

Abstract
Systemic lupus erythematosus (SLE) is a prototype autoimmune disease that affects multiorgan systems. Lupus nephritis is one of the most severe manifestations of SLE whereby immune-mediated inflammation can lead to permanent damage within the glomerular, tubulo-interstitial, and vascular compartments of the kidney, resulting in acute or chronic renal failure. The mechanisms that regulate host inflammatory responses and tissue injury are incompletely understood. Accumulating evidence suggests that hyaluronan and its interaction with its cell surface receptor CD44 plays an important role in mediating pathogenic mechanisms in SLE. This paper discusses the putative mechanisms through which hyaluronan and CD44 contribute to the pathogenesis of SLE, with particular emphasis on lupus nephritis.
1. Introduction
Systemic lupus erythematosus (SLE) is a severe autoimmune disease characterized by a breakdown of immune tolerance and production of autoantibodies. Although the etiology of SLE remains to be fully elucidated, accumulating evidence suggests that genetic, environmental, infectious, and hormonal factors may predispose individuals to the development of SLE [1–3]. This disease predominantly affects females of Afro-American, Hispanic, and Asian descent and can be mild or life threatening depending on the organs involved.
Renal involvement occurs in up to 60% of SLE patients and is a strong predictor of morbidity and mortality [4]. Onset of lupus nephritis is initiated by the deposition of anti-double stranded (ds) DNA antibodies in the kidney parenchyma, which results in complement activation, infiltration of immune cells, and induction of inflammatory and fibrotic processes in the kidney. If these tissue-damaging processes are not sufficiently controlled, destruction of the normal kidney parenchyma and its replacement by fibrous tissue will ensue, which will lead to endstage renal failure [4]. The exact mechanisms through which anti-dsDNA antibodies are deposited in the kidney to mediate kidney injury remains to be fully defined but current knowledge suggests that they can bind directly to mesangial cells through annexin II or α-actinin [5–7] or indirectly to components of the glomerular basement membrane through nucleosomes [8, 9].
The extracellular matrix (ECM) was previously considered to function solely as a structural support that maintained the architecture of tissues and organs, but there is now compelling evidence to show that ECM components also play critical roles during inflammatory processes. Their accumulation and subsequent degradation is a cardinal feature of autoimmune diseases. Hyaluronan (HA) is a major component of the ECM that can directly regulate inflammatory processes through its interaction with CD44, its cell surface receptor [10, 11]. Depending on its molecular weight HA may possess either anti-inflammatory or pro-inflammatory properties. We have demonstrated that serum HA levels in patients with lupus nephritis correlate with disease activity, and that intrarenal HA expression is also increased in lupus nephritis, induced in part by anti-dsDNA antibodies [12]. This paper will discuss the putative roles of HA and CD44 in SLE, with particular emphasis on their roles in mediating inflammatory processes during lupus nephritis.
2. Hyaluronan and CD44
2.1. Synthesis of HA
HA is a nonsulfated, negatively charged glycosaminoglycan that is composed of repeating disaccharide units of D-glucuronic acid and N-acetyl-D-glucosamine [13]. Unlike other glycosaminoglycans, HA is not attached to a protein core and is synthesized on the inner surface of the plasma membrane [13]. HA is synthesized by HA synthases (HAS) and currently three mammalian HAS have been identified, namely, HAS I, HAS II, and HAS III, which utilize UDP-α-N-acetyl-D-glucosamine and UDP-α-glucuronate as substrates for the synthesis of HA [14]. Under physiologic conditions, HA is synthesized as a macromolecule with a MW of 105–107 Da depending on the tissue type [15]. Following its synthesis, HA is directed to the cell surface where it interacts with CD44, or is assembled into pericellular or extracellular matrices [16]. Studies have demonstrated that all three HAS isozymes can contribute to the synthesis of high MW (HMW) HA, but HAS I and HAS III may also produce low MW (LMW) HA depending on the condition of the microenvironment [14].
2.2. Functions of HA under Physiological and Pathological Conditions
Despite its simple chemical structure, HA remains one of the most complex and multifaceted components of the ECM that contributes to diverse biological functions such as the structural stability of basement membranes, maintenance of water balance, plasma protein distribution, sequestration of free radicals, and regulation of cell proliferation, migration, and phenotype [17]. Native HA possesses anti-inflammatory, anti-angiogenic, and immunosuppressive properties [18]. They also provide a protective glycocalyx around endothelial, epithelial, and mesothelial cells that protect these cells from injury, apoptosis, and leukocyte adhesion [19–21]. HA undergoes constant turnover during the daily maintenance of basement membranes. It is degraded into small, nonbiologically active fragments which is rapidly removed through the liver.
The turnover and remodeling of the ECM is a dynamic process that occurs during normal development and tissue repair, and replenishment of ECM components is critical in order to preserve the structural and functional integrity of tissues. These processes become aberrant in pathological conditions associated with chronic inflammation where accumulation of ECM constituents is often observed, which perturbs tissue structure resulting in organ dysfunction. HA accumulates at sites of injury during chronic renal inflammation, where they form long cable-like structures that act as an adhesive matrix for the binding of leukocytes and macrophages. Mesangial cells and proximal tubular epithelial cells have been shown to contribute to the synthesis of these cable-like structures [22, 23]. In line with its anti-inflammatory properties, it has been suggested that binding of leukocytes to HA cables prevents them from interacting with adhesion molecules, thus limiting inflammatory processes in the glomerulus and tubulo-interstitium. Furthermore, it is also conceivable that the HA cable may serve as a temporary scaffold that prevents the loss of ECM components during extreme tissue remodeling [24]. Macrophages have been shown to regulate the clearance of the provisional HA matrix, and this process is essential before a permanent matrix can be synthesized.
