

Abstract
The membrane-active antimicrobial peptide PGLa from Xenopus laevis is known from solid-state 2H-, 15N-, and 19F-NMR spectroscopy to occupy two distinct α-helical surface adsorbed states in membranes: a surface-bound S-state with a tilt angle of ∼95° at low peptide/lipid molar ratio (P/L = 1:200), and an obliquely tilted T-state with a tilt angle of 127° at higher peptide concentration (P/L = 1:50). Using a rapid molecular-dynamics insertion protocol in combination with microsecond-scale simulation, we have characterized the structure of both states in detail. As expected, the amphiphilic peptide resides horizontally on the membrane surface in a monomeric form at a low P/L, whereas the T-state is seen in the simulations to be a symmetric antiparallel dimer, with close contacts between small glycine and alanine residues at the interface. The computed tilt angles and azimuthal rotations, as well as the quadrupolar splittings predicted from the simulations agree with the experimental NMR data. The simulations reveal many structural details previously inaccessible, such as the immersion depth of the peptide in the membrane and the packing of the dimerization interface. The study highlights the ability and limitations of current state-of-the-art multimicrosecond all-atom simulations of membrane-active peptides to complement experimental data from solid-state NMR.
Introduction
Small membrane-active antimicrobial peptides (AMPs) are found in many organisms as part of the innate immune system, defending the host against invading bacteria and other microorganisms (1,2). AMPs have been proposed as a potential source of new antibiotics against increasingly common multiresistant pathogens, as they are unlikely to induce resistance. Most of these peptides kill bacteria by physically interacting with and disrupting their cell membranes. However, the exact molecular mechanism by which this occurs is not fully understood at present. The high affinity of AMPs for lipid bilayers is attributed to their overall amphiphilic structure.
PGLa, from the skin of the African frog Xenopus laevis (Fig. 1) (3–5), is part of the “magainin family” of peptides that exhibit broad antimicrobial activity (6). In addition to antibacterial properties (7), it has been reported that PGLa and other related AMPs show anticancer (8), antiviral (9), and antifungal activity (10). Detailed knowledge of the membrane-bound structure of PGLa is sparse, with most information derived indirectly from activity assays and fluorescence and dye-release experiments (11,12). The antimicrobial activity is generally attributed to the formation of pores in the membrane at high peptide/lipid molar ratios (P/Ls).
Figure 1.
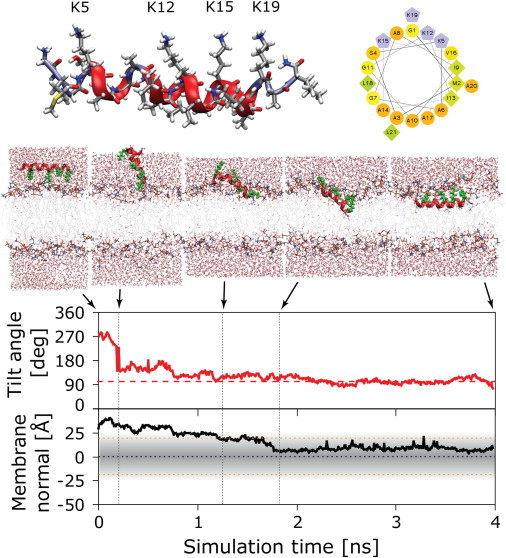
Illustration of a high-temperature peptide insertion simulation for monomeric PGLa in a DMPC bilayer at 207°C. PGLa has an amphiphilic helical structure, with four charged lysines facing one side and hydrophobic residues on the other (helical wheel representation). Insertion can be illustrated using the tilt angle and insertion depth of the monomer: the helically restrained peptide is initially placed into the solvent with the charged lysine residues facing the lipid headgroups. In the course of the simulation, the peptide quickly flips over (0.3 ns), pointing its hydrophobic face toward the bilayer and subsequently burying itself in the membrane (1.3–1.8 ns). After 2.2 ns, a stable interfacial state is reached, and this state is used as the starting configuration for follow-up unrestrained simulations at 35°C.
Recently, substantial progress has been made in the determination of the structure of PGLa and related peptides in oriented lipid bilayers using solid-state 2H-, 15N-, and 19F-NMR spectroscopy (12–23). These measurements have revealed that PGLa is fully helical when membrane-bound. It resides in a well-defined surface-bound S-state in 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayers at P/L = 1:200 and below, at which ratio the peptide is presumed to be monomeric. Under these conditions it has a tilt angle (the angle between the helix long-axis vector, pointing from the N- to the C-terminus, and the membrane normal) of τ ≈ 95° and an azimuthal rotation angle (the angle by which the peptide is rotated around its long axis) of ρ ≈ 116°. This S-state is common for amphipathic helices at low P/L and has been found for many other related antimicrobial peptides (6). Upon increasing the peptide concentration, PGLa is thought to form pores in a transmembrane configuration (16). However, an inserted state could only be detected at artificially low temperatures in gel-phase bilayers (24), or when stabilized by its synergistic partner magainin 2 (MAG2) (16). That is because pure PGLa presumably forms pores only transiently, with lifetimes too short to be studied by NMR (21). Nevertheless, 19F- and 2H-NMR data of PGLa in DMPC have shown a critical realignment step that is considered to be the first functionally relevant event in membrane permeabilization. Upon increasing the PGLa concentration to P/L = 1:50, a transition occurs from the monomeric S-state to an obliquely tilted T-state, with a distinct set of angles, τ ≈ 127° and ρ ≈ 111° (13,16,19,21). Experimentally, the T-state is stable over a wide range of peptide concentrations from 1:100 up to 1:20, and it is independent of lipid charge, being detected also in the presence of DMPG (21). This discrete T-state had not been known before the comprehensive NMR analysis of PGLa, but it was subsequently observed also for many other antimicrobials, such as the related MSI-103 (25–27) and the cell-penetrating peptide MAP (28, and see (52)).
The critical P/Ls of the S-to-T-state transition of these peptides were measured using oriented circular dichroism spectroscopy (OCD) in fully hydrated DMPC bilayers, and they ranged from P/L = 1:80 for PGLa to 1:160 for MAP and 1:240 for MSI-103 (28). Thus, the S-to-T-state realignment upon increasing the peptide concentration appears to be an important general feature of these types of amphiphilic helices. The distinct tilt angle suggests the formation of a stable dimer, with a characteristic helix-association angle. The observation of only one set of NMR splittings suggests that this dimer must be symmetric and antiparallel (21). The interpretation of the NMR data as dimers was supported by earlier observations that magainins and their relatives have a tendency to dimerize, as observed for PGLa in lipid monolayers at the air-water interface (29), MSI-103 (25), and MAG2 (30,31) and suggested for melittin (32).
