggregation between the CTD with intact or partial linker, confirming the role of linker as a lid to protect the hydrophobic residues against exposure to solvent. These observations suggested that the linkers in multidomain proteins could act as IMCs to facilitate the correct folding of the aggregation-prone domains. Furthermore, the intactness of the IMC linker after proteolysis modulates the production of off-pathway aggregates, which may be important to the onset of some diseases caused by the toxic effects of aggregated proteolytic fragments.
Introduction
Structure and stability are key factors that define the function, regulation, and turnover of a protein. It is believed that the information that governs the nascent polypeptide folding to its native structure is encoded in the primary sequence of a protein (1). Although many proteins have been characterized to be able to fold correctly in vitro, misfolding and aggregation are frequently encountered in both laboratory investigations and industrial production of many proteins (2). The folding of multimeric/multidomain proteins are particularly complex and are more prone to compete with the unproductive off-pathway aggregation than the single domain small proteins. Aberrant protein aggregation has also been associated with many severe diseases including neurodegenerative diseases (2) and familial conformational diseases (3). Moreover, the in vivo folding of nascent polypeptides is strongly dependent on the particular environment (2), such as macromolecular crowding (4). To achieve reliable and efficient folding of proteins for various functions, the cells have evolved quality control systems to assist the correct folding, to stabilize the native structure, and to degrade the misfolded proteins. In particular, it is now well established that factors facilitating protein folding include folding catalysts, molecular chaperones, chemical chaperones, and intramolecular chaperones (IMCs) (2,5,6).
Unlike the well-known molecular chaperones, the IMCs are specific domains/segments encoded in the primary structure of proteins (6). IMCs were initially reported in 1987 (7), and have been identified in many proteins. Most of the IMCs were characterized in preproproteins, in which the preregions at the N- or C-terminus play a crucial role in the assistance of correct folding or association of the functional domains (6). These propeptides are usually removed from the matured proteins after the completion of protein folding. In addition to preproproteins, IMCs are also proposed to exist in several other proteins (8–12), in which these regions/domains are not cleaved and play a role in protein functions. These observations strongly suggest that IMCs may be widely distributed in proteins to control the hierarchical folding processes (13). However, this concept needs to be further verified by investigations of more proteins and of detailed mechanisms of IMCs.
Creatine kinase (CK, EC 2.7.3.2), a key enzyme involved in the intracellular energetic homeostasis of vertebrate excitable tissues that require large energy fluxes (14), catalyzes the reversible transfer of the high-energy phosphate between the ATP/ADP and creatine/phosphocreatine systems. It has been well established that in vertebrate cells, the intracellular microdomains associated with ATP production and consumption are mainly connected by the phosphocreatine-CK network (14). Due to its importance in intracellular energetic homeostasis, the dysfunction or deficiency of CK has been associated with many serious diseases (15–18). In particular, CK aggregation has been observed for an acute myocardial infarction patient with inherited D54G mutation (16) and for cells subjected to stress caused by nitrogen-based oxidants (19).
Each subunit of CK contains two domains, the N-terminal domain (NTD) and the C-terminal domain (CTD) connected by a long linker (Scheme 1). The small NTD contains only α-helices and consists of ∼100 residues, whereas the large CTD contains ∼250 residues folding into a core eight-stranded antiparallel β-sheet surrounded by six helices. The linker region has been shown to participate in the muscle-isoform-specific binding to the M-line (20). Recently, we found that the CTD was more likely to be the binding site during rabbit muscle CK (RMCK) aggregation (21) and the linker plays a crucial role in RMCK folding (10,22). These findings led to the hypothesis that the linker might act as an IMC to stabilize CTD. However, the linker between domains is rarely identified to be an IMC or to play a crucial role in folding. To further investigate the role of the linker in CK folding, the isolated CTD was overexpressed in Escherichia coli, with either an intact or a truncated linker (Scheme 1). The results herein strongly suggested that the intact linker exhibited chaperone-like activity to facilitate the folding of the aggregation-prone CTD.
Scheme 1.
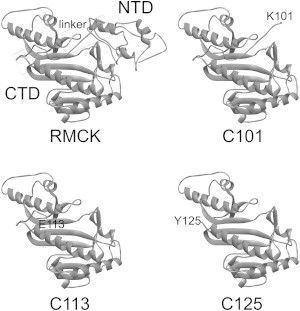
Schematic presentation of the structures of the WT RMCK (PDB ID 2CRK) and the three CTD fragments C101, C113, and C125. The residues at the truncated sites were labeled.
Materials and Methods
Chemicals
Sodium dodecyl sulfate, ultrapure guanidine hydrochloride, Tris, ATP, sequencing-class trypsin, and protease K were purchased from Sigma-Aldrich (St. Louis, MO). Dithiothreitol was a BIOMOL (Plymouth Meeting, PA) product. All other chemicals were local products of analytical grade. The linker peptide L-f (GGFKPTDKHKTDLNHENLKGGDDLDPHY, residues 98–125), L-n (GGFKPTDKHKTDLNH, residues 98–112), and L-c (ENLKGGDDLDPHY, residues 113–125) were synthesized and verified by mass spectrometry.