Unlike other glycosaminoglycans where modifications in their sulfation pattern, deacetylation and epimerization define their biological roles, the functional role in HA is dictated by its molecular weight and its interaction with its binding proteins, the latter termed the hyaladherins. HA undergoes depolymerization either through oxidative stress or enzymatic cleavage by various hyaluronidases during tissue injury and inflammatory processes [10, 11, 25]. LMW HA have biological properties that are distinct from their parent molecule and have been shown to promote inflammatory and angiogenic processes through increased cell proliferation, activation of signaling transduction pathways and induction of chemokine and cytokine secretion in macrophages, dendritic cells, mesothelial cells, mesangial cells, epithelial cells, and chondrocytes [10, 26–32]. The clearance of HA fragments is therefore imperative for the resolution of tissue injury. The removal of LMW HA from sites of injury is dependent on their interaction with CD44 since targeted deletion of CD44 in mice with bleomycin-induced lung injury resulted in the accumulation of HA fragments, unremitting inflammation, and perpetual tissue damage, a finding that was not observed in wild-type mice [33]. The distinct biological roles of HMW and LMW HA thus far identified are summarized (Table 1). An in-depth review of the interaction of HA with hyaladherins and mechanisms of degradation is outside the scope of this paper [10, 11, 13, 16].
Table 1.
Functions of native and depolymerized hyaluronan.
| Native hyaluronan | Hyaluronan fragments |
|---|---|
| Contributes to tissue integrity and maintenance of epithelial cell phenotype | Induces chemokine and cytokine secretion in infiltrating, renal tubular epithelial and endothelial cells |
| Contributes to water balance and regulation of tissue hydration | Induces phosphorylation of signaling pathways, for example, MAPK |
| Contributes to transportation and distribution of plasma proteins | Induces cell proliferation and migration in chondrocytes, endothelial cells and fibroblasts |
| Protects against tissue damage by scavenging free radicals | Activates NFκB |
| Anti-inflammatory-can inhibit activation of inflammatory cells | Induces nitric oxide synthase |
| Protects against apoptosis | Suppresses cell death and apoptosis in cell culture |
| Anti-angiogenic | Promotes angiogenesis |
| Immunosuppressive-prevents ligand binding to surface receptors | Increases matrix protein synthesis, for example, collagen type I |
| Inhibits phagocytosis | Increases transcription of matrix metalloproteinases |
Hyaluronan fragments: range from 4 to 40 saccharide units.
2.3. CD44
CD44 is a transmembrane glycoprotein with a wide tissue distribution and is found on leukocytes, and epithelial, endothelial, and smooth muscle-like cells. The human CD44 gene is located on the short arm of chromosome 11 and consists of 20 exons of which 10 are variant exons (v1–v10) that can undergo alternative splicing to generate multiple CD44 isoforms [34]. The genomic structure of CD44 is shown in Figure 1. Post-translational modifications of the CD44 molecule such as N- and O-glycosylation, and the attachment of heparan sulfate and/or chondroitin sulfate glycosaminoglycan chains may further increase the number of CD44 isoforms. Such post-translational modifications are tissue specific and bestow upon the CD44 molecule an ability to sequester growth factors and cytokines, thereby allowing greater accrual of its variability and functions [35–37]. It has been hypothesized that over one hundred CD44 isoforms can be generated, although to date only 26 have been identified. The predominant form of CD44 expressed in normal tissues does not contain any spliced exons and is designated hematopoetic or standard CD44 (CD44H or CD44s resp.). It can undergo post-translational modifications and has a molecular weight of 80–100 kDa.
Figure 1.

Genomic structure of CD44. The gene encoding for human CD44 consists of 20 exons. The standard form of CD44 contains exons 1–5, 16–18, and 20. Variants forms of CD44 comprise the standard form of CD44 and the insertion of various combinations of variant exons (v1–v10). Exon 19 is normally absent in most CD44 transcripts and its inclusion results in a shorter variant form of CD44.
CD44 can interact with various cell surface and extracellular ligands but its principal ligand is HA [38, 39]. It is noteworthy that binding of HA to CD44 is not constitutive but is activation dependent [40]. In this respect, quiescent leukocytes express inactive forms of CD44 that do not bind to HA and must be activated before it can interact with HA. Recognition of HA by CD44 is dependent on the degree of post-translational modifications, its phosphorylation status, sulfation pattern and ability to form multivalent aggregates on the cell surface [41–44]. Binding of HA to CD44 is a relatively weak interaction in comparison to other cell receptor-ligand interactions such as those that involve integrins or cadherins, but in some instances weak interactions are an advantage particularly when leukocytes require to be in close proximity in order to exchange chemical signals prior to their activation and maturation [45, 46]. The interaction of CD44 with HA has been shown to enhance various cellular functions such as cell proliferation and migration, and activation of PKC, PI3K and MAPK-signaling pathways which have all been shown to induce inflammatory processes in autoimmune diseases including lupus nephritis [47, 48].
CD44 plays an important role in many physiological and pathological processes that include cell-cell and cell-matrix interactions, cell migration, lymphocyte activation and extravasation, and presentation of growth factors, cytokines and chemokines to their cognate receptors. Increased synthesis of CD44 and/or generation of new isoforms is often associated with pathological conditions and CD44 expression can be altered by pro-inflammatory cytokines and chemokines such as TNF-α, IL-1β, IL-8 and RANTES in both lymphoid and non-lymphoid cells. There is increasing evidence to suggest that CD44 plays a pivotal role in autoimmune diseases and its expression is increased in synovial cells in patients with rheumatoid arthritis, which correlates with synovial inflammation [49]. The administration of antibodies against CD44 can significantly reduce inflammatory processes in murine models of collagen- or proteoglycan-induced arthritis and experimental autoimmune encephalomyelitis [50–52]. CD44-HA interactions in normal murine B cells have been shown to induce cell activation, proliferation and differentiation [53]. Readers are referred to reviews by Taylor and Gallo [17] and Jiang et al. [11], which discuss the role of CD44 and HA as immune regulators during pathological disorders.