The goal of the MD simulations performed here is to detect the concentration-dependent realignment of PGLa, to characterize the monomeric S-state and the putative dimeric T-state, and to compare these with the experimental NMR data. In addition, the simulations provide important structural information complementing the solid-state NMR data, such as the depth of insertion of the monomer/dimer, the packing of the dimerization interface, and the detailed peptide-lipid interactions that stabilize PGLa within the lipid bilayer. Recently, high-temperature MD simulations have been shown to be useful in overcoming the sampling limitations that have rendered simulation of membrane-active peptides difficult to achieve in the past (33,34). We present here a straightforward application of this new protocol to the simulation of PGLa in a lipid bilayer under conditions identical to those of the solid-state NMR experiments. This complementary experimental/computational approach is vital to advance our understanding of this important class of membrane-active peptides. The ultimate aim is to identify the specific physicochemical properties that lead to the antimicrobial function, and thus to allow both prediction and improvement of the therapeutic impact of new peptide sequences.
Methods
All simulations were performed and analyzed using Gromacs version 4.0 (www.gromacs.org) (35) and Hippo β (www.biowerkzeug.com), using the CHARMM27 force field (36), the OPLS-AA force field (37), and TIP3P water (38). The united atom lipid parameters for DMPC were taken from Ulmschneider et al. (39). CHARMM36 all-atom lipid parameters were used for the CHARMM simulations (40). OPLS simulations where helical restraints were used are denoted R for full restraints and R-Gly where only critical backbone hydrogen bonds at G7 and G11 were restrained to be helical. No restraints are used for the CHARMM simulations. The production runs were 2 μs for OPLS, and 1 μs for CHARMM, for both the monomeric (S-state) and dimeric (T-state) structures. Electrostatic interactions were computed using particle-mesh Ewald (PME), and a cutoff of 10 Å was used for the van der Waals interactions. Bonds involving hydrogen atoms were restrained using LINCS (41). Simulations were run with a 2-fs time step and neighbor lists were updated every five steps. All simulations were performed in the NPT ensemble, with water, lipids, and the protein coupled separately to a heat bath with T = 35°C and a time constant of τT = 0.1 ps using weak temperature coupling (42). Atmospheric pressure of 1 bar was maintained using weak semi-isotropic pressure coupling with compressibility κz = κxy = 4.6 · 10−5 bar−1 and time constant τP = 1 ps (43). The PGLa peptide (GMASKAGAIAGKIAKVALKAL-amide) was constructed as an ideal α-helix and placed in a box 20 Å from a preformed lipid bilayer made up of 72 lipids and ∼30 water molecules/lipid. During the high-temperature insertion simulations, the intrahelical hydrogen bonds were harmonically restrained to keep the peptide helical. The united-atom lipid molecules in the OPLS simulations result in runs ∼2.5 times faster than the CHARMM simulations, where the lipids are all-atom. All averages in the simulation are obtained over 4000 data points sampled every 500 ps in the case of the 2-μs runs and every 250 ps in the case of the 1-μs runs. Error bars represent the mean ± SD, obtained from block averaging over 10 blocks.
Quadrupolar splittings were obtained from the simulations by calculating the bond order parameter SCD:
Here, the angle θ is between the Cα-Cβ bond of the labeled residue and the membrane normal, which is parallel to the magnetic field (z-direction). The quadrupolar splittings were obtained from
with the quadrupolar coupling constant QCC = 168 kHz and the factor 1/3 coming from the fast rotation of the CD3 group around the Cα-Cβ bond. The splittings can be fitted with a theoretical model to obtain the orientational angles τ and ρ, plus the dynamics parameter Smol, using a procedure described previously (13).
Results
Rapid unbiased determination of the surface-bound state
We have recently shown that the structure of membrane-active peptides can be obtained from direct folding-partitioning simulations, starting from arbitrary unfolded conformations and using fully detailed all-atom models (33,34). Here, we have used an even quicker alternative based on highly elevated temperatures. The key steps are illustrated in Fig. 1. The peptide was helically restrained and initially placed ∼20 Å away from the lipid bilayer. A short, 4-ns simulation was performed at very high temperature (207°C). The peptide rapidly rearranged and integrated itself into the membrane. The final state was reached after 2.2 ns: the amphipathic peptide partitioned into the bilayer interface, with the charged lysine residues pointing into the polar headgroup region, whereas its hydrophobic face was entirely buried in the hydrophobic core of the membrane. This state was stable throughout the rest of the high-temperature run, and no further transitions were observed. Subsequent simulations were performed by cooling the system down to 35°C and removing the helical restraints.
The high-temperature insertion protocol is a computational technique that is not expected to mirror any real-life behavior but is used to overcome kinetic entrapment in the simulations. For example, for T < 200°C, the helix does not partition deeply into the membrane and remains partially solvated in a position much higher than the membrane-buried location. Insertion of the peptide causes a mismatch in bilayer-leaflet density. This density imbalance is a key feature of AMPs at high P/L. However, since the size of the bilayer patch in the simulations is in general much bigger than the size of the peptide, we find that this mass imbalance does not affect the peptide structure, orientation, or insertion depth in any significant way: Simulations for the dimeric T-state where six lipid molecules were deleted in the leaflet containing the peptide revealed no pronounced difference from when the bilayer was left symmetric (Fig. S1 in the Supporting Material).
Monomeric S-state
Starting from the final structure of the high-temperature insertion protocol, monomeric PGLa was simulated at the experimental temperature (T = 35°C) for 2 μs (OPLS) and 1 μs (CHARMM). These runs mimic the low-concentration NMR conditions (P/L = 1:200), under which the peptide is not expected to exist in any oligomeric state. Fig. 2 reveals that the CHARMM simulation shows a full α-helix, whereas PGLa partially unfolds in the OPLS simulation, with an overall ∼73% helicity. The structural reason for the transitions to perturbed-helix states can be found by plotting the overall helicity for each residue averaged over the entire 2 μs. The helix is destabilized at the two glycine positions, G7 and G11, where the backbone hydrogen bonds are open to polar interfacial groups and water molecules. The loss of helical structure is more pronounced than in the NMR experiments, in which some instability was seen only in the C-terminal region of PGLa (residues A17 and A20), whereas an ideal helix was derived between residues A6 and V16 (16). Thus, on the microsecond scale, CHARMM seems to agree with the experimental secondary-structure propensity of PGLa. Three additional control simulations of 2-μs length each were performed to investigate these force-field issues (see Supporting Material).
