

Abstract
Mitochondrial outer membrane permeabilization (MOMP) is a complex multistep process. Studies of MOMP in vivo are limited by the stochastic variability of MOMP between cells and rapid completion of IMS protein release within single cells. In vitro models have provided useful insights into MOMP. We have investigated the dynamics of Bax-mediated MOMP in isolated mitochondria using ionic strength as a tool to control the rate of MOMP. We find that Bax can induce both transient permeabilization, detected by protein release, and more substantial long-lasting permeabilization, measured by the rate of oxidation of added cytochrome c. We found that higher ionic strength causes Bax to form small channels quickly but the expansion of these early channels is impeded. This inhibitory effect of ionic strength is independent of tBid. Channels formed under low ionic strength are not destabilized by raising the ionic strength. Increase in ionic strength also increases the ability of Bcl-xL to inhibit Bax-mediated MOMP. Ionic strength does not affect Bax insertion into mitochondria. Thus, ionic strength influences the assembly of Bax molecules already in membrane into channels. Ionic strength can be used as an effective biophysical tool to study Bax-mediated channel formation.
Introduction
Apoptosis is a form of programmed cell death that is crucial to the elimination of damaged or unwanted cells. Its proper execution is vital for appropriate development. A key event in the apoptotic pathway is increased permeability of the mitochondrial outer membrane (MOM), which leads to the release of intermembrane space (IMS) proteins. These proteins trigger the activation of downstream caspases, which carry out the repackaging of the cell into apoptotic bodies. The rate-limiting increase in MOM permeabilization (MOMP) leading to the release of IMS proteins is well known to be modulated by Bcl-2 family of proteins (1–3). The Bcl-2 proteins can be categorized broadly on the basis of their mode of action into pro-apoptotic and anti-apoptotic proteins. Pro-apoptotic BH1-3 proteins act directly to enhance MOMP by forming protein-permeable pores. BH3-only proteins increase MOMP indirectly by activating BH1-3 proteins or inhibiting anti-apoptotic Bcl-2 proteins (or both). Anti-apoptotic BH1-4 type proteins inhibit the action of the pro-apoptotic proteins (4–6).
The control of MOMP by the Bcl-2 proteins has been studied extensively. Bax, normally cytosolic, upon activation by the BH3 only protein, tBid, targets the MOM, inserts, and forms oligomeric pores (7–10). Bcl-xL inhibits MOMP by either out-competing Bax for tBid and/or interacting directly with Bax to inhibit its insertion into the MOM (10,11). The molecular roles of different domains of Bax in membrane insertion are well known (12–16). Some amino acids have also been identified that either enhance or eliminate the pro-apoptotic function of Bax (14–16) but the properties of the oligomeric channel such as size, stoichiometry, and kinetics of channel assembly are only partially understood. We sought to study the dynamics of Bax-induced permeabilization of the MOM to proteins in isolated mitochondria using ionic strength as a modulating tool.
Because MOMP is a rapid, irreversible, essentially all-or-none process where most IMS proteins are released almost simultaneously within a few minutes (17–20), in vitro models have been very useful in studying the regulation of MOMP by Bcl-2 family proteins (8,11,13,21,22). We use ionic strength as a diagnostic tool to provide mechanistic insights into the dynamics of Bax mediated permeabilization.
Materials and Methods
The sources of materials used, the procedure for isolating mitochondria from rat liver, the details of the Western blotting, the enzymatic assays for protein release and cytochrome c accessibility, and the purification procedure for the recombinant Bcl-2 proteins are all described in the Supporting Material.
Preparation of mitochondrial samples for cytochrome c oxidation and IMS protein release assays
Isolated rat liver mitochondria were incubated in buffers of varying salt concentrations with the total osmotic pressure kept constant at 300 mOs by addition of the appropriate amount of mannitol. A combination of Bcl-2 family proteins (Bax, tBid, and Bcl-xL) was added to this mitochondrial suspension as needed. Before each experiment, the mitochondria were diluted to a protein concentration of 160 μg/mL in FH (300 mM mannitol, 0.1 mM EGTA, and 5 mM HEPES, pH 7.4) buffer. Because mitochondrial function degrades once diluted, the mitochondria were used within 10 min of dilution. In all experiments, 600 μL of this mitochondrial dilution was added to 400 μL of isoosmotic buffer (with either 25.1 mM KCl or 150 mM KCl) premixed with appropriate amounts of Bcl-2 family proteins to achieve a final mitochondrial concentration of 96 μg/mL (and final [KCl] of either 10 mM or 60 mM, respectively).