3. Hyaluronan and CD44 in the Pathogenesis of SLE
Alterations in the distribution pattern of HA and CD44 have been shown to play an important role in the development of SLE. Elevated serum HA levels have been observed in patients and mice with active lupus nephritis, and murine anti-dsDNA antibodies have been shown to cross-react with HA [12, 54–56]. In the next section, we will discuss the contributing role of HA and CD44 in SLE with particular emphasis of their roles in the progression of lupus nephritis.
3.1. HA, CD44, and Immune Cells
An important step in the initiation and propagation of lupus nephritis is the recruitment of immune cells, namely T cells, B cells, macrophages, and dendritic cells, to sites of injury including the kidney [57–62]. Polyclonal B-cell activation precedes the development of clinical nephritis [63], thereby highlighting the crucial role of leukocytes in the pathogenesis of disease. The mechanism of local immune regulation and leukocyte-mediated kidney injury is not well delineated and is a topic of much interest. HA can induce chemokine and cytokine secretion in both lymphoid and nonlymphoid cells and therefore assumes an important role in the activation, recruitment, and retention of lymphocytes at sites of injury [26, 64–66]. We have demonstrated that in patients with active lupus nephritis subpopulations of glomerular lymphocytic infiltrates possess cell surface HA, a finding that is not observed in healthy individuals [12]. Although the mechanism through which HA regulates the activities of immune cells in the kidney during lupus nephritis remains to be defined, studies have shown that through its interaction with CD44, HA can induce murine B-cell activation, T cell, and macrophage effector functions and dendritic cell maturation [53, 67, 68]. Siegelman et al. demonstrated that CD44-HA interactions contributes to leukocyte rolling [69], a process that is essential for their extravasation to sites of injury. These researchers further observed that a subpopulation of circulating peripheral blood T cells strongly expressed CD44-dependent adhesion in SLE patients and their existence correlated with disease activity [70]. T cells that possess increased expression of CD44 have an enhanced capacity to infiltrate the kidney and induce inflammation [71], and this is dependent on the colocalization of CD44 with F-actin and phosphorylated ezrin, radixin, and moesin (ERM) at their polar caps, resulting in their polarization and conversion from freely circulating lymphocytes to those that can adhere to the endothelium and migrate into injured tissues, a process mediated through Rho-associated, coiled coil containing protein kinase (ROCK) activation (Figure 2) [71]. Genetic deletion of CD44 or inhibition of CD44 expression using a peptide based on the CDR1 sequence of a human anti-DNA antibody inhibited lymphoproliferation in lupus-prone mice and non-autoimmune mice immunized with a monoclonal anti-DNA antibody, respectively [72, 73], thereby highlighting the importance of CD44 in the pathogenesis of SLE. Crispín et al. [74] demonstrated that CD44v3 and CD44v6 expression are increased on CD4+ and CD8+ T cells isolated from patients with SLE, which correlated with disease activity, whereas CD44v6 on T cells was associated with lupus nephritis and positivity for anti-dsDNA antibodies [74].
Figure 2.
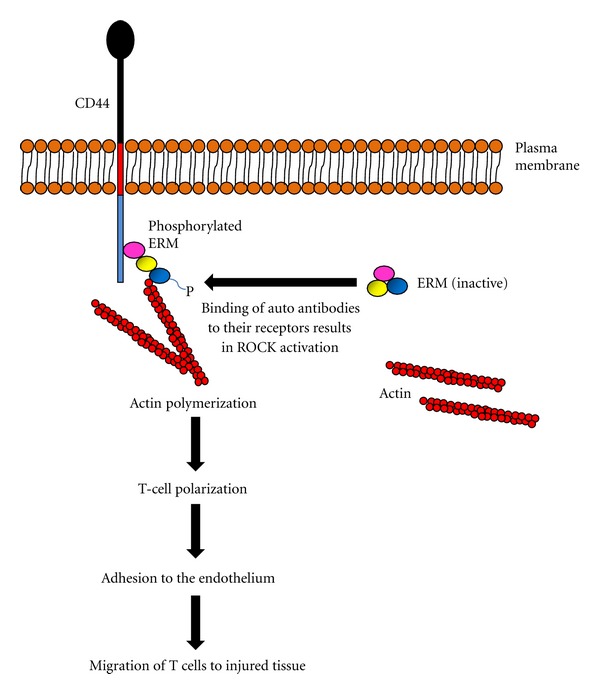
Schematic diagram showing the effect of ERM activation on T-cell function in SLE patients. Autoantibodies such as anti-CD3/T-cell receptor (TCR) antibodies bind to CD3/TCR complex in circulating T cells and induce ROCK activation, which in turn mediates ERM phosphorylation. Once activated, ERM directly interacts with CD44 and F actin resulting in their colocalization at the polar caps of T cells, leading to actin polymerization, T-cell polarization, adhesion to the endothelium and subsequent chemotactic migration to sites of injury in the kidney.
Apoptosis and the phagocytic clearance of apoptotic cells from sites of injury are tightly regulated processes that are essential for the maintenance of tissue structure and function. The recognition and removal of apoptotic bodies is mediated by macrophages. Studies have demonstrated that CD44 on the surface of macrophages plays an important role in the clearance of apoptotic bodies and this process is dependent on the prior activation of intracellular pathways such as tyrosine phosphorylation of p561ck and interaction with cytoskeletal proteins [75]. Defective clearance of apoptotic cells is a cardinal feature of SLE that results in persistent inflammation and autoimmunity, since chromatin fragments and cellular components that escape from nondigested apoptotic cells can serve as immunogens that will further exacerbate disease pathogenesis [75, 76]. Studies have demonstrated that the expression of variant CD44 isoforms is induced in activated macrophages that are present at sites of inflammation and this may alter the repertoire of CD44 ligands [77]. Furthermore, studies have demonstrated that CD44 expression is reduced on monocytes/macrophages in SLE patients, which inversely correlate with the percentage of apoptotic neutrophils [78]. Therefore, a reduction in CD44 expression together with a change in CD44 isoform on monocytes/macrophages will impair their ability to recognize and remove apoptotic cells from sites of injury. Although the mechanism through which CD44 expression is altered in SLE patients remains to be investigated, it is possible that changes in cytokine expression in the microenvironment may contribute.