Figure 2.
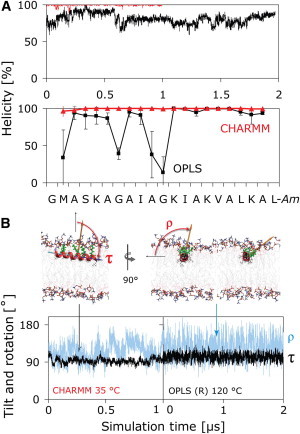
Structure (A) and orientation (B) of PGLa in the S-state. (A) The CHARMM simulation (red) reveals a full α-helix, whereas PGLa partially unfolds in the OPLS simulation (black), with loss of helicity prominent at G7 and G11. (B) The orientation is described by two angles: the tilt angle (τ) is the angle between the membrane normal and the helix long axis, which is defined to point from the N- to the C-terminus. The azimuthal rotation angle (ρ) is defined as a right-handed rotation around the helix long axis, with 0° defined to place the vector from the helix axis to the Cα atom of K12 parallel to the membrane plane. Both angles show similar behavior in the CHARMM and helically restrained (R) OPLS simulations. The rotation angle slowly fluctuates over a wide range at 35°C (orange vector, blue curves), resulting in large uncertainties in ρ. Convergence can be accelerated by use of high temperature (120°C) using a helically restrained peptide (R), which yields rapid convergence of τ and ρ without noticeably changing the averages obtained at 35°C.
Fig. 3 shows the time-averaged orientation of PGLa in the S-state simulations, which is described in terms of the helix tilt angle, τ, and the azimuthal rotation angle, ρ. A 2D histogram plot as a function of these angles reveals overall averages that are very close to the 2H-NMR results of τ ≈ 95° and ρ ≈ 116° indicated by the red dots. It is apparent from the simulations that the peptide cannot deviate much from these ideal values, as tilting or rotating further would result in a deeper burial of one of the charged lysine side chains into the hydrophobic core of the membrane, which is energetically strongly disfavored. Although the tilt angle has a narrow distribution close to the NMR result, ρ slowly transits over a wide range (Fig. 2 B, lower), resulting in slow convergence of the average value at 35°C.
Figure 3.
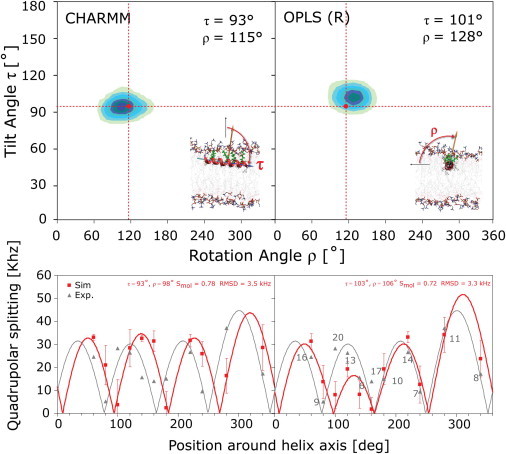
Orientation and quadrupolar splittings |Δvq| obtained from the MD simulation of PGLa in the S-state. Two-dimensional histogram plots of τ and ρ, averaged over the entire simulation are shown. The averages for the 1- to 2-μs simulations are close to the NMR results (red dots) of τ ≈ 95° and ρ ≈ 116°. |Δνq| from Ala-d3-labeled PGLa in DMPC at P/L = 1:200 are shown as gray triangles (16), with the number of the labeled residue indicated and the gray line corresponding to the fitted helical wave. Computed |Δνq| values are shown in red, with the best fit represented by a red line. RMSD values represent the quality of the fit to the computational data. The computational values have huge standard deviations (σ|Δνq| ≈ 20 kHz) due to the large fluctuation of ρ. However, the averages of |Δνq| converge to very accurate values, as illustrated by the small error bars σ|Δνq|mean obtained from block averaging.
Insight can be gained by computing the orientational angles for the Cα-Cβ vectors of the individual residues, which are proportional to the quadrupolar splitting of the corresponding Ala-d3 label, and comparing them to the 2H-NMR data. Plots of |Δνq| predicted from the simulations of monomeric PGLa in the S-state are shown in the lower part of Fig. 3 (red squares). Also shown are the solid-state 2H-NMR splittings from Ala-d3-labeled PGLa in DMPC at P/L = 1:200 (16,19,21) (Fig. 3, gray triangles). The best fit to a helical wave is also plotted, with the computed splittings yielding reasonable fits to the model (∼2.9–3.3 kHz RMSD). Achieving a better match of quadrupolar splitting between experiment and simulation is challenging, as has been noted before (44,45). The standard deviations of the computed splittings, σ|Δνq| are large (∼20 kHz) due to the big fluctuation of ρ. However, the averages of |Δνq| converge to accurate values, which can be seen by the small error bars (σ|Δνq|mean) obtained from block averaging.
Concentration-dependent alignment of PGLa in the lipid bilayer
Solid-state 2H-, 15N-, and 19F-NMR measurements of PGLa in DMPC indicate that upon an increase of peptide concentration to P/L = 1:50, a transition occurs from the monomeric S-state to a tilted T-state, with τ ≈ 127° and a rotation angle of ρ ≈ 111° (13,16,19,21). The exact structure of this state is not known. Experimentally, it is stable over a wide range of peptide concentrations from 1:100 up to 1:20 and independent of lipid charge (21). This hints at the formation of a peptide dimer with a characteristic helix-association angle that could explain the increased tilt. Dimerization is also supported by the stepwise change of the NMR splittings with peptide concentration (and with temperature (24)), ruling out a gradual realignment that might be observed if the peptide remained monomeric (19). A loss of helical structure can be excluded from the NMR data and CD experiments, indicating that the observed changes cannot be attributed to any conformational transitions either (19,21). Thus, the T-state is very likely dimeric. The presence of a single set of NMR splittings indicates that this dimer must be rotationally symmetric with respect to the membrane normal, suggesting an antiparallel dimer (19).