These isoosmotic solutions are referred to as “KCl buffer” prefixed with the [KCl]. The same methodology was followed when NaCl or potassium lactobionate was tested. For 90 mM buffer, the mitochondrial stock was diluted 50–100 fold in 150 mM KCl buffer to a protein concentration of 160 μg/mL. A quantity of 600 μL of this suspension was then diluted with 400 μL of FH buffer containing the Bcl-2 family proteins to achieve a final ionic strength of 90 mM. Except for time-course experiments, the mitochondria were preincubated at 30°C with the Bcl-2 family proteins for 30 min. For all experiments using Bcl-xL, the Bcl-2 proteins were incubated together for 10 min before the addition of mitochondria. Once the incubation period was over, 100 μL of the mitochondrial suspension was used for the cytochrome c accessibility assay, while the rest was centrifuged and the supernatant used for adenylate kinase and sulfite oxidase assays. In corresponding experiments, 50 μL of cyclosporin A (dissolved in dimethyl sulfoxide to a stock concentration of 1 mg/mL) was added to 1 mL of mitochondrial suspension to a final concentration of 50 μM just before the preincubation process.
Normalization of data
Normalization of data was performed for two reasons:
-
1.
The activity varied from one mitochondrial preparation to another.
-
2.
The activity of some enzymes was sensitive to ionic strength.
The latter was especially important because the relevant parameters sought were the degree of permeabilization of the outer membrane and the fractional release of a particular protein. By normalizing, the results obtained at different ionic strengths could be compared directly. Normalization involved subtracting the activity of untreated mitochondria (control) from the experimental value and dividing the difference by the maximal activity after hypotonic shock (control subtracted) measured under the same conditions (e.g., the same ionic strength).
Statistics
The results are reported as mean ± SE of at least three independent experiments. Significance was determined by Student's t-test. The single asterisk (∗), double asterisk (∗∗), and triple asterisk (∗∗∗) symbols indicate significance with P values <0.05, <0.01, and <0.001, respectively.
N/S indicates P values >0.05 and thus judged to be not significant.
Results
Increase in ionic strength decreases real-time MOMP but the extent of IMS protein release increases
In isolated mitochondria, MOMP can be measured in two ways:
The first method is one whereby release of IMS proteins can be measured. This provides information on the minimum size of the MOMP pore as well as the fraction of the population of mitochondria that have been permeabilized, but it does not define the status of the pore at any given time.
The second method records the real-time flux of cytochrome c across the outer membrane by measuring the rate of oxidation of exogenously added cytochrome c. MOM is impermeable to cytochrome c. However, once the mitochondria are permeabilized with Bax, cytochrome c crosses the MOM, accesses the cytochrome oxidase complex in the inner membrane, and becomes oxidized. The rate of oxidation is directly proportional to the extent of permeabilization, i.e., the size of the MOMP pore and the number of pores in the mitochondrial population.
Both methods were used to observe the ability of Bax to permeabilize the outer membrane at different KCl concentrations. Protein release from the IMS was monitored by measuring the release of adenylate kinase (AK), 24 kDa. Real-time permeabilization was measured by measuring the rate of oxidation of exogenously added cytochrome c. Neither Bax alone (up to 50 nM) nor tBid alone (up to 120 nM) resulted in any significant MOMP by either assay method (see Fig. S2, A and B, in the Supporting Material). However, when mitochondria were incubated with Bax and tBid at either low salt (10 mM KCl) or high salt (90 mM KCl) buffer, permeabilization was achieved (Fig. 1 A and Fig. S2, A and B). After treatment, a fraction of the mitochondrial suspension was assayed for cytochrome c oxidation; the remaining suspension was centrifuged and the supernatant was assayed for AK activity. At the lower KCl concentration (10 mM), a substantial amount of both AK release and real-time permeabilization was observed.
Figure 1.
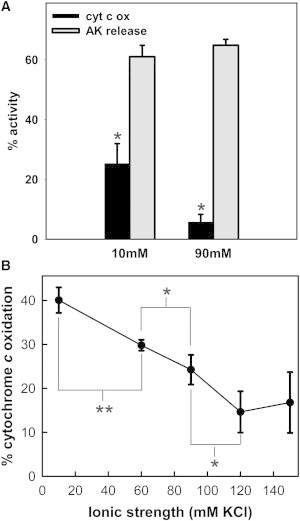
Increase in ionic strength inhibits real-time permeabilization by Bax without inhibiting IMS protein release. (A) Mitochondrial suspension was treated with 17 nM Bax and 120 nM tBid at either 10 mM or 90 mM KCl buffer and rates of cytochrome c oxidation (black bar) and AK release (gray bar) were measured. The asterisk (∗) refers to the statistically significant difference between the black bars. (B) The measured rates of cytochrome c oxidation by mitochondria exposed to tBid/Bax at the indicated KCl concentrations. The results are means ± SE of three experiments.
However, at the higher KCl concentration (90 mM), the release of AK was comparable to (e.g., in Fig. 1 A), or sometimes more than that at low salt, depending on the Bax concentration, but the extent of real-time permeabilization was always much lower (Fig. 1 A). The real-time permeabilization decreased linearly with increase in [KCl]. (Fig. 1 B) This behavior is not specific to KCl but rather an ionic strength effect because substituting KCl with either NaCl or potassium lactobionate resulted in a similar decline in cytochrome c oxidation rate with increasing salt concentration during the Bax/tBid treatment (see Fig. S2 C). In these experimental conditions, PTP did not influence MOMP, as we found that CsA did not alter the rate of cytochrome c oxidation or AK release by Bax and tBid at either ionic strength (see Fig. S3).