Increased expression of interferon-inducible genes is a prominent feature in SLE. Recent analysis of the interferon pathway showed an association between CD44 and SLE [79]. In a recent study, CD44 has also been linked to thrombocytopenia in SLE patients [80–82].
3.2. HA, CD44, and Resident Renal Cells
In the normal kidney, HA is found solely in the medullary and papillary interstitium of the kidney where it contributes to the mechanical stability of tubules and blood vessels, and also in the concentration of urine, whilst the expression of CD44 is restricted to passenger leukocytes and resident macrophages [83–85]. Accumulation of HA in the renal cortex is observed in patients and mice with active lupus nephritis and in autoimmune crescentic glomerulonephritis [12, 86]. In vitro studies have demonstrated that mesangial cells, proximal tubular epithelial cells and interstitial fibroblasts are able to synthesize HA and it is likely that these cells all contribute to the synthesis of HA in renal diseases [12, 87–91]. We and others have demonstrated that HA and CD44 expression is increased in the glomerular and tubulo-interstitial compartments of the kidneys, with predominant expression of HA and CD44 in the periglomerular area and in atrophic tubules of patients and mice with active lupus nephritis [12, 54, 85, 92, 93]. The accumulation of HA in the kidney was shown to correlate with the infiltration of lymphocytes in the tubulo-interstitium and tissue damage [92]. In vitro studies have shown that proinflammatory mediators involved in the pathogenesis of lupus nephritis such as TNF-α and IFN-γ can increase HA synthesis in proximal tubular epithelial cells [92], and therefore may contribute to increased synthesis of HA in lupus patients.
We have previously demonstrated that human polyclonal anti-dsDNA antibodies can induce IL-1β, IL-6, and TNF-α in cultured human mesangial cells and proximal tubular epithelial cells [12, 94]. We further demonstrated that anti-dsDNA antibodies can induce HA synthesis in human mesangial cells and proximal tubular epithelial cells, with the production of both HMW and LMW HA, and this induction was dependent on increased synthesis of HAS II mRNA, and IL-1β and IL-6 secretion [12, 95]. Our observation that increased circulating HA levels in patients with lupus nephritis correlated with anti-dsDNA antibodies substantiates the likelihood that anti-dsDNA antibodies contribute to increased HA synthesis during pathogenesis of disease [12]. Considering that LMW HA possesses pro-inflammatory properties, that anti-dsDNA antibodies can induce LMW HA in resident renal cells may represent a pathogenic mechanism through which anti-dsDNA antibodies induce inflammatory processes in the kidney parenchyma during lupus nephritis.
Exogenous LMW, but not HMW HA, has been shown to induce de novo synthesis of MCP-1 mRNA and protein secretion in proximal tubular epithelial cells, and this induction was dependent on the interaction of HA with CD44 [64]. Intrarenal MCP-1 expression is increased in both the glomerular and tubulo-interstitial compartments of the kidney during lupus nephritis and precedes leukocyte infiltration, proteinuria, and renal damage [96]. The importance of MCP-1 in the pathogenesis of lupus nephritis is underscored by studies by Tesch et al. [97], which demonstrated that lupus-prone mice rendered genetically deficient in MCP-1 showed less severe renal histology and proteinuria [97]. Studies have also demonstrated that exogenous LMW HA can induce ICAM-1 and VCAM-1 in murine cortical tubular epithelial cells, suggesting that HA may play a role in the adhesion of leukocytes to resident renal cells [31]. We have demonstrated that inhibition of HA synthesis in NZBWF1/J mice is associated with an improvement in clinical parameters of disease and decreased intrarenal expression of IL-6 and TNF-α [54].
4. Conclusion
Despite its simple structure, HA is a multifaceted macromolecule that, depending on its molecular weight, is involved in tissue homeostasis and pathological processes. Through its interaction with CD44, HA regulates leukocyte infiltration, secretion of inflammatory mediators, and clearance of apoptotic cells processes that dictate the severity of lupus nephritis. Although studies have demonstrated that the interaction of HA with toll-like receptors can modulate inflammatory processes in animal models of bleomycin-induced lung injury, there is currently no data on the interaction of HA and toll-like receptors in the pathogenesis of lupus nephritis. Further research into the interaction of HA with other binding proteins will provide us with a better understanding of their roles in the pathophysiology of lupus nephritis and whether targeting HA or CD44 may serve as a novel therapeutic strategy.
Acknowledgments
This study was supported by the Hong Kong Research Grants Council General Research Fund (HKU 7812/08M), UGC Matching Grant Schemes (Phases IV and V), and the Estate of the late Mr. Chan Wing Hei. S. Yung is partially supported by the Wai Hung Charitable Foundation Limited and the Endowment Fund established for the Yu Chiu Kwong Professorship in Medicine awarded to T. M. Chan.