Can such a dimer directly be found using current microsecond-scale MD simulations? To explore this question, two monomers were inserted into a DMPC bilayer using the same high-temperature protocol as for the S-state (Fig. S4). Ideally, dimer formation would be studied by allowing the two peptides to spontaneously assemble until a stable contact free-energy minimum is reached. However, currently, it is computationally challenging to perform such simulations, as illustrated in Fig. 4. At ambient temperature, the two peptides (initially placed 20 Å from each other) diffuse very slowly in the membrane plane, with little change of their distance over ∼100 ns (Fig. 4 A). The slow speed of the lateral movement can be overcome by heating the system to 150°C, where the peptides drift and reorient more rapidly (Fig. 4 B). However, dimer formation is not observed, presumably because dimers are not stable at this temperature. In light of these results, the preferred strategy for predicting dimers is to start the simulations from various self-assembled states and judge the structures according to their fitness to retain a thermodynamically stable conformation. This strategy was pursued for two initial structures (for details of the dimer setup, see Fig. S5). The parallel dimer is shown in Fig. 4 C. This structure was not stable: the two helices slowly minimized their contact surface, and the center-of-mass distance continuously increased. Indeed, such an arrangement can be excluded from the NMR data, in which only one set of NMR splittings is observed, pointing to an antiparallel dimer (19). PGLa is unlikely to form parallel dimers due to its sequence: the two lateral sides of the membrane-bound helix are not symmetric, with one face consisting of small residues (G7, A10, G11, and A14), whereas the opposite face is made up of bulky hydrophobic residues (I9, I13, and V16) (Fig. 1). Close packing is thus much more likely with an antiparallel setup involving an interface consisting of the Ala and Gly residues. Were the dimers to assemble via packing of the bulky hydrophobic side chains of I9 and I13, this packing would have been disturbed by their mutations to Ala-d3 in the NMR experiments, leading to a strongly deviating splitting, which was not observed (19,21).
Figure 4.
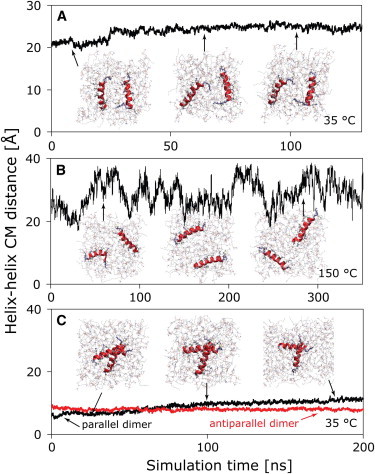
Dimerization simulations of two PGLa peptides in a DMPC bilayer, viewed from above the membrane surface. Representative structures are shown, with the center-of-mass distance of the two peptides plotted versus simulation time. (A) Spontaneous aggregation is slow and not observed at 35°C, with the two peptides remaining at a constant distance. (B) Upon heating to 150°C, the peptides diffuse and rotate rapidly in their monolayer, but aggregation is disfavored. (C) An initially parallel dimer is not thermodynamically stable and quickly dissolves (red, reference distance of the stable antiparallel dimer).
Dimeric T-state
When constructed as a symmetric antiparallel dimer, a stable structure was found in the simulations (Figs. 5 and 6). The quadrupolar splittings, |Δνq|, for the T-state at 35°C are shown in Fig. 5 and compared with the solid-state 2H-NMR data from Ala-d3-labeled PGLa in DMPC at P/L = 1:50. The overall match is not as good as for the S-state, which can be traced to a lower tilt angle encountered in the simulations compared to the experiments. Similar to the NMR results, MD simulations show that the helices increase their tilt angle in the T-state, though the increase to τ ≈ 104°–107° is not as high as the τ ≈ 127° found in the NMR analysis. Two sets of splittings exist from the two chains, but the values are almost identical, agreeing with the experimental hypothesis. Despite this, the fitting procedure can give quite distinct results (CHARMM), demonstrating the difficulty of arriving at a perfect match between simulation and experiment. The stable dimerization interface exhibits close van der Waals contacts of G11-G11, flanked on either side by two A10-A14 pairings (Fig. 6). These small residues allow the two helices to pack very closely together, whereas the bulky hydrophobic residues on the opposing face (I9, I13, and V16) make contact with the lipid hydrocarbon tails of the membrane. The CHARMM dimer is slightly shifted from the OPLS dimer, but without alteration of the close G11-G11 and A10-A14 contacts. Given the large differences in the force fields, small dissimilarities like these are expected. All lysine side chains face upwards into the polar lipid headgroup region, as in the S-state. This finding is fully compatible with the marginal change observed for the azimuthal rotation angle, from ρ ≈ 116° to ρ ≈ 111°. Positional root mean-square fluctuations (RMSF) of each residue over the course of the simulation indicate generally larger fluctuations for C-terminal residues than for residues 1–20 (Fig. 6).
Figure 5.
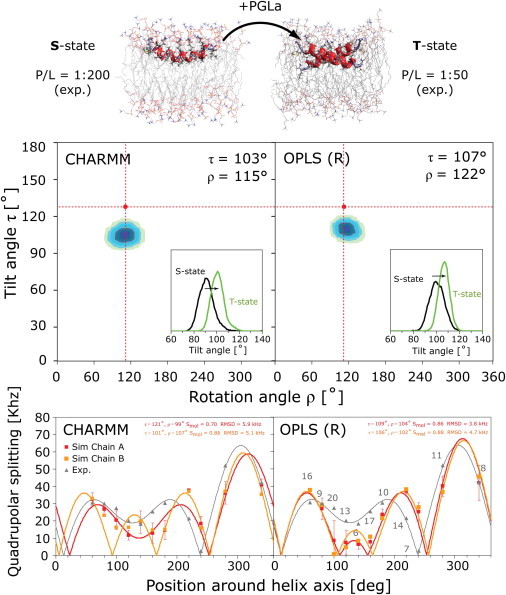
Concentration-dependent realignment and dimerization of PGLa in the lipid bilayer. Solid-state NMR measurements have revealed a change of orientation from the monomeric surface-bound S-state at low P/L molar ratio (P/L = 1:200) to an obliquely tilted T-state with increasing peptide concentration (P/L = 1:50). The T-state is presumed to be an antiparallel dimer. The final structures, as well as the tilt angles from the 2-μs MD simulations of the S-state and T-state at 35°C, are shown (upper). Two-dimensional histogram plots as a function of the tilt (τ) and rotation (ρ) angle of the T-state are shown below, as are the quadrupolar splittings |Δνq| and fitted helical waves (gray triangles, experimental data; red/orange squares, simulation results). Similar to the NMR results (τ ≈ 127° and ρ ≈ 111° (red dots)), the helices are found to tilt more in the T-state than in the S-state (histogram insets), though the increase is not as large as that found in the NMR analysis. The error bars of |Δνq| represent σ|Δνq|mean obtained from block averaging. The RMSD of the fit is large, and tiny differences in the splittings between the two chains result in very different fits for the CHARMM simulations.