Protein expression and purification
The gene of the wild-type (WT) RMCK and the pET-21b expression vector were the same as those described elsewhere (23). Five isolated domains: N100 (residues 1–100), N112 (residues 1–112), C101 (residues 101–381), C113 (residues 113–381), and C125 (residues 125–381) and six mutants were constructed using the primers listed in Table S1 in the Supporting Material. Details regarding protein expression and purification were the same as those described previously (23). All protein samples were in 10 mM Tris-HCl buffer, pH 8.0. The protein concentration was determined according to the Bradford method by using bovine serum albumin as a standard (24). The final concentration of the dimeric WT protein was 3 μM, whereas those of the monomeric mutants were 6 μM for all experiments unless otherwise indicated.
Activity assay
The activity of the WT and mutated enzymes was determined according to the pH-colorimetry method (25) at 25°C. The activity of the cell lysates was determined using a coupled enzyme reaction system as described previously (26).
Spectroscopy
Intrinsic fluorescence spectra were recorded on an F2500 spectrophotometer (Hitachi, Tokyo, Japan) using a 0.2 ml cuvette with an excitation wavelength of 295 nm. Parameter A, which is the characteristic of the shape and position of the fluorescence spectrum, was calculated by dividing the intensity at 320 nm by that at 365 nm (I320/I365) (27). Far-ultraviolet (UV) circular dichroism (CD) spectra were measured on a Jasco-715 spectrophotometer (Jasco, Tokyo, Japan) using a cell with a path length of 0.1 cm. All resultant spectra were obtained by the subtraction of the spectra of the buffer.
Protein thermal unfolding and aggregation
The thermal unfolding of the proteins was investigated by incubating the samples at the given temperature for 2 min, and the data were recorded every 2°C. The time-course aggregation was monitored by measuring A400 with a 4300 pro UV/Visible spectrophotometer (Ultraspec, Amersham Biosciences, Piscataway, NJ) using a 0.2 ml cuvette at 50 or 46°C. The aggregation kinetics was analyzed using a first-order reaction as proposed previously (28), and the data fitting were performed by nonlinear regression analysis using the following equation:
| (1) |
where Alim is the A400 value at the infinite time, t is the time of incubation at a given temperature, and kagg is the rate constant of the first-order reaction. The thermal transition curves from CD were analyzed by a two-state thermal unfolding model, and the following equation was used for the nonlinear least square analysis to obtain the thermodynamic parameters:
| (2) |
where y is the mean residue molar ellipticity at 222 nm or parameter A of Trp fluorescence, T is the temperature, Tm is the temperature where half unfolding is achieved, R is the Boltzmann constant and ΔHm is the enthalpy of unfolding of Tm. The pre- and posttransitional baselines were defined by (yf + mfT) and (yu + muT). For the transition curve of WT RMCK from CD spectroscopy, the three-state process was analyzed by the sum of two sequential two-state processes because a stable intermediate could be observed.
Molecular dynamics (MD) simulation
CK structures were simulated by atomistic molecular dynamics (MD) simulation, using the CHARMM22 force field in the program NAMD2.7 (29). The x-ray crystal structure of RMCK (PDB code 2CRK) was adopted as the starting structure. By removing the NTD at residues 101, 113, or 125, respectively, three models of the RMCK CTD (C101, C113, and C125) were built with decreasing length of the linker. Another model (C113 + L-n) was built by breaking the peptide bond between residues 112 and 113 of C101 to further analyze the protection role of the linker. Each protein was explicitly solvated by TIP3 water molecules, and 0.01 mol/L Na+ and Cl− ions were placed into the water box to neutralize the system. Each model was equilibrated for 11 ns and simulated for 10 ns at high temperature (400 K) and room temperature (300 K), respectively. All simulations were conducted under constant temperature (300 K/400 K) and pressure (1 atm), controlled by Langevin dynamics and Langevin piston, respectively. To quantify the aggregation tendency of each residue, spatial aggregation propensity (SAP) (30) value was calculated for each system. This parameter was scaled between −0.5 and +0.5, to evaluate the hydrophobicity of the microenvironment around a specific residue in a given protein structure. A large positive SAP score typically indicates a greater aggregation tendency. The SAP score for all residues were calculated by averaging over all structural snapshots in the 10 ns productive simulations. The aggregation propensity for each residue was also calculated by the Zyggregator method (31) using the online computational algorithm (http://www-vendruscolo.ch.cam.ac.uk/zyggregator_test.php).
Results
The linker is vital to CK assembly, but not to domain structure
To evaluate the role of the linker in domain folding and structure, five fragments of RMCK (N100, N112, C101, C113, and C125) were overexpressed in E. coli. The five domain fragments showed quite different behavior: N100 and N112 mainly existed in the supernatants, C101 and C113 were in both the supernatants and precipitations, whereas C125 mainly existed in the precipitant (Fig. S1). However, the yields of N100 and N112 were very low and the purity was unsatisfactory for structural analysis via spectroscopic experiments. Nonetheless, the CD spectrum of N112 with ∼90% purity indicated that the NTD fragments were more likely to be well folded (Fig. S2). The corresponding NTD and CTD fragments were mixed with an equal molar ratio, equilibrated for 24 h at 25°C, and then the activity was measured. No CK activity was detectable for the mixed solutions. The inability of the isolated NTD and CTD to assemble to an active form was also verified by the lack of CK activity in the soluble extracts of the E. coli cells that coexpressed the N100/C101 or N112/C113 fragments, which indicated that the intactness of the linker was essential to RMCK assembly.