References
- 1.Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nature Reviews Rheumatology. 2010;6(12):683–692. doi: 10.1038/nrrheum.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.González DA, Díaz BB, Rodríguez Pérez MDC, Hernández AG, Chico BND, de León AC. Sex hormones and autoimmunity. Immunology Letters. 2010;133(1):6–13. doi: 10.1016/j.imlet.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Sarzi-Puttini P, Atzeni F, Iaccarino L, Doria A. Environment and systemic lupus erythematosus: an overview. Autoimmunity. 2005;38(7):465–472. doi: 10.1080/08916930500285394. [DOI] [PubMed] [Google Scholar]
- 4.Cameron JS. Lupus nephritis. Journal of the American Society of Nephrology. 1999;10(2):413–424. doi: 10.1681/ASN.V102413. [DOI] [PubMed] [Google Scholar]
- 5.Yung S, Cheung KF, Zhang Q, Chan TM. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. Journal of the American Society of Nephrology. 2010;21(11):1912–1927. doi: 10.1681/ASN.2009080805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostoslavsky G, Fischel R, Yachimovich N, et al. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. European Journal of Immunology. 2001;31(4):1221–1227. doi: 10.1002/1521-4141(200104)31:4<1221::aid-immu1221>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 7.Deocharan B, Qing X, Lichauco J, Putterman C. α-Actinin is a cross-reactive renal target for pathogenic anti-DNA aptibodies. Journal of Immunology. 2002;168(6):3072–3078. doi: 10.4049/jimmunol.168.6.3072. [DOI] [PubMed] [Google Scholar]
- 8.Kalaaji M, Fenton KA, Mortensen ES, et al. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kidney International. 2007;71(7):664–672. doi: 10.1038/sj.ki.5002133. [DOI] [PubMed] [Google Scholar]
- 9.Kalaaji M, Mortensen E, Jørgensen L, Olsen R, Rekvig OP. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. American Journal of Pathology. 2006;168(6):1779–1792. doi: 10.2353/ajpath.2006.051329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annual Review of Cell and Developmental Biology. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 11.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiological Reviews. 2011;91(1):221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yung S, Tsang RCW, Leung JKH, Chan TM. Increased mesangial cell hyaluronan expression in lupus nephritis is mediated by anti-DNA antibody-induced IL-1β . Kidney International. 2006;69(2):272–280. doi: 10.1038/sj.ki.5000042. [DOI] [PubMed] [Google Scholar]
- 13.Laurent TC, Fraser JRE. Hyaluronan. The FASEB Journal. 1992;6(7):2397–2404. [PubMed] [Google Scholar]
- 14.Itano N, Sawai T, Yoshida M, et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. The Journal of Biological Chemistry. 1999;274(35):25085–25092. doi: 10.1074/jbc.274.35.25085. [DOI] [PubMed] [Google Scholar]
- 15.Lee JY, Spicer AP. Hyaluronan: a multifunctional, megaDalton, stealth molecule. Current Opinion in Cell Biology. 2000;12(5):581–586. doi: 10.1016/s0955-0674(00)00135-6. [DOI] [PubMed] [Google Scholar]
- 16.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nature Reviews Cancer. 2004;4(7):528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 17.Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. The FASEB Journal. 2006;20(1):9–22. doi: 10.1096/fj.05-4682rev. [DOI] [PubMed] [Google Scholar]
- 18.Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information-rich system. European Journal of Cell Biology. 2006;85(8):699–715. doi: 10.1016/j.ejcb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Yung S, Thomas GJ, Davies M. Induction of hyaluronan metabolism after mechanical injury of human peritoneal mesothelial cells in vitro. Kidney International. 2000;58(5):1953–1962. doi: 10.1111/j.1523-1755.2000.00367.x. [DOI] [PubMed] [Google Scholar]
- 20.Yung S, Chan TM. Pathophysiology of the peritoneal membrane during peritoneal dialysis: the role of hyaluronan. Journal of Biomedicine and Biotechnology. 2011;2011:11 pages. doi: 10.1155/2011/180594.180594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagy N, Freudenberger T, Melchior-Becker A, et al. Inhibition of hyaluronan synthesis accelerates murine atherosclerosis: novel insights into the role of hyaluronan synthesis. Circulation. 2010;122(22):2313–2322. doi: 10.1161/CIRCULATIONAHA.110.972653. [DOI] [PubMed] [Google Scholar]
- 22.Wang A, Hascall VC. Hyaluronan structures synthesized by rat mesangial cells in response to hyperglycemia induce monocyte adhesion. The Journal of Biological Chemistry. 2004;279(11):10279–10285. doi: 10.1074/jbc.M312045200. [DOI] [PubMed] [Google Scholar]
- 23.Selbi W, de la Motte CA, Hascall VC, Day AJ, Bowen T, Phillips AO. Characterization of hyaluronan cable structure and function in renal proximal tubular epithelial cells. Kidney International. 2006;70(7):1287–1295. doi: 10.1038/sj.ki.5001760. [DOI] [PubMed] [Google Scholar]
- 24.Day AJ, de la Motte CA. Hyaluronan cross-linking: a protective mechanism in inflammation? Trends in Immunology. 2005;26(12):637–643. doi: 10.1016/j.it.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Noble PW. Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biology. 2002;21(1):25–29. doi: 10.1016/s0945-053x(01)00184-6. [DOI] [PubMed] [Google Scholar]
- 26.McKee CM, Penno MB, Cowman M, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages: the role of HA size and CD44. Journal of Clinical Investigation. 1996;98(10):2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKee CM, Lowenstein CJ, Horton MR, et al. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor κB-dependent mechanism. The Journal of Biological Chemistry. 1997;272(12):8013–8018. doi: 10.1074/jbc.272.12.8013. [DOI] [PubMed] [Google Scholar]