Figure 6.
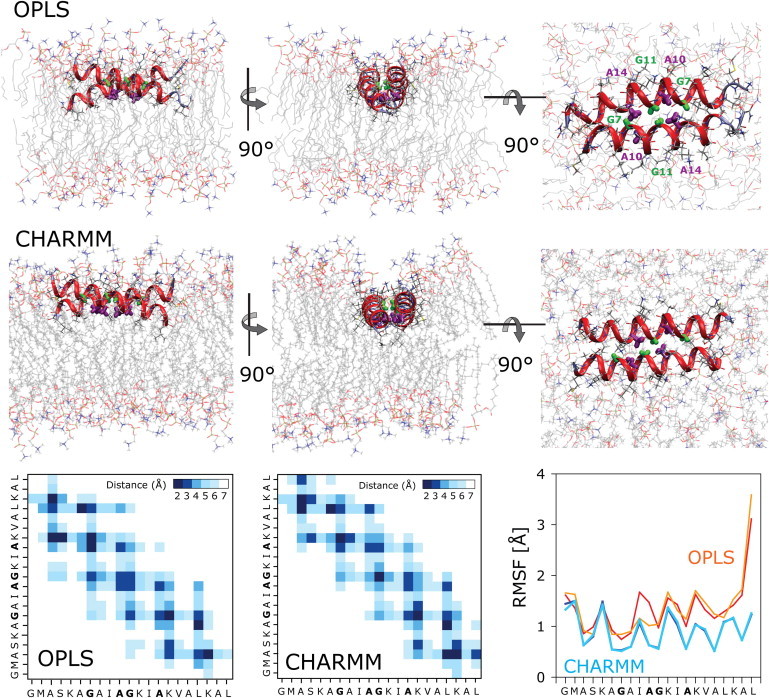
(A) Dimeric T-state of PGLa. Shown are representative structures of the antiparallel dimer taken from the simulations. The dimerization interface is made up of the small central residues G11 and G7 (green) and A10 and A14 (purple). This allows the two helices to closely pack together in a stable structure, which does not change during 2 μs for the OPLS (R-Gly) and 1 μs for the CHARMM simulation. A contact map shows the time-averaged distance of the interhelix residue contacts. Positional root-mean-square fluctuations (RMSF) of each residue over the course of the simulations reveal that the peptide is much more flexible in the OPLS (R-Gly) than in the CHARMM simulation.
As shown in Fig. 7, the simulations show monomeric PGLa to be deeply buried below the lipid phosphate and glycerol/carbonyl region. The peptide remains at this buried position in the membrane throughout the simulation, with a center of mass zCM = 9.4 Å from the center of the bilayer and only a small fluctuation of ±1 Å along the membrane normal. This location embeds the hydrophobic residues firmly in the membrane interior, whereas the charged lysine side chains are fully extended into the polar lipid phosphocholine region. Peptide positions any higher up in the bilayer are not observed in the simulations. A similar deep burial is seen for the T-state dimer. The introduction of such a large and highly charged complex into the membrane causes a considerable surface distortion of the lipid phase (Fig. 7) and a severe local thinning (−5 Å) of the bilayer is seen in the bilayer leaflet containing the peptide.
Figure 7.
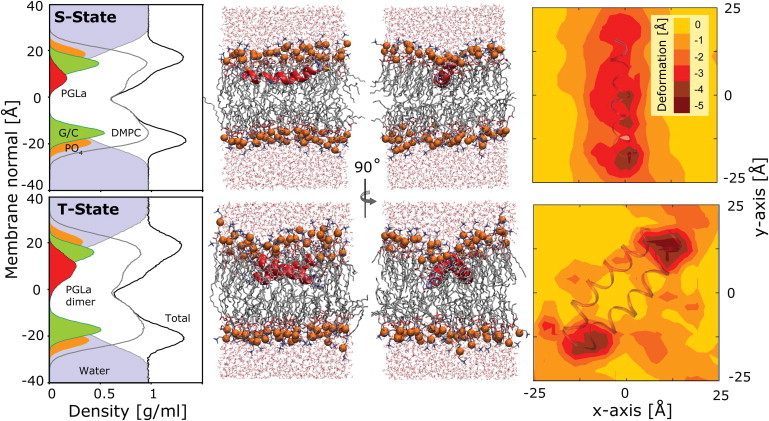
Insertion depth and bilayer deformation of PGLa in a DMPC lipid bilayer in the S-state and the T-state (OPLS runs). Plots of the density cross section reveal the peptide to be deeply inserted, below the lipid phosphate (PO4) and glycerol/carbonyl (G/C) groups. Representative snapshots are shown to scale, with the phosphate groups highlighted as orange beads. The bilayer surface distortion induced by the PGLa peptide is illustrated on the right. The contour plots show the rotationally adjusted deviation in the time-averaged phosphate distance from the center of the bilayer for the leaflet containing the peptide. The peptides cause a significant local thinning (up to −5 Å) of the membrane. Results for the CHARMM simulations are similar.