The CD spectra of the two CTD fragments exhibited a typical shape of α+ β structure (Fig. 1). The mean residue ellipticity at 222 nm was similar to the value of the WT protein, indicating that secondary structures are well preserved in both fragments. RMCK contains four Trp residues, and all of them are located in the CTD. The emission maximum wavelengths (Emax) of the Trp fluorescence were red-shifted from 334 to 337 nm by mutations, suggesting that the Trp residues in C101 and C113 were more exposed to solvent when compared to the WT protein. Thus, the removal of the NTD did not significantly affect the regular secondary structure contents, but resulted in a relative loose tertiary structure of the CTD.
Figure 1.
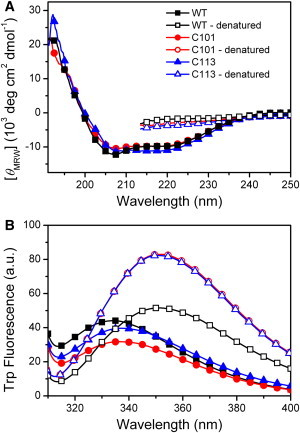
Structural properties of the WT RMCK and the CTD fragments C101 and C113 evaluated by far-UV CD (A) and Trp fluorescence spectroscopy (B). The spectra of the guanidine hydrochloride-denatured state are also presented as open symbols.
The linker does not affect the thermal unfolding but protects CTD against aggregation
Consistent with the previous studies (10,21), the thermal unfolding of the WT RMCK was dominated by an irreversible two-state process (Fig. 2), and the midpoint of the main thermal transition (Tm) was around 63°C (Table S2). A minor transition at around the body temperature could also be identified from the CD spectroscopy. The large discrepancy between the Tm values obtained from the CD and Trp fluorescence transition curves suggested that the thermal unfolding of the WT RMCK involves noncooperative events, which coincides with the previous observation (21). The minor transition in the WT protein thermal unfolding was invisible for the two CTD fragments, revealing that this transition correlated to the structural changes of the NTD or modification of the domain/subunit interactions. Nonetheless, the similar transition curves of two CTD fragments indicated that the existence of the intact or truncated linker did not significantly affect the thermal transitions of the CTD.
Figure 2.
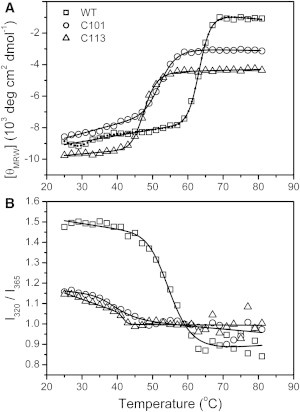
Thermal transition curves of the WT RMCK, C101 and C113 by the mean residue molar ellipticity at 222 nm (A) and I320/I365 of the Trp fluorescence spectra (B). The transition curves were fitted by Eq. 2, and the thermodynamic parameters are listed in Table S2. For the transition curve of WT RMCK from CD spectroscopy, both two-state and three-state models were applied for fitting. The three-state fitting was performed by regarding the transition as the sum of two sequential two-state processes because a stable intermediate could be observed.
A close inspection of the overexpression of the three CTD fragments in the E. coli cells (Fig. S1) indicated that the longer was the linker in the CTD fragments, the higher content of soluble fractions. Turbidity measurements were carried out to investigate the time-course aggregation of the WT and truncated proteins at 50°C (Fig. 3). Only minor aggregation could be characterized by A400 for the WT protein and C101, whereas C113 aggregated seriously and quickly. The transmission electron microscopy images indicated that the WT protein did not aggregate, C101 formed small aggregates, whereas C113 was converted to large amorphous aggregates after 10 min incubation at 50°C (Fig. S3). Aggregation studies were also conducted for the two isolated NTDs (with purity around 90%) with or without the segment from residues K101 to H112. Neither N100 nor N112 aggregated at 46°C, which coincided with the previous result that the CTD is responsible for RMCK aggregation (21). When fitted by Eq. 1, the kagg value of C101 was similar to the WT RMCK, whereas that of C113 was ∼14-fold larger than that of the WT protein (Table S2). These results suggested that the intactness of the linker significantly affected the thermal aggregation of the isolated CTD, although contributed little to the thermal unfolding transition.
Figure 3.
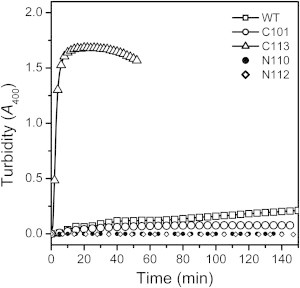
Thermal aggregation of the WT and mutated RMCKs. The aggregation kinetics were measured by turbidity at 50°C. The protein stock solutions were mixed with the preheated buffers, and then the absorbance at 400 nm was recorded every 1 s.
The results in Fig. 3 suggested that the intact linker behaved as a chaperone-like effect on CTD thermal aggregation. To further verify this proposal, the intact linker (L-f) and the truncated linker (L-n and L-c) peptides were synthesized and provided additionally in the thermal aggregation assay (Fig. 4). L-f could successfully protect both C113 and C101 in a concentration-dependent manner, and the aggregation could be fully suppressed with the addition of >300 μM L-f. The truncated linkers L-n and L-c also had the properties of preventing C113 and C101 aggregation, although the efficiency is much weaker than that of L-f. It is worth noting that L-c was more efficient than L-n, indicating that residues 113–125 was the major contributor of the chaperone-like effect of the linker. A quantitative evaluation was achieved by fitting the aggregation kinetics using the first-order kinetics (Fig. 4, D and F). The lag time, which reflected the conformational change before aggregation, was not significantly affected by the addition of the linker peptide. The amounts of aggregation monitored by Alim was slightly suppressed by L-n and L-c, but was dramatically suppressed by L-f in a saturation manner. All three peptides could successfully slow down the aggregation rate of C113. These observations suggested that the linker peptides did not interfere with the conformational transition of C113 before aggregation, but inhibit aggregation by blocking the adhesion rate of the molecules.