- 28.Haslinger B, Mandl-Weber S, Sellmayer A, Sitter T. Hyaluronan fragments induce the synthesis of MCP-1 and IL-8 in cultured human peritoneal mesothelial cells. Cell and Tissue Research. 2001;305(1):79–86. doi: 10.1007/s004410100409. [DOI] [PubMed] [Google Scholar]
- 29.Fieber C, Baumann P, Vallon R, et al. Hyaluronan-oligosaccharide-induced transcription of metalloproteases. Journal of Cell Science. 2004;117(2):359–367. doi: 10.1242/jcs.00831. [DOI] [PubMed] [Google Scholar]
- 30.Guo N, Li X, Mann MM, Funderburgh ML, Du Y, Funderburgh JL. Hyaluronan synthesis mediates the fibrotic response of keratocytes to transforming growth factor β . The Journal of Biological Chemistry. 2010;285(42):32012–32019. doi: 10.1074/jbc.M110.127183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oertli B, Beck-Schimmer B, Fan X, Wüthrich RP. Mechanisms of hyaluronan-induced up-regulation of ICAM-1 and VCAM-1 expression by murine kidney tubular epithelial cells: hyaluronan triggers cell adhesion molecule expression through a mechanism involving activation of nuclear factor-κB and activating protein-1. Journal of Immunology. 1998;161(7):3431–3437. [PubMed] [Google Scholar]
- 32.Ohno S, Im HJ, Knudson CB, Knudson W. Hyaluronan oligosaccharides induce matrix metalloproteinase 13 via transcriptional activation of NFκB and p38 MAP kinase in articular chondrocytes. The Journal of Biological Chemistry. 2006;281(26):17952–17960. doi: 10.1074/jbc.M602750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teder P, Vandivier RW, Jiang D, et al. Resolution of lung inflammation by CD44. Science. 2002;296(5565):155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein LA, Butcher EC. Identification of mRNA that encodes an alternative form of H-CAM(CD44) in lymphoid and nonlymphoid tissues. Immunogenetics. 1990;32(6):389–397. doi: 10.1007/BF00241632. [DOI] [PubMed] [Google Scholar]
- 35.Lesley J, Hyman R, Kincade PW. CD44 and its interaction with extracellular matrix. Advances in Immunology. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- 36.Greenfield B, Wang WC, Marquardt H, et al. Characterization of the heparan sulfate and chondroitin sulfate assembly sites in CD44. The Journal of Biological Chemistry. 1999;274(4):2511–2517. doi: 10.1074/jbc.274.4.2511. [DOI] [PubMed] [Google Scholar]
- 37.Jones M, Tussey L, Athanasou N, Jackson DG. Heparan sulfate proteoglycan isoforms of the CD44 hyaluronan receptor induced in human inflammatory macrophages can function as paracrine regulators of fibroblast growth factor action. The Journal of Biological Chemistry. 2000;275(11):7964–7974. doi: 10.1074/jbc.275.11.7964. [DOI] [PubMed] [Google Scholar]
- 38.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61(7):1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 39.Lesley J, Hascall VC, Tammi M, Hyman R. Hyaluronan binding by cell surface CD44. The Journal of Biological Chemistry. 2000;275(35):26967–26975. doi: 10.1074/jbc.M002527200. [DOI] [PubMed] [Google Scholar]
- 40.Lesley J, Hyman R. CD44 can be activated to function as an hyaluronic acid receptor in normal murine T cells. European Journal of Immunology. 1992;22(10):2719–2723. doi: 10.1002/eji.1830221036. [DOI] [PubMed] [Google Scholar]
- 41.Hathcock KS, Hirano H, Murakami S, Hodes RJ. CD44 expression on activated B cells: differential capacity for CD44- dependent binding to hyaluronic acid. Journal of Immunology. 1993;151(12):6712–6722. [PubMed] [Google Scholar]
- 42.Lesley J, English N, Perschl A, Gregoroff J, Hyman R. Variant cell lines selected for alterations in the function of the hyaluronan receptor CD44 show differences in glycosylation. Journal of Experimental Medicine. 1995;182(2):431–437. doi: 10.1084/jem.182.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lesley J, Howes N, Perschl A, Hyman R. Hyaluronan binding function of CD44 is transiently activated on T cells during an in vivo immune response. Journal of Experimental Medicine. 1994;180(1):383–387. doi: 10.1084/jem.180.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katoh S, Zheng Z, Oritani K, Shimozato T, Kincade PW. Glycosylation of CD44 negatively regulates its recognition of hyaluronan. Journal of Experimental Medicine. 1995;182(2):419–429. doi: 10.1084/jem.182.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Underhill C. CD44: the hyaluronan receptor. Journal of Cell Science. 1992;103(2):293–298. doi: 10.1242/jcs.103.2.293. [DOI] [PubMed] [Google Scholar]
- 46.Denning SM, Le PT, Singer KH, Haynes BF. Antibodies against the CD44 p80, lymphocyte homing receptor molecule augment human peripheral blood T cell activation. Journal of Immunology. 1990;144(1):7–15. [PubMed] [Google Scholar]
- 47.Barber DF, Bartolomé A, Hernandez C, et al. PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nature Medicine. 2005;11(9):933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 48.Yung S, Zhang Q, Zhang CZ, Chan KW, Lui L, Chan TM. Anti-DNA antibody induction of protein kinase C phosphorylation and fibronectin synthesis in human and murine lupus and the effect of mycophenolic acid. Arthritis and Rheumatism. 2009;60(7):2071–2082. doi: 10.1002/art.24573. [DOI] [PubMed] [Google Scholar]
- 49.Haynes BF, Hale LP, Patton KL, Martin ME, McCallum RM. Measurement of an adhesion molecule as an indicator of inflammatory disease activity: up-regulation of the receptor for hyaluronate (CD44) in rheumatoid arthritis. Arthritis and Rheumatism. 1991;34(11):1434–1443. doi: 10.1002/art.1780341115. [DOI] [PubMed] [Google Scholar]
- 50.Mikecz K, Brennan FR, Kim JH, Glant TT. Anti-CD44 treatment abrogates tissue oedema and leukocyte infiltration in murine arthritis. Nature Medicine. 1995;1(6):558–563. doi: 10.1038/nm0695-558. [DOI] [PubMed] [Google Scholar]