Discussion
The S- to T-state realignment of PGLa with increased peptide concentration was first found experimentally and is studied here via MD simulations. This transition is likely described as the formation of antiparallel dimers. The simulation-derived tilt and rotation angles, as well as the quadrupolar splittings for both the monomeric S-state and the dimeric T-state, match those derived from solid-state 2H-, 15N-, and 19F-NMR spectroscopy (16,19,21), with an overall better agreement for the S-state than the T-state. What the simulations demonstrate is that PGLa can form such dimers, and that dimerization increases the tilt angle (independent of chosen force field), so this is the likely cause of the increase seen in the NMR experiments at high P/L. Ab initio prediction of dimers in membranes is very challenging and has been recently attempted for the TM dimerization of glycophorin A (46–49) and integrins (50). These studies employed helically restrained coarse-grained models and the final dimeric structure was verified using atomistic MD for only very short sampling times (∼50 ns). Our study goes beyond those efforts in verifying that the candidate structure is stable for 1–2 μs of MD. Current computational limitations unfortunately prevent a more thorough conformational search, so it is possible that other antiparallel dimeric structures might exist that are also stable. These issues will be addressed in the future by even longer simulations in the ∼10–20 μs range.
Dimer formation appears to be an important property of this class of long helical AMPs, and it has also been observed for the model peptide MSI-103 (25,26), for MAG2 (30), for melittin (32), for PGLa in lipid monolayers at the air-water interface (29), in 1:1 mixtures of PGLa with MAG2 (51), and for the cell-penetrating peptide MAP (52). The transition to the dimeric state occurs at high peptide concentration, with critical values ranging from P/L = 1:80 to 1:240 for these AMPs in DMPC bilayers, as determined by oriented CD (28). Interestingly, at intermediate peptide concentrations (P/L = 1:100) in DMPC bilayers, PGLa monomers and dimers can coexist as an equilibrium with fast exchange between the states (21). For the T-state dimer of PGLa, we find a crossing angle of ∼29–33° for the two antiparallel helices, with a close contact involving strong van der Waals interactions between the small hydrophobic amino acid residues G7, G11, A10, and A14. Such an interface is supported by the 2H-NMR measurements, which found no structural disturbance when mutating the residues I9 and I13 on the opposite face of the peptide (13). Similar contacts involving the A10 and A14 residues were reported for the proposed parallel dimer of the analogous peptide MSI-103, which has a sequence derived from PGLa (25). The crossing angle in this case was ∼20°.
Recently, several computational studies of related antimicrobial peptides have been reported, for example for magainin (MAG-H2) (53), melittin (54), and cateslytin (55), in which a variety of disordered structural aggregates were found both for interfacial and membrane-inserted pore states. No specific interfacial assemblies such as dimeric T-states were encountered. In addition, the peptides were found to be only marginally α-helical: For example, 40–50% helicity for melittin was reported, compared to the experimental value of ∼50–70% (54). These results differ qualitatively from the NMR data for PGLa, in which it appears highly helical, with well-ordered, distinct structures exhibiting clearly defined tilt and rotation angles (16). The formation of large peptide aggregates at high P/L—indicative of a carpetlike model (56)—is not supported by the observed fast rotational diffusion of PGLa around the membrane normal, which instead supports formation of dimers (13,19). Only at the highest peptide concentrations (P/L > 1:20) does PGLa seem to become immobilized in DMPC by the lateral crowding, but these assemblies nevertheless retain the well-defined orientation of the dimeric T-state (21).
We have shown here that some of the disparity in the various simulation results of AMPs can be traced to the simulation force field. The CHARMM simulations show perfectly helical PGLa in both S- and T- states, in agreement with the NMR data, whereas the OPLS simulations reveal partial loss of secondary structure. These results highlight the importance of choosing both balanced simulation parameters and long sampling times (μs) if accurate comparison to experimental data is the goal. Recent findings also show that the outcome of the simulations of membrane-active peptides is greatly influenced by the choice of the electrostatic long-range models, and by the inclusion or omission of counterions (57). Moreover, studies of hydrophobic WALP peptides (which are of similar length to AMPs) in lipid bilayers have revealed that unphysical helical unfolding can occur on the microsecond scale if protein and lipid parameters are not properly balanced (33,39,58,59). Even if parameters are good, it is challenging to achieve quantitative agreement between experimental and computed quadrupolar splittings for membrane-bound peptides such as WALP (44,45).
The T-state may represent an intermediate stage in the formation of the proposed transmembrane pores (or fully inserted I-states) of PGLa. These are expected to consist of groups of such dimers assembled into larger toroidal pores, in which the cationic residues of the peptides together with anionic lipid headgroups line a water-filled channel (11,16,21). No such state has yet been observed in liquid crystalline bilayers, possibly because the pores are transient and not stable enough to be studied by NMR, though they are still able to cause antimicrobial activity (21). However, recent data demonstrate that PGLa can insert into liquid crystalline DMPC/DMPG (3:1) bilayers with a tilt angle of ∼158°, when prepared as a synergistic 1:1 mixture with MAG2 at P/L = 1:50 (22). Interestingly, proton-decoupled 15N solid-state NMR measurements on similar mixtures indicate that MAG2 remains in a stable in-plane alignment in this arrangement (23). Short-lived pore structures can be considered part of a general mechanism, in which the initial imbalance of mass and charge on one bilayer leaflet leads to assembly and temporary porelike structures in the bilayer until enough peptide has accumulated on the opposing side of the membrane (60). Once the peptide is equilibrated across the membrane, the pores are then thought to dissipate, as evidenced by leakage assays (60). However, such behavior has not yet been detected in the MD simulations of AMPs (53–55), where the disordered membrane-inserted porelike aggregates seemed to be the dominant state above a certain peptide concentration and were not observed to dissolve. Further studies will be required to answer these questions.
MD simulations, coupled with quantitative comparisons with solid-state NMR data, offer a practical means of addressing many of the questions concerning the mechanism of these peptides that can otherwise only be indirectly observed through functional leakage assays. Once a reliable protocol has been established for any one type of system on the basis of experimental NMR data, the effect of mutations and chemical modifications can be elucidated in a very efficient manner by MD simulations.
Acknowledgments
The authors acknowledge the Deutsche Forschungsgemeinschaft Center for Functional Nanotechnology (project E1.2).
This research was supported by a Center for Modeling and Simulation in the Biosciences Fellowship (BIOMS) to J.P.U, a Marie Curie International Fellowship to M.B.U, and a Laboratory Directed Research and Development award from the U.S. Department of Energy to J.C.S.