Figure 4.
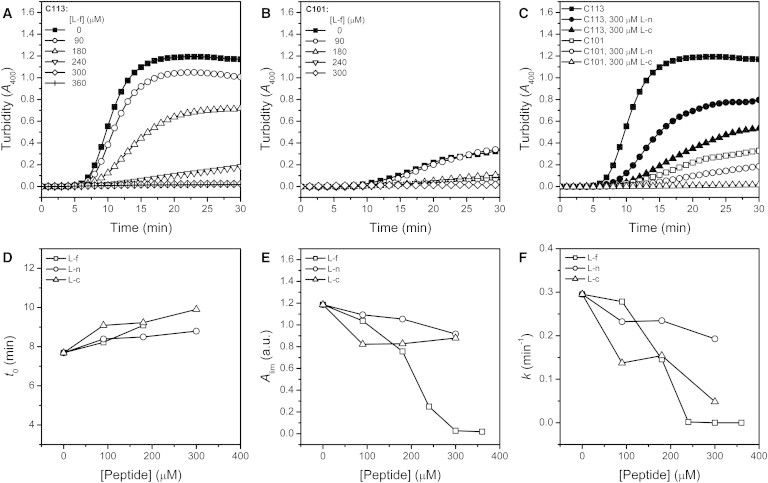
Effect of the synthetic linker peptide on the thermal aggregation of C113 and C101. (A) Effect of L-f on C113 aggregation. (B) Effect of L-f on C101 aggregation. (C) Effect of L-n and L-c on C113 and C101 aggregation. The experimental data were fitted by Eq. 1. The peptide concentration dependence of the kinetic parameters of C113 aggregation is presented in D–F. The data of C101 were not fitted due to the failure of reaching the equilibrium within the experimental time. Time-course aggregation at 46C was measured by turbidity. L-f, L-n, and L-c are the synthetic peptides with sequence corresponding to the fragments 98–125, 98–112, and 113–125 in the WT RMCK, respectively.
Role of the linker in CTD stability by MD simulations and mutational analysis
To identify how the linker worked out as an IMC, we performed MD simulations for C101, C113, and C125 (Fig. 5). The average SAP values ranging from residues 125 to 381 were −0.0462, −0.0465, and −0.420 at 300 K and −0.0458, −0.0402, and −0.0410 at 400 K for C101, C113, and C125, respectively. The largest SAP (Fig. 5) and Zyggregator (Fig. S5) of C125 at room temperature indicated it had the strongest aggregation propensity. Meanwhile, the low SAP and Zyggregator scores of the linker indicated that the linker was aggregation-resistant. The key residues responsible for CTD aggregation were characterized by comparing the SAP scores based on the dissimilar properties of the three proteins. Three hydrophobic residues, Phe-250, Val-255, and Val-347, were therefore identified. As shown in Table S3, in the presence of the full linker (C101), neither Phe-250 nor Val-347 exhibit significantly different SAP scores after temperature rising (from 300 to 400 K). However, the significance appeared when the linker was partially removed (in C113). In other words, Phe-250 and Val-347 are significantly more aggregation prone in the lack of the protection from the integral linker. Notably, the significance of Val-255 also increases after the partial removal of the linker, as suggested by the p-values of C101 and C113. Furthermore, in the simulation system (C113 + L-n) where the peptide bond between residues 112 and 113 was cleaved in C101 and L-n was retained, these three residues were still aggregation prone (with positive SAP scores: 0.071, 0.19, and 0.105), consistent with the previous conclusion that the L-n was not the main contributor for the protection effect of the linker (Fig. 4). Meanwhile, the linker residue Leu-115 was found to become more exposed to solvent when the linker is progressively truncated: its SAP scores at 300 and 400 K grew from −0.002 and −0.01 in C101 to 0.09 and 0.17 in C113; simultaneously its SAP score in C113 + L-n (0.11) was positive and close to that in C113 at high temperatures. Consistently, our previous research has shown that the mutation of Leu-115 dramatically promoted RMCK aggregation during heating and refolding (10).
Figure 5.
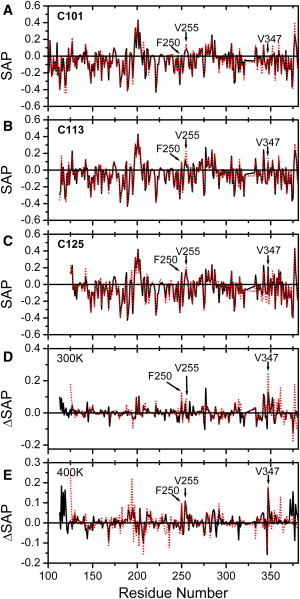
Role of the linker in CTD stability by MD simulations. (A–C) SAP scores of the truncated forms C101, C113, and C125 at 300 K (solid black lines) and 400 K (red dotted lines). The SAP score evaluates the surface hydrophobicity of a residue based on three-dimensional structures. Usually a residue with a high SAP score is highly prone to aggregation because it creates a hydrophobic locus on the protein surface. (D and E) The difference profiles of SAP scores at 300 and 400 K obtained by subtracting the scores of C113 (solid lines) and C125 (dotted lines) by those of C101. The residues responsible for CTD aggregation were characterized based on the following two criteria: i), a low SAP score in C101 and C113 but a high score in C125 at 300 K; ii), a high SAP score in C113 and C125 but a low score in C101 at 400 K.