It was reported that tBid causes remodeling of the inner membrane and ionic strength could affect this process (23). This remodeling might explain the different rates of cytochrome c oxidation observed at different ionic strengths if restricted diffusion of cytochrome c between the cristae is rate-limiting in our system and changed by the remodeling. To investigate whether the cristae folds limit the cytochrome c oxidation rate, we compared the maximal rates of cytochrome c oxidation after osmotic lysis where swelling unfolds the cristae and MOM permeabilization with digitonin where the cristae structures remain intact at doses that do not permeabilize the inner membrane (23,24). Under our conditions, 0.01% digitonin selectively permeabilized the MOM without releasing the matrix enzyme, fumarase (see Fig. S4 A). Whether mitochondria were treated with digitonin or hypotonic shock, in the presence of 10- or 90-mM KCl buffer, the rates of cytochrome c oxidation were not significantly different. In another set, mitochondria were preincubated for 30 min with tBid to allow remodeling of the inner membrane at either ionic strength and then treated with digitonin and similar oxidation rates were observed (see Fig. S4 B). Thus, any potential changes in the inner membrane due to changes in the ionic strength did not affect the rates of cytochrome c oxidation observed. The permeability of the outer membrane seems to be the rate-limiting factor in the oxidation of exogenous cytochrome c by mitochondria.
The different results obtained with the two methods of measurement of permeability (AK release and cytochrome c oxidation) could be explained in three ways:
-
1.
Change in the energetics. The higher ionic strength makes the channels flicker, allowing release without much permeabilization.
-
2.
Transient permeabilization. At the higher ionic strength, the channels form for some time and then close, resulting in restoration of a protein impermeable outer membrane.
-
3.
Kinetic delay. Bax channel formation begins with small or flickering channels but with time, these become larger and more stable. At high ionic strength, this process is slow.
Experiments were performed to distinguish among these possibilities.
MOMP induced at lower [KCl] is not reversed by increasing the salt concentration
One interpretation of the ionic strength effect is that Bax channels are in equilibrium between an open and a closed state and the ionic strength influences the position of the equilibrium. Perhaps the equilibrium favors the open state at low ionic strength and the closed at high ionic strength. To test this, Bax mediated MOMP was induced under low ionic strength (10 mM KCl buffer) for 30 min by treating mitochondria with Bax and tBid. Under these conditions, a significant level of MOMP was achieved. The permeabilized mitochondria were sedimented (14,000 × g, 5 min) and resuspended in higher ionic strength medium (90 mM KCl buffer) and incubated for another 30 min.
We found that after switching from low salt to high salt (90 mM), the MOMP did not decrease but remained at the high value found at low salt (Fig. 2). This demonstrates that the process is far from equilibrium. Thus, ionic strength is not merely shifting a fast equilibrium between conducting states of Bax channels. Instead, once Bax channels achieve a highly permeable state, that state is not reversible. Thus, the first option is not correct. A time course of growth of real-time MOMP with Bax and tBid shows that the rate of permeabilization is monotonic. There is no indication of a transient permeabilization because at all times tested, the real-time permeability remained low at higher ionic strength. Thus, the second option is incorrect. However, it is also clear that the permeabilization increases much faster at low salt than at high salt (see Fig. S5). Thus, the difference between the two methods is partly explained by a difference in kinetics.
Figure 2.
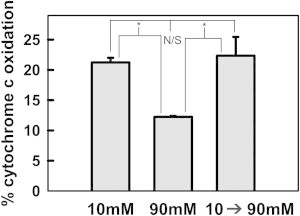
Bax permeability induced at lower ionic strength was not reversed by raising ionic strength. Mitochondrial suspensions were treated with 8 nM Bax and 120 nM tBid for 30 min at 30°C in either 10 mM or 90 mM KCl buffer. At the end of 30 min, the rate of cytochrome c oxidation by these mitochondria was determined. Simultaneously, the mitochondrial suspension incubated in 10 mM KCl buffer was centrifuged and mitochondrial pellet resuspended in 90 mM KCl buffer to raise the ionic strength. This suspension was incubated for 30 min at 30°C, and at the end of the incubation, the rate of cytochrome c oxidation was measured. To correct for any loss of mitochondria during centrifugation, control and lysed samples with equivalent amount of mitochondria were also treated in an identical manner and the pellet resuspended in 90 mM KCl buffer (control without Bax or tBid) or H2O (lysed) and rates measured. The results are means ± SE of three experiments.