Supporting Material
References
- 1.Epand R.M., Vogel H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- 2.van 't Hof W., Veerman E.C., Amerongen A.V. Antimicrobial peptides: properties and applicability. Biol. Chem. 2001;382:597–619. doi: 10.1515/BC.2001.072. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann W., Richter K., Kreil G. A novel peptide designated PYLa and its precursor as predicted from cloned mRNA of Xenopus laevis skin. EMBO J. 1983;2:711–714. doi: 10.1002/j.1460-2075.1983.tb01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richter K., Aschauer H., Kreil G. Biosynthesis of peptides in the skin of Xenopus laevis: isolation of novel peptides predicted from the sequence of cloned cDNAs. Peptides. 1985;6(Suppl 3):17–21. doi: 10.1016/0196-9781(85)90345-6. [DOI] [PubMed] [Google Scholar]
- 5.Andreu D., Aschauer H., Merrifield R.B. Solid-phase synthesis of PYLa and isolation of its natural counterpart, PGLa [PYLa-(4-24)] from skin secretion of Xenopus laevis. Eur. J. Biochem. 1985;149:531–535. doi: 10.1111/j.1432-1033.1985.tb08957.x. [DOI] [PubMed] [Google Scholar]
- 6.Maloy W.L., Kari U.P. Structure-activity studies on magainins and other host defense peptides. Biopolymers. 1995;37:105–122. doi: 10.1002/bip.360370206. [DOI] [PubMed] [Google Scholar]
- 7.Soravia E., Martini G., Zasloff M. Antimicrobial properties of peptides from Xenopus granular gland secretions. FEBS Lett. 1988;228:337–340. doi: 10.1016/0014-5793(88)80027-9. [DOI] [PubMed] [Google Scholar]
- 8.Hoskin D.W., Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chinchar V.G., Bryan L., Rollins-Smith L. Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology. 2004;323:268–275. doi: 10.1016/j.virol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 10.Helmerhorst E.J., Reijnders I.M., Nieuw Amerongen A.V. A critical comparison of the hemolytic and fungicidal activities of cationic antimicrobial peptides. FEBS Lett. 1999;449:105–110. doi: 10.1016/s0014-5793(99)00411-1. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzaki K., Mitani Y., Miyajima K. Mechanism of synergism between antimicrobial peptides Magainin 2 and PGLa. Biochemistry. 1998;37:15144–15153. doi: 10.1021/bi9811617. [DOI] [PubMed] [Google Scholar]
- 12.Wieprecht T., Apostolov O., Seelig J. Membrane binding and pore formation of the antibacterial peptide PGLa: thermodynamic and mechanistic aspects. Biochemistry. 2000;39:442–452. doi: 10.1021/bi992146k. [DOI] [PubMed] [Google Scholar]
- 13.Strandberg E., Wadhwani P., Ulrich A.S. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys. J. 2006;90:1676–1686. doi: 10.1529/biophysj.105.073858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bechinger B., Kim Y., Opella S.J. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J. Biomol. NMR. 1991;1:167–173. doi: 10.1007/BF01877228. [DOI] [PubMed] [Google Scholar]
- 15.Bechinger B., Zasloff M., Opella S.J. Structure and dynamics of the antibiotic peptide PGLa in membranes by solution and solid-state nuclear magnetic resonance spectroscopy. Biophys. J. 1998;74:981–987. doi: 10.1016/S0006-3495(98)74021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strandberg E., Tremouilhac P., Ulrich A.S. Synergistic transmembrane insertion of the heterodimeric PGLa/magainin 2 complex studied by solid-state NMR. Biochim. Biophys. Acta. 2009;1788:1667–1679. doi: 10.1016/j.bbamem.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Bechinger B., Gierasch L.M., Opella S.J. Orientations of helical peptides in membrane bilayers by solid state NMR spectroscopy. Solid State Nucl. Magn. Reson. 1996;7:185–191. doi: 10.1016/0926-2040(95)01224-9. [DOI] [PubMed] [Google Scholar]
- 18.Glaser R.W., Sachse C., Ulrich A.S. Orientation of the antimicrobial peptide PGLa in lipid membranes determined from 19F-NMR dipolar couplings of 4-CF3-phenylglycine labels. J. Magn. Reson. 2004;168:153–163. doi: 10.1016/j.jmr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Glaser R.W., Sachse C., Ulrich A.S. Concentration-dependent realignment of the antimicrobial peptide PGLa in lipid membranes observed by solid-state 19F-NMR. Biophys. J. 2005;88:3392–3397. doi: 10.1529/biophysj.104.056424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Afonin S., Mikhailiuk P.K., Ulrich A.S. Evaluating the amino acid CF3-bicyclopentylglycine as a new label for solid-state 19 F-NMR structure analysis of membrane-bound peptides. J. Pept. Sci. 2007;13:614–623. doi: 10.1002/psc.854. [DOI] [PubMed] [Google Scholar]
- 21.Tremouilhac P., Strandberg E., Ulrich A.S. Conditions affecting the re-alignment of the antimicrobial peptide PGLa in membranes as monitored by solid state 2H-NMR. Biochim. Biophys. Acta. 2006;1758:1330–1342. doi: 10.1016/j.bbamem.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 22.Tremouilhac P., Strandberg E., Ulrich A.S. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J. Biol. Chem. 2006;281:32089–32094. doi: 10.1074/jbc.M604759200. [DOI] [PubMed] [Google Scholar]
- 23.Salnikov E.S., Bechinger B. Lipid-controlled peptide topology and interactions in bilayers: structural insights into the synergistic enhancement of the antimicrobial activities of PGLa and magainin 2. Biophys. J. 2011;100:1473–1480. doi: 10.1016/j.bpj.2011.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afonin S., Grage S.L., Ulrich A.S. Temperature-dependent transmembrane insertion of the amphiphilic peptide PGLa in lipid bilayers observed by solid state 19F NMR spectroscopy. J. Am. Chem. Soc. 2008;130:16512–16514. doi: 10.1021/ja803156d. [DOI] [PubMed] [Google Scholar]
- 25.Toke O., O'Connor R.D., Schaefer J. Structure of (KIAGKIA)3 aggregates in phospholipid bilayers by solid-state NMR. Biophys. J. 2004;87:675–687. doi: 10.1529/biophysj.103.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strandberg E., Kanithasen N., Ulrich A.S. Solid-state NMR analysis comparing the designer-made antibiotic MSI-103 with its parent peptide PGLa in lipid bilayers. Biochemistry. 2008;47:2601–2616. doi: 10.1021/bi701944r. [DOI] [PubMed] [Google Scholar]