The impact of Phe-250, Val-255, and Val-347 on CTD aggregation was further investigated by mutational analysis (Fig. 6). When substituted the three hydrophobic residues by the polar residue Ser, the thermal aggregation of C113 was suppressed, and the effect of Phe-250-Ser was the most significant with a dramatic decrease of the aggregation rate and amounts of aggregation. To speculate out whether these three residues interacted with the linker, the aggregation behavior of the C101 and WT RMCK mutants were also studied. For both C101 and RMCK, all of the three mutations enhanced protein thermal aggregation. As for C101, the impairing of Phe-250-Ser was the most serious, followed by Val-347-Ser and Val-255-Ser. The opposing effects of these mutations between C101 and C113 confirmed that these three residues were the adhesion sites of aggregation protected by the intact linker. However, the promoting effect of Val-347-Ser was the most significant for the WT RMCK, followed by Phe-250-Ser and Val-255-Ser. The kinetic parameter analysis indicated that the mutants Val-347-Ser and Phe-250-Ser of RMCK had similar aggregation rates, but different Alim values. Further analysis of the thermal denaturation transition of RMCK and the mutants indicated that Phe-250-Ser and Val-255-Ser had the same Tm values (54 ± 1°C) as the WT RMCK, whereas that of Val-347-Ser was ∼3°C lower (Fig. 6 G). This suggested that the former two mutations did not interfere with the thermal transition, whereas Val-347-Ser slightly decreased the thermal stability of RMCK. Nonetheless, these results indicated that Phe-250 and Val-347 were two crucial residues in mediating the chaperone-like effect of the linker, whereas the role of Val-255 was moderate.
Figure 6.
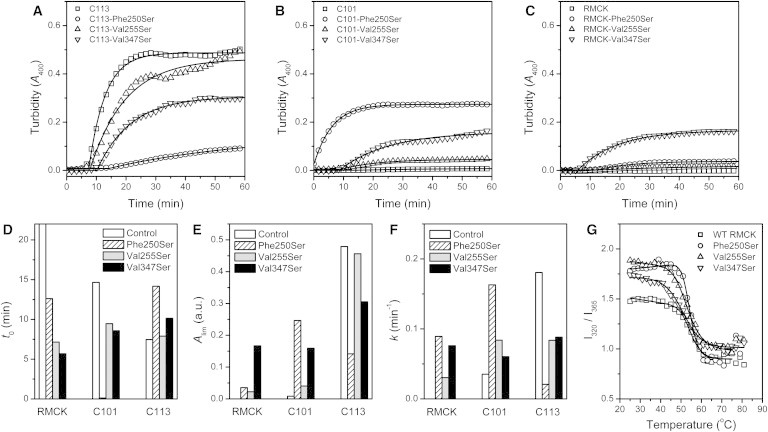
Mutational analysis of the role of Phe-250, Val-255, and Val-347 in RMCK thermal unfolding and aggregation. (A–C) Thermal aggregation of C113 (A), C101 (B), and RMCK (C) at 46°C. (D–F) Comparison of the kinetic parameters of thermal aggregation. (G) Thermal unfolding transition curves of the WT RMCK and the mutants. The raw data (symbols) were fitted by Eq. 2, and the fitted data are presented as solid lines.
Discussion
Implications to CK folding mechanism
CK, a multimeric two-domain protein, has been proposed to fold via a hierarchy process involving several intermediates (32,33). The folding of CTD is thought to be independent and to happen before the folding of the NTD (34). Mutational analysis indicated that the modifications of intra- or interdomain interactions affect both the equilibrium and kinetic folding pathways of CK (16,23,35). Several reports have studied the structure and folding of isolated segments or proteolytic products of CK, and have suggested that the stability of NTD relies on domain interactions (34,36,37). However, most of the previous work separated the two domains within the CTD instead of the linker region. In this research, we found that both the NTD and CTD could fold to a well-structured state, which strongly suggested that the domain and subunit interactions were crucial to CK assembly into an active enzyme but not to the autonomous folding of the domains. During the cotranslational folding in vivo, the folded or partially folded NTD and the linker protects the aggregation-prone CTD against nonspecific intermolecular interactions and facilitates CK folding and assembly. The opinion that the NTD enhances the solubilization of the CTD has also been proposed by studies of the three E. coli multidomain proteins (38).