At high ionic strength, Bax forms smaller channels more rapidly
To better understand the kinetics of Bax channel formation, the Bax-induced release of AK (24 kDa) and that of a larger protein sulfite oxidase (SOX, 120 kDa) were measured as a function of time and ionic strength using different Bax concentrations (Fig. 3). At all Bax concentrations tested, the rate of release of AK was faster at 90 mM than at 10 mM KCl. At the smallest Bax concentration tested (8 nM), the release of SOX lagged behind the release of AK at both salt concentrations (Fig. 3, A and B). Because the flux of proteins across the membrane through the Bax pore is not rate-limiting (see Discussion), the increase in the amount of release corresponds to the formation of channels in more mitochondria. The kinetic delay between the release of AK and SOX suggests that the channels grow in size with time. At a higher Bax concentration (17 nM), the rates of release of AK and SOX were identical for 10 mM KCl but at 90 mM, the release of AK was faster than release of SOX (Fig. 3, C and D).
Figure 3.
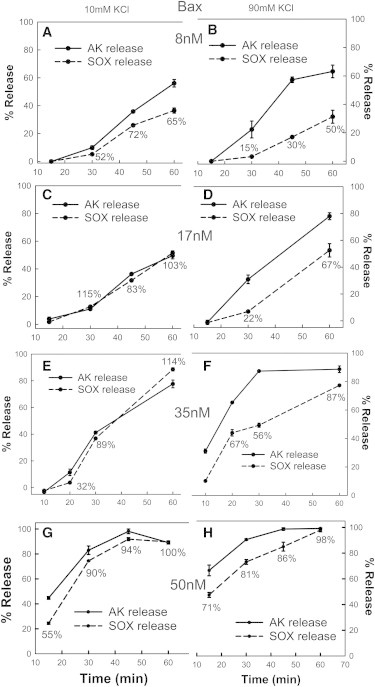
Bax channels expand in size with time but this expansion is slower at higher ionic strength. Mitochondrial suspensions were incubated in 10 mM KCl buffer (A, C, E, and G) or 90 mM KCl buffer (B, D, F, and H) with 120 nM tBid and either 8 nM Bax (A and B) or 17 nM Bax (C and D) or 34 nM Bax (E and F) or 50 nM Bax (G and H) for various time points at 30°C. Then the mitochondria were centrifuged and the supernatants were assayed for either AK release (solid lines) or SOX release (dashed lines). Percentage values next to SOX data indicate % of AK permeable mitochondria that have been permeabilized to SOX. Note that in some cases the error bars are smaller than the data point. The results are means ± SE of three experiments.
It must be noted again that the rate of AK release was higher with 90 mM KCl than with 10 mM KCl. At 34 nM, with 90 mM KCl, the amount of release of AK saturated at 30 min, indicating that essentially all the mitochondria were permeabilized but only about half of the channels were large enough to allow to flux of SOX. The release of SOX increased with time and by 60 min, 90% of the AK permeable mitochondria had channels large enough to release SOX, clearly indicating slow channel growth. In contrast, at 10 mM KCl, the release of AK and SOX increased synchronously (Fig. 4, E and F), indicating slower channel formation but fast growth to a large size capable of releasing both proteins at the same time. Clearly, both the initial permeabilization of mitochondria, as indicated by the AK release, and the growth of channels, as indicated by the % of permeabilized mitochondria that released SOX, are augmented by increasing the Bax concentration. The extent of SOX release did not change very much comparing low and high salt, but that can be explained by the conflicting effects of ionic strength: augmenting the formation of new channels and inhibiting their rate of growth.
Figure 4.
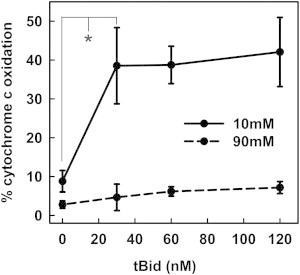
Ionic strength effect on Bax channel dynamics is not mediated by tBid. Mitochondrial suspensions were incubated in 10 mM KCl buffer (solid line) or 90 mM KCl buffer (dashed line) with 34 nM Bax and varying concentrations of tBid (24, 60, and 120 nM) for 30 min at 30°C. Then, the real-time permeability was determined by measuring the cytochrome c oxidation rate. There was no significant difference in cytochrome c oxidation rates between any treatments at 90 mM KCl buffer. The results are mean ± SE of three experiments.
tBid does not alter the sensitivity of Bax-induced MOMP to ionic strength
tBid is necessary for Bax to permeabilize the MOM. Thus, we hypothesized that the interaction between tBid and Bax may be sensitive to ionic strength, i.e., changing the tBid concentration could change the sensitivity of Bax-induced MOMP to ionic strength. Increasing the level of tBid might enhance channel expansion at 90 mM. To test this, real-time MOMP induced by Bax was measured in either 10 mM or 90 mM KCl buffer at three different tBid concentrations (Fig. 4). The permeabilization did not change over a wide concentration range of tBid at either ionic strength. This is consistent with a catalytic function of tBid in initiating Bax-mediated MOMP (25,26), but not in the growth process. Thus, the influence of ionic strength on MOMP is not due to an effect on tBid.