- 27.Strandberg E., Tiltak D., Ulrich A.S. Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim. Biophys. Acta. 2012;1818:1764–1776. doi: 10.1016/j.bbamem.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Bürck J., Roth S., Ulrich A.S. Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophys. J. 2008;95:3872–3881. doi: 10.1529/biophysj.108.136085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konovalov O., Myagkov I., Lohner K. Lipid discrimination in phospholipid monolayers by the antimicrobial frog skin peptide PGLa. A synchrotron X-ray grazing incidence and reflectivity study. Eur. Biophys. J. 2002;31:428–437. doi: 10.1007/s00249-002-0233-3. [DOI] [PubMed] [Google Scholar]
- 30.Wakamatsu K., Takeda A., Matsuzaki K. Dimer structure of magainin 2 bound to phospholipid vesicles. Biopolymers. 2002;64:314–327. doi: 10.1002/bip.10198. [DOI] [PubMed] [Google Scholar]
- 31.Matsuzaki K., Murase O., Miyajima K. Orientational and aggregational states of magainin 2 in phospholipid bilayers. Biochemistry. 1994;33:3342–3349. doi: 10.1021/bi00177a027. [DOI] [PubMed] [Google Scholar]
- 32.Hristova K., Dempsey C.E., White S.H. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys. J. 2001;80:801–811. doi: 10.1016/S0006-3495(01)76059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulmschneider M.B., Doux J.P.F., Ulmschneider J.P. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc. 2010;132:3452–3460. doi: 10.1021/ja909347x. [DOI] [PubMed] [Google Scholar]
- 34.Ulmschneider J.P., Smith J.C., Ulmschneider M.B. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 2011;133:15487–15495. doi: 10.1021/ja204042f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;95:43–56. [Google Scholar]
- 36.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 38.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 39.Ulmschneider J.P., Ulmschneider M.B. United atom lipid parameters for combination with the Optimized Potentials for Liquid Simulations all-atom force field. J. Chem. Theory Comput. 2009;5:1803–1813. doi: 10.1021/ct900086b. [DOI] [PubMed] [Google Scholar]
- 40.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 42.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 43.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 44.Ozdirekcan S., Etchebest C., Fuchs P.F. On the orientation of a designed transmembrane peptide: toward the right tilt angle? J. Am. Chem. Soc. 2007;129:15174–15181. doi: 10.1021/ja073784q. [DOI] [PubMed] [Google Scholar]
- 45.Holt A., Killian J.A. Orientation and dynamics of transmembrane peptides: the power of simple models. Eur. Biophys. J. 2010;39:609–621. doi: 10.1007/s00249-009-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Psachoulia E., Fowler P.W., Sansom M.S. Helix-helix interactions in membrane proteins: coarse-grained simulations of glycophorin a helix dimerization. Biochemistry. 2008;47:10503–10512. doi: 10.1021/bi800678t. [DOI] [PubMed] [Google Scholar]
- 47.Psachoulia E., Marshall D.P., Sansom M.S.P. Molecular dynamics simulations of the dimerization of transmembrane α-helices. Acc. Chem. Res. 2010;43:388–396. doi: 10.1021/ar900211k. [DOI] [PubMed] [Google Scholar]
- 48.Sengupta D., Marrink S.J. Lipid-mediated interactions tune the association of glycophorin A helix and its disruptive mutants in membranes. Phys. Chem. Chem. Phys. 2010;12:12987–12996. doi: 10.1039/c0cp00101e. [DOI] [PubMed] [Google Scholar]
- 49.Janosi L., Prakash A., Doxastakis M. Lipid-modulated sequence-specific association of glycophorin A in membranes. Biophys. J. 2010;99:284–292. doi: 10.1016/j.bpj.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalli A.C., Hall B.A., Sansom M.S.P. A helix heterodimer in a lipid bBilayer: prediction of the structure of an integrin transmembrane domain via multiscale simulations. Structure. 2011;19:1477–1484. doi: 10.1016/j.str.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hara T., Mitani Y., Matsuzaki K. Heterodimer formation between the antimicrobial peptides magainin 2 and PGLa in lipid bilayers: a cross-linking study. Biochemistry. 2001;40:12395–12399. doi: 10.1021/bi011413v. [DOI] [PubMed] [Google Scholar]
- 52.Wadhwani P., Bürck J., Ulrich A.S. Using a sterically restrictive amino acid as a 19F NMR label to monitor and to control peptide aggregation in membranes. J. Am. Chem. Soc. 2008;130:16515–16517. doi: 10.1021/ja804928q. [DOI] [PubMed] [Google Scholar]
- 53.Leontiadou H., Mark A.E., Marrink S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006;128:12156–12161. doi: 10.1021/ja062927q. [DOI] [PubMed] [Google Scholar]
- 54.Sengupta D., Leontiadou H., Marrink S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta. 2008;1778:2308–2317. doi: 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 55.Jean-François F., Elezgaray J., Dufourc E.J. Pore formation induced by an antimicrobial peptide: electrostatic effects. Biophys. J. 2008;95:5748–5756. doi: 10.1529/biophysj.108.136655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shai Y., Oren Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides. 2001;22:1629–1641. doi: 10.1016/s0196-9781(01)00498-3. [DOI] [PubMed] [Google Scholar]
- 57.Gurtovenko A.A., Anwar J., Vattulainen I. Defect-mediated trafficking across cell membranes: insights from in silico modeling. Chem. Rev. 2010;110:6077–6103. doi: 10.1021/cr1000783. [DOI] [PubMed] [Google Scholar]
- 58.Ulmschneider M.B., Ulmschneider J.P. Folding peptides into lipid bilayer membranes. J. Chem. Theory Comput. 2008;4:1807–1809. doi: 10.1021/ct800100m. [DOI] [PubMed] [Google Scholar]
- 59.Ulmschneider J.P., Doux J.P.F., Ulmschneider M.B. Peptide partitioning and folding into lipid bilayers. J. Chem. Theory Comput. 2009;5:2202–2205. doi: 10.1021/ct900256k. [DOI] [PubMed] [Google Scholar]
- 60.Rathinakumar R., Wimley W.C. Biomolecular engineering by combinatorial design and high-throughput screening: small, soluble peptides that permeabilize membranes. J. Am. Chem. Soc. 2008;130:9849–9858. doi: 10.1021/ja8017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.