The previous mutational analysis has indicated that the correct positioning of the linker is important to RMCK folding (10). However, mutations in the linker affect the native structure of RMCK. The results herein clearly indicated that the linker acts as an IMC (referred as IMC linker). We found that the linker specifically protects the CTD but not the NTD against aggregation, consistent with the fact that the CTD is the adhesion sites of RMCK aggregation (21). The existence of the intact linker did not affect the thermal unfolding pathway (Fig. 2) of the CTD, implying that its function was mainly through influence on the aggregation pathway. The action of the linker was verified by mutational analysis of the key residues, Phe-250, Val-255, and Val-347, characterized by MD simulations. The substitution of these hydrophobic residues by the polar residue Ser could successfully inhibit the aggregation of CTD with the truncated linker, but accelerated the thermal aggregation of both the WT enzyme and CTD with the intact linker. This opposing effect of the mutations strongly suggested that the existence of the linker in the native protein actually acted as an IMC to prevent the protein against aggregation when subject to stresses. Structurally, the linker acts as a lid to protect the hydrophobic side chains of Phe-250 and Val-347, but not Val-255 against exposure to solvent (Fig. S6, Scheme 2). This can explain why the effect of mutations at Phe-250 and Val-347 were much more pronounced than that of Val-255. When the lid (linker) is easily opened with a large dissociation constant at elevated temperatures, the protein is prone to aggregate. Meanwhile, the addition of extrinsic lid could successfully rescue the proteins from the aggregation pathway.
Scheme 2.
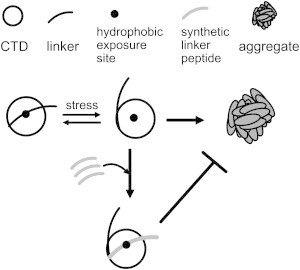
Proposed open-and-close model of the action of the IMC linker. The linker acts as a lid to protect the hydrophobic site against exposure to solvent. At elevated temperatures, the lid (linker) is easily opened with a large dissociation constant, and the protein is prone to aggregate by the adhesion of the hydrophobic site. The addition of extrinsic lid (synthetic linker peptide) can prevent the hydrophobic exposure, and thus rescue the proteins from the aggregation pathway.
Proposed actions of IMC linkers in protein folding and aggregation
Our current understanding of IMCs is mainly based on extensive studies of preproproteins (6). Recently, the functional regions have also been characterized as IMCs in several proteins (8–13), implying that IMCs might be much more universal than previously expected (13). However, it is seldom found that the linkers in multidomain proteins can act as IMCs. One well-studied case is the linker, also called C-peptide, in human proinsulin, which can act as an IMC to facilitate the formation of disulfide bonds by correctly positioning the N- and C-terminal fragments (39). Although the in vitro folding of multidomain proteins usually competes with aggregation and needs the help of molecular chaperones or foldases, many others such as CK can refold independently in vitro. The fact that at least some linkers can behave as a chaperone-like activity may be important to the understanding of how multidomain proteins fold after translation in vivo and how they avoid off-pathway aggregation during unfolding upon stresses or other physiological processes. That is, besides the widely studied chaperones and foldases, the cells may have evolved intramolecular mechanism such as IMCs to fight against the unproductive aggregation pathway (6). The characterization of potential IMCs, particular IMC linkers, may facilitate the recombinant production of isolated domains because there are no general guidelines for designing construction coding for a single domain of a multidomain protein (40).
The distinct features of IMCs from intermolecular chaperones and foldases have been thoroughly discussed by Ma et al. previously (13). In particular, they have proposed that the uncleaved IMCs not only play a role in the folding pathway, but also have their own physiological importance in protein function. This is exactly the case for the linker of RMCK, which is essential to the binding of RMCK to the M-line (20). On the basis of those proposed by Ma et al. (13) and the observations herein, the following features are proposed for an IMC linker: i), the existence of the IMC linker facilitates the folding of the protein by assisting the correct positioning of adjacent domains/fragments, but does not significantly affect the thermodynamics of protein folding; ii), the IMC linker covalently linked to the protein protects the aggregation-prone domain, mostly at the C-terminal part of the IMC linker, against aggregation during refolding and unfolding; iii), the disruption of the IMC linker either by mutations or by deletions promotes protein aggregation; iv), the IMC linker provided additionally prevents protein aggregation in a concentration dependent manner. The essential concentration of the linker fragment to fully block the aggregation depends on the dissociation constants of both the linker covalently linked to the protein and provided non-covalently. Thus, the behavior of the linker is not stoichiometric, but is between the intermolecular chaperones/foldases and the non-specific chemical chaperones.
The characterization of IMC linkers in proteins is important not only to the understanding of protein folding but also to protein aggregation. In particular, the results of the two truncated CTD forms mimicked the different properties of various proteolytic products. The relative flexible linker region is prone to be cleaved by proteases. Actually, RMCK was very sensitive to proteolysis by trypsin or protease K (Fig. S4). The dissimilar behaviors of C101 and C113 suggested that the intact but not truncated IMC is crucial to the chaperone-like effect on the aggregation of protein segments. The disruption of IMC linkers might be vital to the onset of some aggregation diseases because the deposition of the proteolytic fragments is frequently observed in the pathology of many diseases (41). Although it is unclear yet how much proteolysis contributes to the diseases accompanied with CK inactivation or deficiency, it is clear that CK aggregation is important to the onset of some diseases such as acute myocardial infarction caused by the inherited D54G mutation (16) and Alzheimer’s disease (42). CK is also found to form aggresomes in cells subjected to nitrogen-based oxidative stress (19).