Ionic strength affects Bcl-xL-mediated inhibition of MOMP by Bax-tBid
Bcl-xL inhibits MOMP by multiple processes. We tested whether the inhibition of Bax-mediated MOMP by Bcl-xL is also sensitive to ionic strength. We hypothesized that because decreasing the ionic strength resulted in more stable permeabilization (a pro-apoptotic effect), a higher ionic strength will facilitate Bcl-xL-mediated MOMP inhibition. Consistent with this hypothesis, for a given amount of tBid and Bax, less Bcl-xL was needed to inhibit MOMP at 90 mM KCl than at 10 mM KCl (Fig. 5). Unlike what was observed for the action of tBid on Bax, inhibition of MOMP by Bcl-xL changed with the tBid concentration (see Fig. S6). Presumably, tBid reduced the amount of Bcl-xL available to interact with and inhibit Bax.
Figure 5.
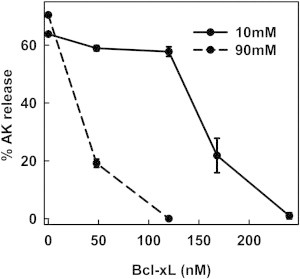
Increase in ionic strength increases the efficiency of Bcl-xL in inhibiting Bax-mediated MOMP. Mitochondrial suspensions were incubated with 17 nM Bax, 120 nM tBid, and varying concentrations of purified Bcl-xL (48, 120, 160, and 240 nM) at either 10 mM KCl buffer (solid line) or 90 mM KCl buffer (dashed line) for 30 min at 30°C. Then, the mitochondria were centrifuged and supernatant assayed for AK release. The results are means ± SE of three experiments.
Change in ionic strength has little effect on membrane insertion of Bax
It is possible that ionic strength could interfere with the extent of Bax insertion into the membrane, causing differences in the degree of permeability. Our initial hypothesis was that more Bax inserts into mitochondria at higher ionic strength, causing more mitochondria to be permeabilized; hence, greater release of AK. To test this hypothesis, mitochondria were incubated with Bax and tBid and the amount of AK released was measured at 10, 20, and 30 min of incubation. We found that, although there was an almost threefold difference in the amount of AK released at 10 min between 10 and 90 mM KCl, the amount of Bax inserted was about the same (Fig. 6, A and B). In our hands, Bax insertion was essentially complete by 10 min. Although there was no significant difference in the amount of Bax inserted across the time points tested, AK release increased with time (Fig. 6 B), indicating that new channel formation in more mitochondria continued with time.
Figure 6.
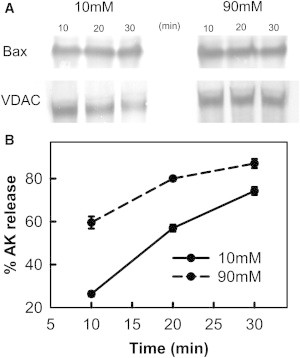
Bax insertion into mitochondrial outer membranes is not affected by ionic strength. (A) Mitochondrial suspensions (960 μg mitochondrial protein/mL) were treated with 50 nM Bax and 240 nM tBid for increasing amounts of time (10, 20, and 30 min) in either 10 mM or 90 mM KCl buffer. Then the mitochondria were centrifuged and supernatant assayed for AK release. The pellets were subjected to carbonate treatment and probed for Bax insertion by Western blot. The carbonate treatment Western blots were performed as described in the Materials and Methods in the Supporting Material. VDAC served as loading control. (B) Extent of AK release at 10 (solid line) and 90 mM KCl buffer (dashed line) for time points corresponding to those in A.
Thus, ionic strength affected the channel formation process rather than Bax insertion. This is consistent with the observation made with purified Bak whose insertion into membranes is not affected by salt concentration (27). To test whether ionic strength affected Bcl-xL-mediated inhibition of Bax insertion into membranes, mitochondria were incubated with Bax and tBid with or without Bcl-xL. We found that Bcl-xL had little effect on Bax insertion, although there was a complete inhibition of AK release (see Fig. S7). Therefore, under these conditions, Bcl-xL inhibits MOMP by interfering with Bax channel formation.
Discussion
Bax activation and subsequent MOMP occur in multiple steps that are only partially understood. MOMP is a rapid and irreversible process and very stochastic in cell populations (17–20). Thus, in vitro models have been used to gain insights into this process. Bax is a cytosolic monomeric protein in normal cells, but upon apoptotic induction, translocates to MOM, forms homo-oligomers and hetero-oligomers with Bak and some other proteins (28–34), and induces MOMP.
We show that increasing the ionic strength influences the size of the Bax channels, consequently leading to smaller channels.
In summary, our results are consistent with the following conclusions:
-
1.
Bax insertion into MOM is fast and is followed by channel formation and then a slow growth in channel size.
-
2.
The rate of formation of channels, as measured by the kinetics of AK release at different salt concentrations, is faster at higher salt concentration.
-
3.
The rate of expansion of the size of the initial channels is faster at the lower salt concentration, as measured by comparing the kinetics of release of AK and SOX and the time course of growth of real-time MOMP.
-
4.
This effect of salt concentration on the rate of growth of Bax channels explains the differences in the extent of real-time MOMP seen with the cytochrome c oxidation assay at different salt concentrations.
-
5.