- 51.Nedvetzki S, Walmsley M, Alpert E, Williams RO, Feldmann M, Naor D. CD44 involvement in experimental collagen-induced arthritis (CIA) Journal of Autoimmunity. 1999;13(1):39–47. doi: 10.1006/jaut.1999.0294. [DOI] [PubMed] [Google Scholar]
- 52.Brocke S, Piercy C, Steinman L, Weissman IL, Veromaa T. Antibodies to CD44 and integrin α4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(12):6896–6901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rafi A, Nagarkatti M, Nagarkatti PS. Hyaluronate-CD44 interactions can induce murine B-cell activation. Blood. 1997;89(8):2901–2908. [PubMed] [Google Scholar]
- 54.Chan TM, Tse WW, Chau M, Yung S. Suppression of hyaluronan synthesis with 4-methylumbelliferone in NZB/W F1 mice is associated with reduced renal inflammation and renal function improvement. Journal of the American Society of Nephrology. 2011;22, article 134A [Google Scholar]
- 55.Hansen C, Otto E, Kuhlemann K, Förster G, Kahaly GJ. Glycosaminoglycans in autoimmunity. Clinical and Experimental Rheumatology. 1996;14(supplement 15):S59–S67. [PubMed] [Google Scholar]
- 56.Faaber P, Capel J, Rijke G, Vierwinden G, van de Putte LB, Koene RA. Cross-reactivity of anti-DNA antibodies with proteoglycans. Clinical and Experimental Immunology. 1984;55(3):502–508. [PMC free article] [PubMed] [Google Scholar]
- 57.Klinman DM, Eisenberg RA, Steinberg AD. Development of the autoimmune B cell repertoire in MRL-lpr/lpr mice. Journal of Immunology. 1990;144(2):506–511. [PubMed] [Google Scholar]
- 58.Moreth K, Brodbeck R, Babelova A, et al. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. Journal of Clinical Investigation. 2010;120(12):4251–4272. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. Journal of Clinical Investigation. 2005;115(2):407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bagavant H, Deshmukh US, Wang H, Ly T, Shu MF. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. Journal of Immunology. 2006;177(11):8258–8265. doi: 10.4049/jimmunol.177.11.8258. [DOI] [PubMed] [Google Scholar]
- 61.Chang A, Henderson SG, Brandt D, et al. In situ B Cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. Journal of Immunology. 2011;186(3):1849–1860. doi: 10.4049/jimmunol.1001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schiffer L, Bethunaickan R, Ramanujam M, et al. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. Journal of Immunology. 2008;180(3):1938–1947. doi: 10.4049/jimmunol.180.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klinman DM. Polyclonal B cell activation in lupus-prone mice precedes and predicts the development of autoimmune disease. Journal of Clinical Investigation. 1990;86(4):1249–1254. doi: 10.1172/JCI114831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beck-Schimmer B, Oertli B, Pasch T, Wüthrich RP. Hyaluronan induces monocyte chemoattractant protein-1 expression in renal tubular epithelial cells. Journal of the American Society of Nephrology. 1998;9(12):2283–2290. doi: 10.1681/ASN.V9122283. [DOI] [PubMed] [Google Scholar]
- 65.Hodge-Dufour J, Noble PW, Norton MR, et al. Induction of IL-12 and chemokines by hyaluronan requires adhesion-dependent priming of resident but not elicited macrophages. Journal of Immunology. 1997;159(5):2492–2500. [PubMed] [Google Scholar]
- 66.Horton MR, McKee CM, Bao C, et al. Hyaluronan fragments synergize with interferon-γ to induce the C-X-C chemokines mig and interferon-inducible protein-10 in mouse macrophages. The Journal of Biological Chemistry. 1998;273(52):35088–35094. doi: 10.1074/jbc.273.52.35088. [DOI] [PubMed] [Google Scholar]
- 67.Galandrini R, Albi N, Tripodi G, et al. Antibodies to CD44 trigger effector functions of human T cell clones. Journal of Immunology. 1993;150(10):4225–4235. [PubMed] [Google Scholar]
- 68.Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. Journal of Experimental Medicine. 2002;195(1):99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siegelman MH, DeGrendele HC, Estess P. Activation and interaction of CD44 and hyaluronan in immunological systems. Journal of Leukocyte Biology. 1999;66(2):315–321. doi: 10.1002/jlb.66.2.315. [DOI] [PubMed] [Google Scholar]
- 70.Estess P, DeGrendele HC, Pascual V, Siegelman MH. Functional activation of lymphocyte CD44 in peripheral blood is a marker of autoimmune disease activity. Journal of Clinical Investigation. 1998;102(6):1173–1182. doi: 10.1172/JCI4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y, Harada T, Juang YT, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. Journal of Immunology. 2007;178(3):1938–1947. doi: 10.4049/jimmunol.178.3.1938. [DOI] [PubMed] [Google Scholar]
- 72.Weber GF. The absence of CD44 ameliorates Faslpr/lpr disease. Autoimmunity. 2004;37(1):1–8. doi: 10.1080/08916930310001630316. [DOI] [PubMed] [Google Scholar]
- 73.Sela U, Mauermann N, Hershkoviz R, et al. The inhibition of autoreactive T cell functions by a peptide based on the CDR1 of an anti-DNA autoantibody is via TGF-β-mediated suppression of LFA-1 and CD44 expression and function. Journal of Immunology. 2005;175(11):7255–7263. doi: 10.4049/jimmunol.175.11.7255. [DOI] [PubMed] [Google Scholar]
- 74.Crispín JC, Keenan BT, Finnell MD, et al. Expression of CD44 variant isoforms CD44v3 and CD44v6 is increased on T cells from patients with systemic lupus erythematosus and is correlated with disease activity. Arthritis and Rheumatism. 2010;62(5):1431–1437. doi: 10.1002/art.27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nature Reviews Immunology. 2002;2(12):965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 76.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis and Rheumatism. 1998;41(7):1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 77.Levesque MC, Haynes BF. In vitro culture of human peripheral blood monocytes induces hyaluronan binding and up-regulates monocyte variant CD44 isoform expression. Journal of Immunology. 1996;156(4):1557–1565. [PubMed] [Google Scholar]