The fact that the disruption of IMCs will greatly accelerate the nonnative aggregation of the corresponding proteolytic species also provides the possibility of the involvement of uncleaved IMCs in the onset of protein aggregation diseases. This opinion is well documented in preproproteins. For example, mutations in the IMC prodomain have been characterized in several diseases, which are named as protein-memory diseases (6,43). The C-terminus of α-synuclein is characterized to be an IMC and its removal will accelerate α-synuclein fibrilization, which is suggested to be a critical factor in several neurodegenerative diseases (44). The IMC function of BRICHOS, a novel domain located in the propeptide region of several proteins, is proposed to be related to dementia, respiratory distress, and cancer (45). However, the relationship between the uncleaved IMCs and aggregation in diseases remains elusive due to the limited reports of IMCs in the other proteins apart from preproproteins. The observation that IMC linker added additionally could prevent protein aggregation also provides a valuable strategy to fight against protein aggregation diseases.
Acknowledgments
The authors thank Drs. Lingpeng Cheng and Yanxia Jia from the Institute of Biophysics, Chinese Academy of Science, for assisting with the electron microscopy observations.
This research was supported in part by grant 30970559 from the National Natural Science Foundation of China.
Footnotes
Zhe Chen’s present address is National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD.
Hua-Wei He’s present address is Southwest University, Chongqing, China.
Supporting Material
References
- 1.Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Crowther D.C. Familial conformational diseases and dementias. Hum. Mutat. 2002;20:1–14. doi: 10.1002/humu.10100. [DOI] [PubMed] [Google Scholar]
- 4.Zhou B.R., Liang Y., Chen J. Mixed macromolecular crowding accelerates the oxidative refolding of reduced, denatured lysozyme: implications for protein folding in intracellular environments. J. Biol. Chem. 2004;279:55109–55116. doi: 10.1074/jbc.M409086200. [DOI] [PubMed] [Google Scholar]
- 5.Burg M.B., Ferraris J.D. Intracellular organic osmolytes: function and regulation. J. Biol. Chem. 2008;283:7309–7313. doi: 10.1074/jbc.R700042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y.J., Inouye M. The intramolecular chaperone-mediated protein folding. Curr. Opin. Struct. Biol. 2008;18:765–770. doi: 10.1016/j.sbi.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Ikemura H., Takagi H., Inouye M. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J. Biol. Chem. 1987;262:7859–7864. [PubMed] [Google Scholar]
- 8.Bhattacharyya J., Santhoshkumar P., Sharma K.K. A peptide sequence-YSGVCHTDLHAWHGDWPLPVK [40-60]-in yeast alcohol dehydrogenase prevents the aggregation of denatured substrate proteins. Biochem. Biophys. Res. Commun. 2003;307:1–7. doi: 10.1016/s0006-291x(03)01116-1. [DOI] [PubMed] [Google Scholar]
- 9.Brunger A.T., Breidenbach M.A., Montal M. Botulinum neurotoxin heavy chain belt as an intramolecular chaperone for the light chain. PLoS Pathog. 2007;3:1191–1194. doi: 10.1371/journal.ppat.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He H.-W., Feng S., Yan Y.B. Role of the linker between the N- and C-terminal domains in the stability and folding of rabbit muscle creatine kinase. Int. J. Biochem. Cell Biol. 2007;39:1816–1827. doi: 10.1016/j.biocel.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 11.Liu W.-F., Zhang A., Yan Y.B. The R3H domain stabilizes poly(A)-specific ribonuclease by stabilizing the RRM domain. Biochem. Biophys. Res. Commun. 2007;360:846–851. doi: 10.1016/j.bbrc.2007.06.139. [DOI] [PubMed] [Google Scholar]
- 12.Markossian K.A., Golub N.V., Kurganov B.I. Mechanism of thermal aggregation of yeast alcohol dehydrogenase I: role of intramolecular chaperone. Biochim. Biophys. Acta. 2008;1784:1286–1293. doi: 10.1016/j.bbapap.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 13.Ma B., Tsai C.J., Nussinov R. Binding and folding: in search of intramolecular chaperone-like building block fragments. Protein Eng. 2000;13:617–627. doi: 10.1093/protein/13.9.617. [DOI] [PubMed] [Google Scholar]
- 14.Wallimann T., Wyss M., Eppenberger H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992;281:21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bürklen T.S., Schlattner U., Wallimann T. The creatine kinase/creatine connection to Alzheimer’s disease: CK-inactivation, APP-CK complexes and focal creatine deposits. J. Biomed. Biotechnol. 2006;2006:35936. doi: 10.1155/JBB/2006/35936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng S., Zhao T.-J., Yan Y.B. Effects of the single point genetic mutation D54G on muscle creatine kinase activity, structure and stability. Int. J. Biochem. Cell Biol. 2007;39:392–401. doi: 10.1016/j.biocel.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Streijger F., Pluk H., Van der Zee C.E. Mice lacking brain-type creatine kinase activity show defective thermoregulation. Physiol. Behav. 2009;97:76–86. doi: 10.1016/j.physbeh.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlattner U., Tokarska-Schlattner M., Wallimann T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta. 2006;1762:164–180. doi: 10.1016/j.bbadis.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z., Li J., Zhou H.M. Metallothioneins protect cytosolic creatine kinases against stress induced by nitrogen-based oxidants. Biochem. J. 2012;441:623–632. doi: 10.1042/BJ20111565. [DOI] [PubMed] [Google Scholar]