Increase in ionic strength also makes Bax-mediated MOMP more sensitive to inhibition by Bcl-xL.
-
6.
Ionic strength does not affect the insertion of Bax into membranes, suggesting that subsequent steps in channel formation are impeded by an elevated ionic strength.
Key to gaining insight into the dynamics of Bax channel formation and the influence of ionic strength on this dynamics is the measurement of outer membrane permeabilization in two ways: 1), IMS protein release, and 2), the rate of cytochrome c oxidation. Because of the small volume of the intermembrane space and the long timescale of our experiments, once a channel is formed in one mitochondrion that is large enough to allow the translocation of a particular protein, e.g., AK, all the AK will be released from that mitochondrion within a few milliseconds. Thus, the time-dependent release of AK or SOX from a population of mitochondria does not measure the rate of release from each individual mitochondrion, but the rate of permeabilization of each of the mitochondria in the population. Therefore, partial release of AK signifies that only a portion of the mitochondrial population has Bax channels large enough to allow AK release. A difference in the extent of release of AK and SOX in the population means that some mitochondria have channels that are too small to release SOX but are large enough to release AK. It is important to note that release of AK and SOX is independent of ionic strength (35,36); hence, these proteins may be more suitable for measuring MOMP than cytochrome c release. Cytochrome c, being a charged protein, binds membranes, especially the inner membrane of mitochondria that is rich in cardiolipin, and a separate dissociation step is necessary to facilitate cytochrome c release apart from pore formation (35,37). Thus, assessing permeabilization of the outer membrane becomes more complicated if measuring cytochrome c release.
A change in ionic strength is not a very specific tool, and therefore other possible effects of ionic strength that may influence MOMP were examined. Over a wide concentration range of tBid, the sensitivity to ionic strength did not change—suggesting that the interaction between tBid and Bax is not affected by changes in ionic strength. tBid has been reported to remodel cristae in a PTP-dependent manner (23), releasing loosely bound IMS proteins from inner membrane into the IMS. Given the catalytic rather than stoichiometric nature of tBid in facilitating Bax-mediated MOMP (25,26) and that Bax-mediated cytochrome c oxidation rate did not change with tBid at either ionic strength, this is consistent with a model in which the effect of ionic strength is directly on the ability of Bax to form channels in the MOM rather than on the structure of the inner membrane. The inner membrane remodeling induced by tBid was inhibitable by CsA (23) but, in our hands, the exogenous cytochrome c oxidation induced by Bax-tBid at either ionic strength was independent of the presence of CsA. This is consistent with other reports (38–41) that found MOMP induced by Bax and tBid to be independent of PTP (see Fig. S2).
These observations strongly signify that the cytochrome c oxidation rates observed after Bax-tBid treatment are limited by MOM permeability rather than other effects on the structure of inner membrane. Though we do not rule out that changes are caused to the inner membrane by tBid under our conditions, these changes do not affect the interpretation of MOM permeability from the cytochrome c oxidation assay. Thus, the rate of oxidation of exogenously-added reduced cytochrome c measures the actual permeability of the MOM at any time (real-time permeabilization) and depends on the number of channels and the size of channels present. However, it does not distinguish between many small channels spread out among many mitochondria or a few large channels in just a few mitochondria. Thus, each type of assay measures different properties of the permeabilizing pore. By combining the results obtained from the three assays (AK release, SOX release, and cytochrome c oxidation) one obtains a good picture of the nature of the permeabilization of the MOM.
At low ionic strength, there is general congruency between the permeability measurements (cytochrome c oxidation rates) and the amount of protein release but, nevertheless, the % permeabilization is quite a bit smaller (half in Fig. 1 A) than the % AK release. Fig. 3 shows that at this level of Bax, the release of AK and SOX are the same. Thus, the Bax channels are large enough to release SOX, but not large enough to keep from limiting the rate of cytochrome c oxidation. To allow SOX release, the Bax pore must be >3.3 nm in radius. Simple calculations show that such a pore would limit cytochrome c translocation rates under the conditions of our experiments. Electron microscopy of Bax channels formed in liposomes show that Bax channel can reach much larger sizes (42,43), and under those conditions they would certainly not be rate-limiting. Thus, channel growth requires time both at low and high ionic strength, and this is also seen at 8 nM Bax (Fig. 3) when comparing the release of AK and SOX. The quicker release of AK at higher salt suggests that the initial pore formation by Bax is faster at higher ionic strength. However, the release of SOX showed a greater time lag for release at higher ionic strength. This indicates that the Bax channels start out small and grow in size and the expansion is delayed by increasing ionic strength. Thus, the Bax channel-forming process seems to consist of two stages: 1), initial formation of channels capable of releasing AK (stimulated by high ionic strength), and 2), growth of channels (inhibited by high ionic strength). The small channels limit the rate of cytochrome c oxidation, resulting in low real-time permeabilization and thus low oxidation rates.