- 78.Cairns AP, Crockard AD, McConnell JR, Courtney PA, Bell AL. Reduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: relations with apoptotic neutrophils and disease activity. Annals of the Rheumatic Diseases. 2001;60(10):950–955. doi: 10.1136/ard.60.10.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramos PS, Williams AH, Ziegler JT, et al. Genetic analyses of interferon pathway-related genes reveal multiple new loci associated with systemic lupus erythematosus. Arthritis and Rheumatism. 2011;63(7):2049–2057. doi: 10.1002/art.30356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaufman KM, Rankin J, Harley IT, Kelly JA, Harley JB, Scofield RH. A genetic marker within the CD44 gene confirms linkage at 11p13 in African-American families with lupus stratified by thrombocytopenia, but genetic association with CD44 is not present. Genes and Immunity. 2002;3(1):S86–S88. doi: 10.1038/sj.gene.6363887. [DOI] [PubMed] [Google Scholar]
- 81.Scofield RH, Bruner GR, Kelly JA, et al. Thrombocytopenia identifies a severe familial phenotype of systemic lupus erythematosus and reveals genetic linkages at 1q22 and 11p13. Blood. 2003;101(3):992–997. doi: 10.1182/blood-2002-04-1003. [DOI] [PubMed] [Google Scholar]
- 82.Lessard CJ, Adrianto I, Kelly JA, et al. Identification of a systemic lupus erythematosus susceptibility locus at 11p13 between PDHX and CD44 in a multiethnic study. American Journal of Human Genetics. 2011;88(1):83–91. doi: 10.1016/j.ajhg.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hansell P, Goransson V, Odlind C, Gerdin B, Hallgren R. Hyaluronan content in the kidney in different states of body hydration. Kidney International. 2000;58(5):2061–2068. doi: 10.1111/j.1523-1755.2000.00378.x. [DOI] [PubMed] [Google Scholar]
- 84.Roy-Chaudhury P, Khong TF, Williams JH, et al. CD44 in glomerulonephritis: expression in human renal biopsies, the Thy 1.1 model, and by cultured mesangial cells. Kidney International. 1996;50(1):272–281. doi: 10.1038/ki.1996.312. [DOI] [PubMed] [Google Scholar]
- 85.Benz PS, Fan X, Wüthrich RP. Enhanced tubular epithelial CD44 expression in MRL-lpr lupus nephritis. Kidney International. 1996;50(1):156–163. doi: 10.1038/ki.1996.298. [DOI] [PubMed] [Google Scholar]
- 86.Jun Z, Hill PA, Lan HY, et al. CD44 and hyaluronan expression in the development of experimental crescentic glomerulonephritis. Clinical and Experimental Immunology. 1997;108(1):69–77. doi: 10.1046/j.1365-2249.1997.d01-977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ren J, Hascall VC, Wang A. Cyclin D3 mediates synthesis of a hyaluronan matrix that is adhesive for monocytes in mesangial cells stimulated to divide in hyperglycemic medium. The Journal of Biological Chemistry. 2009;284(24):16621–16632. doi: 10.1074/jbc.M806430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jones S, Phillips AO. Regulation of renal proximal tubular epithelial cell hyaluronan generation: implications for diabetic nephropathy. Kidney International. 2001;59(5):1739–1749. doi: 10.1046/j.1523-1755.2001.0590051739.x. [DOI] [PubMed] [Google Scholar]
- 89.Michael DR, Phillips AO, Krupa A, et al. The human hyaluronan synthase 2 (HAS2) gene and its natural antisense RNA exhibit coordinated expression in the renal proximal tubular epithelial cell. The Journal of Biological Chemistry. 2011;286(22):19523–19532. doi: 10.1074/jbc.M111.233916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pedagogos E, Hewitson TD, Nicholls KM, Becker GJ. Hyaluronan and rat renal fibroblasts: in vitro studies. Nephron. 2001;88(4):347–353. doi: 10.1159/000046019. [DOI] [PubMed] [Google Scholar]
- 91.Takeda M, Babazono T, Nitta K, Iwamoto Y. High glucose stimulates hyaluronan production by renal interstitial fibroblasts through the protein kinase C and transforming growth factor-β cascade. Metabolism. 2001;50(7):789–794. doi: 10.1053/meta.2001.24207. [DOI] [PubMed] [Google Scholar]
- 92.Feusi E, Sun L, Sibalic A, Beck-Schimmer B, Oertli B, Wüthrich RP. Enhanced hyaluronan synthesis in the MRL-Fas(lpr) kidney: role of cytokines. Nephron. 1999;83(1):66–73. doi: 10.1159/000045475. [DOI] [PubMed] [Google Scholar]
- 93.Daniel L, Sichez H, Giorgi R, et al. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney International. 2001;60(6):2215–2221. doi: 10.1046/j.1523-1755.2001.00055.x. [DOI] [PubMed] [Google Scholar]
- 94.Yung S, Tsang RCW, Sun Y, Leung JKH, Chan TM. Effect of human anti-DNA antibodies on proximal renal tubular epithelial cell cytokine expression: implications on tubulointerstitial inflammation in lupus nephritis. Journal of the American Society of Nephrology. 2005;16(11):3281–3294. doi: 10.1681/ASN.2004110917. [DOI] [PubMed] [Google Scholar]
- 95.Chan TM, Tsang RCW, Chan KW, Yung S. Anti-DNA antibodies stimulate hyaluronan synthesis in proximal tubular epithelial cells through the induction of IL-6 and IL-1β . Journal of the American Society of Nephrology. 2006;17, article 352A [Google Scholar]
- 96.Pérez de Lema G, Maier H, Nieto E, et al. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. Journal of the American Society of Nephrology. 2001;12(7):1369–1382. doi: 10.1681/ASN.V1271369. [DOI] [PubMed] [Google Scholar]
- 97.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. Journal of Experimental Medicine. 1999;190(12):1813–1824. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]