- 20.Hornemann T., Kempa S., Wallimann T. Muscle-type creatine kinase interacts with central domains of the M-band proteins myomesin and M-protein. J. Mol. Biol. 2003;332:877–887. doi: 10.1016/s0022-2836(03)00921-5. [DOI] [PubMed] [Google Scholar]
- 21.He H.-W., Zhang J., Yan Y.B. Conformational change in the C-terminal domain is responsible for the initiation of creatine kinase thermal aggregation. Biophys. J. 2005;89:2650–2658. doi: 10.1529/biophysj.105.066142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y.-M., Feng S., Yan Y.B. Mutation of the conserved Asp-122 in the linker impedes creatine kinase reactivation and refolding. Int. J. Biol. Macromol. 2009;44:271–277. doi: 10.1016/j.ijbiomac.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 23.Zhao T.-J., Feng S., Yan Y.B. Impact of intra-subunit domain-domain interactions on creatine kinase activity and stability. FEBS Lett. 2006;580:3835–3840. doi: 10.1016/j.febslet.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 24.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Yao Q.-Z., Zhou H.-M., Zou C.G. A comparison of denaturation and inactivation rates of creatine kinase in guanidine solutions. Sci. Sin. [B] 1982;25:1296–1802. [PubMed] [Google Scholar]
- 26.Rosalki S.B. An improved procedure for serum creatine phosphokinase determination. J. Lab. Clin. Med. 1967;69:696–705. [PubMed] [Google Scholar]
- 27.Turoverov K.K., Haitlina S.Y., Pinaev G.P. Ultra-violet fluorescence of actin. Determination of native actin content in actin preparations. FEBS Lett. 1976;62:4–6. doi: 10.1016/0014-5793(76)80003-8. [DOI] [PubMed] [Google Scholar]
- 28.Kurganov B.I. Kinetics of protein aggregation. Quantitative estimation of the chaperone-like activity in test-systems based on suppression of protein aggregation. Biochemistry (Mosc.) 2002;67:409–422. doi: 10.1023/a:1015277805345. [DOI] [PubMed] [Google Scholar]
- 29.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chennamsetty N., Voynov V., Trout B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA. 2009;106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tartaglia G.G., Pawar A.P., Vendruscolo M. Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 2008;380:425–436. doi: 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 32.Fan Y.X., Zhou J.M., Tsou C.L. Unfolding and refolding of dimeric creatine kinase equilibrium and kinetic studies. Protein Sci. 1998;7:2631–2641. doi: 10.1002/pro.5560071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuznetsova I.M., Stepanenko O.V., Uversky V.N. Unraveling multistate unfolding of rabbit muscle creatine kinase. Biochim. Biophys. Acta. 2002;1596:138–155. doi: 10.1016/s0167-4838(02)00212-1. [DOI] [PubMed] [Google Scholar]
- 34.Gross M., Wyss M., Furter R. Reconstitution of active octameric mitochondrial creatine kinase from two genetically engineered fragments. Protein Sci. 1996;5:320–330. doi: 10.1002/pro.5560050216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perraut C., Clottes E., Marcillat O. Role of quaternary structure in muscle creatine kinase stability: tryptophan 210 is important for dimer cohesion. Proteins. 1998;32:43–51. doi: 10.1002/(sici)1097-0134(19980701)32:1<43::aid-prot6>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 36.Morris G.E., Frost L.C., Hudson N. Monoclonal antibody studies of creatine kinase. Antibody-binding sites in the N-terminal region of creatine kinase and effects of antibody on enzyme refolding. Biochem. J. 1987;248:53–59. doi: 10.1042/bj2480053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webb T., Jackson P.J., Morris G.E. Protease digestion studies of an equilibrium intermediate in the unfolding of creatine kinase. Biochem. J. 1997;321:83–88. doi: 10.1042/bj3210083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim C.W., Han K.S., Seong B.L. N-terminal domains of native multidomain proteins have the potential to assist de novo folding of their downstream domains in vivo by acting as solubility enhancers. Protein Sci. 2007;16:635–643. doi: 10.1110/ps.062330907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Min C.Y., Qiao Z.S., Feng Y.M. Unfolding of human proinsulin. Intermediates and possible role of its C-peptide in folding/unfolding. Eur. J. Biochem. 2004;271:1737–1747. doi: 10.1111/j.1432-1033.2004.04079.x. [DOI] [PubMed] [Google Scholar]
- 40.Bhaskara R.M., Srinivasan N. Stability of domain structures in multi-domain proteins. Sci. Rep. 2011;1:40. doi: 10.1038/srep00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westermark P. Aspects on human amyloid forms and their fibril polypeptides. FEBS J. 2005;272:5942–5949. doi: 10.1111/j.1742-4658.2005.05024.x. [DOI] [PubMed] [Google Scholar]
- 42.David S., Shoemaker M., Haley B.E. Abnormal properties of creatine kinase in Alzheimer’s disease brain: correlation of reduced enzyme activity and active site photolabeling with aberrant cytosol-membrane partitioning. Brain Res. Mol. Brain Res. 1998;54:276–287. doi: 10.1016/s0169-328x(97)00343-4. [DOI] [PubMed] [Google Scholar]
- 43.Kim M., Suh J., Tanzi R.E. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum. Mol. Genet. 2009;18:3987–3996. doi: 10.1093/hmg/ddp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uversky V.N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 45.Sánchez-Pulido L., Devos D., Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem. Sci. 2002;27:329–332. doi: 10.1016/s0968-0004(02)02134-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.