Kluck et al. (44) first observed a dichotomy between extent of IMS protein release and real-time permeabilization in mitochondria treated with Bax or tBid. The apparent paradoxical finding that the extent of MOMP depended on the technique used to make the measurement is resolved by the experiments reported in this article. MOMP measurement is complicated by the growth in Bax channel size and the marker of permeabilization is also influenced by channel size. We find that the real-time permeability induced by Bax and tBid increases with time and is influenced by ionic strength. Larger channels formed at lower ionic strength allow greater flux of exogenous cytochrome c, causing its faster rate of oxidation. A high rate of cytochrome c oxidation does not imply unphysiologically high channel sizes as we found that such permeabilities happened when release of AK or SOX was submaximal. In cells, release of most IMS proteins is rapid and complete (17). It is an all-or-none event (17–20). Thus, one can conclude that large channels are present in vivo, and these have been observed in reconstituted systems (42,43).
We found that Bcl-xL was essentially ineffective at preventing Bax insertion even at concentrations of Bcl-2 proteins when MOMP was completely inhibited. Thus, in the experimental conditions and concentrations of Bcl-2 proteins we used, Bcl-xL may be acting at the level of Bax channel formation. Such an experimental setting provides a platform to study the mechanics of Bax channel assembly downstream of Bax insertion into membranes.
Conclusion
In summary, we have identified that decreasing ionic strength results in the formation of large channels that allow high flux rates of cytochrome c and synchronously release both small and large proteins. High ionic strength favors the initial formation of small channels and these develop more slowly into the larger structures that were observed much sooner at low ionic strength. Higher ionic strength also increases the sensitivity of Bax-mediated MOMP to inhibition by Bcl-xL. Ionic strength can be used as a diagnostic tool to dissect the different steps in Bax-mediated MOMP.
Acknowledgments
We thank Meenu N. Perera for purification of the recombinant proteins. We also thank Soumya Samanta for early help and useful discussions. We thank Dr. Antonella Antignani for providing the Bax plasmid. We thank Dr. Donald Newmeyer for providing the plasmid for tBid. We extend our gratitude to Dr. Marie Hardwick for providing the Bcl-xL plasmid.
This work was supported by National Science Foundation grant MCB-1023008.
Supporting Material
References
- 1.Reed J.C., Jurgensmeier J.M., Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim. Biophys. Acta. 1998;1366:127–137. doi: 10.1016/s0005-2728(98)00108-x. [DOI] [PubMed] [Google Scholar]
- 2.Chipuk J.E., Green D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antignani A., Youle R.J. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 2006;18:685–689. doi: 10.1016/j.ceb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 5.Petros A.M., Olejniczak E.T., Fesik S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Shamas-Din A., Brahmbhatt H., Andrews D.W. BH3-only proteins: orchestrators of apoptosis. Biochim. Biophys. Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Korsmeyer S.J., Wei M.C., Schlesinger P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 8.Dejean L.M., Martinez-Caballero S., Kinnally K.W. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol. Biol. Cell. 2005;16:2424–2432. doi: 10.1091/mbc.E04-12-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonsson B., Montessuit S., Martinou J.C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- 10.Eskes R., Desagher S., Martinou J.C. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Billen L.P., Kokoski C.L., Andrews D.W. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavathiotis E., Suzuki M., Walensky L.D. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heimlich G., McKinnon A.D., Jürgensmeier J.M. Bax-induced cytochrome c release from mitochondria depends on α-helices-5 and -6. Biochem. J. 2004;378:247–255. doi: 10.1042/BJ20031152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Upton J.P., Valentijn A.J., Gilmore A.P. The N-terminal conformation of Bax regulates cell commitment to apoptosis. Cell Death Differ. 2007;14:932–942. doi: 10.1038/sj.cdd.4402092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valentijn A.J., Upton J.P., Gilmore A.P. Bax targeting to mitochondria occurs via both tail anchor-dependent and -independent mechanisms. Cell Death Differ. 2008;15:1243–1254. doi: 10.1038/cdd.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H., Hou Q., Hsu Y.T. Complete activation of Bax by a single site mutation. Oncogene. 2007;26:7092–7102. doi: 10.1038/sj.onc.1210517. [DOI] [PubMed] [Google Scholar]
- 17.Muñoz-Pinedo C., Guío-Carrión A., Green D.R. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc. Natl. Acad. Sci. USA. 2006;103:11573–11578. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldstein J.C., Muñoz-Pinedo C., Green D.R. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- 19.Bhola P.D., Simon S.M. Determinism and divergence of apoptosis susceptibility in mammalian cells. J. Cell Sci. 2009;122:4296–4302. doi: 10.1242/jcs.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhola P.D., Mattheyses A.L., Simon S.M. Spatial and temporal dynamics of mitochondrial membrane permeability waves during apoptosis. Biophys. J. 2009;97:2222–2231. doi: 10.1016/j.bpj.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christenson E., Merlin S., Schlesinger P. Cholesterol effects on BAX pore activation. J. Mol. Biol. 2008;381:1168–1183. doi: 10.1016/j.jmb.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montessuit S., Somasekharan S.P., Martinou J.C. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scorrano L., Ashiya M., Korsmeyer S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb E., Armour S.M., Thompson C.B. Mitochondrial respiratory control is lost during growth factor deprivation. Proc. Natl. Acad. Sci. USA. 2002;99:12801–12806. doi: 10.1073/pnas.202477599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bleicken S., Classen M., Bordignon E. Molecular details of Bax activation, oligomerization, and membrane insertion. J. Biol. Chem. 2010;285:6636–6647. doi: 10.1074/jbc.M109.081539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lovell J.F., Billen L.P., Andrews D.W. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Landeta O., Landajuela A., Basañez G. Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J. Biol. Chem. 2011;286:8213–8230. doi: 10.1074/jbc.M110.165852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Llambi F., Moldoveanu T., Green D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valentijn A.J., Upton J.P., Gilmore A.P. Analysis of endogenous Bax complexes during apoptosis using blue native PAGE: implications for Bax activation and oligomerization. Biochem. J. 2008;412:347–357. doi: 10.1042/BJ20071548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens T.W., Valentijn A.J., Gilmore A.P. Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ. 2009;16:1551–1562. doi: 10.1038/cdd.2009.102. [DOI] [PubMed] [Google Scholar]
- 31.Dejean L.M., Martinez-Caballero S., Kinnally K.W. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim. Biophys. Acta. 2006;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Westphal D., Dewson G., Kluck R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta. 2011;1813:521–531. doi: 10.1016/j.bbamcr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 33.Annis M.G., Soucie E.L., Andrews D.W. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikhailov V., Mikhailova M., Saikumar P. Bcl-2 prevents Bax oligomerization in the mitochondrial outer membrane. J. Biol. Chem. 2001;276:18361–18374. doi: 10.1074/jbc.M100655200. [DOI] [PubMed] [Google Scholar]
- 35.Ott M., Robertson J.D., Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uren R.T., Dewson G., Kluck R.M. Mitochondrial release of pro-apoptotic proteins: electrostatic interactions can hold cytochrome c but not Smac/DIABLO to mitochondrial membranes. J. Biol. Chem. 2005;280:2266–2274. doi: 10.1074/jbc.M411106200. [DOI] [PubMed] [Google Scholar]
- 37.Yuan H., Williams S.D., Gottlieb R.A. Cytochrome c dissociation and release from mitochondria by truncated Bid and ceramide. Mitochondrion. 2003;2:237–244. doi: 10.1016/S1567-7249(02)00106-X. [DOI] [PubMed] [Google Scholar]
- 38.Doran E., Halestrap A.P. Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. Biochem. J. 2000;348:343–350. [PMC free article] [PubMed] [Google Scholar]
- 39.Finucane D.M., Bossy-Wetzel E., Green D.R. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 40.Zhuang J., Cohen G.M. Release of mitochondrial cytochrome c is upstream of caspase activation in chemical-induced apoptosis in human monocytic tumor cells. Toxicol. Lett. 1998;102-103:121–129. doi: 10.1016/s0378-4274(98)00296-3. [DOI] [PubMed] [Google Scholar]
- 41.Li T., Brustovetsky T., Brustovetsky N. Dissimilar mechanisms of cytochrome c release induced by octyl glucoside-activated BAX and by BAX activated with truncated BID. Biochim. Biophys. Acta. 2010;1797:52–62. doi: 10.1016/j.bbabio.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuwana T., Mackey M.R., Newmeyer D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 43.Schafer B., Quispe J., Kuwana T. Mitochondrial outer membrane proteins assist Bid in Bax-mediated lipidic pore formation. Mol. Biol. Cell. 2009;20:2276–2285. doi: 10.1091/mbc.E08-10-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kluck R.M., Esposti M.D., Newmeyer D.D. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J. Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siskind L.J., Kolesnick R.N., Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 2002;277:26796–26803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki M., Youle R.J., Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 47.Basañez G., Zhang J., Zimmerberg J. Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J. Biol. Chem. 2001;276:31083–31091. doi: 10.1074/jbc.M103879200. [DOI] [PubMed] [Google Scholar]
- 48.Thuduppathy G.R., Terrones O., Hill R.B. The N-terminal domain of Bcl-xL reversibly binds membranes in a pH-dependent manner. Biochemistry. 2006;45:14533–14542. doi: 10.1021/bi0616652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siskind L.J., Feinstein L., Colombini M. Anti-apoptotic Bcl-2 proteins disassemble ceramide channels. J. Biol. Chem. 2008;283:6622–6630. doi: 10.1074/jbc.M706115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teijido O., Dejean L. Upregulation of Bcl2 inhibits apoptosis-driven Bax insertion but favors Bax relocalization in mitochondria. FEBS Lett. 2010;584:3305–3310. doi: 10.1016/j.febslet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed H. CRC Press; Boca Raton, FL: 1959. Principles and Reactions of Protein Extraction, Purification, and Characterization. [Google Scholar]
- 52.Hill R.L., Bradshaw R.A. Fumarase. Methods Enzymol. 1969;13:91–99. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.