

Abstract
Fibroblast growth factors (FGFs) orchestrate a variety of cellular functions by binding to their transmembrane tyrosine-kinase receptors (FGFRs) and activating downstream signalling pathways, including RAS/MAPK, PLCγ1, PI3K, and STATs. In the last ten years, it has become clear that FGF signalling is altered in a high proportion of bladder tumours. Activating mutations and/or overexpression of FGFR3 are common in urothelial tumours with low malignant potential and low-stage and -grade urothelial carcinomas (UCs) and are associated with a lower risk of progression and better survival in some subgroups. FGFR1 is not mutated in UC, but overexpression is frequent in all grades and stages and recent data indicate a role in urothelial epithelial-mesenchymal transition. In vitro and in vivo studies have shown that FGFR inhibition has cytotoxic and/or cytostatic effects in FGFR-dependent bladder cancer cells and FGFR-targeted agents are currently being investigated in clinical studies for the treatment of UC. Urine-based tests detecting common FGFR3 mutations are also under development for surveillance of low-grade and -stage tumours and for general population screening. Overall, FGFRs hold promise as therapeutic targets, diagnostic and prognostic markers, and screening tools for early detection and clinical management of UC.
1. Urothelial Carcinoma: Clinical Management and Challenges
Bladder cancer is a common malignancy with over 70,000 estimated new cases and 14,000 deaths per year in the USA alone [1]. In western countries, around 90% of bladder tumours are transitional cell carcinoma, with rare cases of squamous cell carcinoma and adenocarcinoma [2]. Bladder tumours are classified using the TNM classification system [3] according to their invasiveness (stage Ta: confined to the urothelium; T1: invading the lamina propria; T2: invading the muscular layer; T3: invading the submuscular layers; T4: disseminating to other organs) and their differentiation state (1973 WHO grading system: grade 1, 2, or 3 [4]; 2004 WHO grading system: PUNLMP: papillary urothelial neoplasm of low-malignant potential, low grade: well-differentiated neoplasms, high grade: poorly differentiated neoplasms [2]). At presentation, the vast majority of urothelial carcinomas (UC) (~70%) are low-grade superficial papillary tumours with a relatively benign prognosis. Their conventional treatment involves surgical resection and intravesical chemo- or immunotherapy [5]. One of the major challenges in the management of these tumours is their propensity to recur, therefore requiring frequent and often life-long surveillance with cystoscopy and urine cytology. This, coupled with a relatively long life expectancy (5-year survival rate >90%), makes superficial bladder cancer the most expensive and time-consuming malignancy to treat [6, 7]. A minority of superficial tumours (~15%) will eventually progress to become invasive. Despite treatment with radical cystectomy, radiotherapy, and adjuvant or neoadjuvant chemotherapy, newly diagnosed invasive bladder tumours and superficial tumours that have progressed to invasion often metastasize and the 5-year survival rate is poor (<40%) [8].
Currently there are no validated prognostic molecular biomarkers to guide the clinical management of UC. Crucial therapeutic decisions are based on risk tables that include tumour size and number, and previous history [9], in addition to histopathological criteria, which are often limited by inter- and intraobserver variability and have relatively low reproducibility [10, 11]. Overall, UC management would greatly benefit from rapid cost-effective and noninvasive methods for screening and surveillance, and reproducible and objective molecular biomarkers to predict the risks of recurrence and progression so that more aggressive therapeutic regimes and intensive monitoring could be focussed on patients at higher risk. Furthermore, novel therapeutic approaches and related predictive biomarkers are needed, for use alone or in combination with conventional treatment, to reduce recurrence rate and progression of superficial tumours and prolong survival and quality of life in patients with invasive and metastatic tumours.
2. Structure and Function of Fibroblast Growth Factor Receptors
In humans, fibroblast growth factors are a family comprising 18 growth factors (FGFs) and 4 FGF-homologous factors (FHFs), many of which play a crucial role during both normal physiological processes, such as embryogenesis, development, and wound healing, and a range of pathological conditions [12–14]. The effects of FGFs are mediated by a family of four fibroblast growth factor receptors (FGFR1–4). FGFRs are transmembrane glycoproteins with a conserved structure comprising an extracellular portion with two to three immunoglobulin-like domains (IgI–III), a transmembrane domain, and an intracellular split tyrosine-kinase domain. IgI and IgII are separated by a short negatively charged serine-rich segment, termed the “acid box”, followed by a heparin-binding domain with high affinity for heparan sulphate proteoglycans (HSPGs) [12, 13]. IgI and the acid box are thought to have an auto-inhibitory function [15], while IgII and IgIII bind to FGFs in association with HSPGs. FGF binding to the monomeric receptor triggers its dimerization and subsequent transphosphorylation of tyrosine residues in the kinase domain. This initiates a phosphorylation cascade involving a number of docking proteins, resulting in signalling through various downstream pathways, including PLCγ1, RAS-MAPK, and PI3K and STATs [16]. These pathways regulate a variety of cellular functions, including proliferation, migration, and differentiation [16].
Affinity for specific FGFs varies between receptors [17, 18] and a further layer of complexity is added by the fact that FGFRs are subject to alternative splicing, generating isoforms with different ligand-binding specificity in different cell lineages. For example, two alternative isoforms of FGFR3, denoted “b” and “c” are produced by mutually exclusive splicing of exon 8 and exon 9, affecting IgIII [19]. FGFR3b is expressed in epithelial cells and has high affinity only for FGF1. FGFR3c is expressed in cells of mesenchymal origin and has affinity for FGF1, FGF2, FGF4, and other FGFs [17, 19]. Similarly, an alternative FGFR1 isoform, denoted FGFR1β, lacking the IgI domain and with increased affinity to FGF1 and heparin compared to FGFR1α, has been described [20]. Secreted isoforms of FGFRs have also been reported [21, 22].
A fifth FGF receptor has been described [23]. FGFR5 is homologous to the other four receptors in the extracellular portion, but lacks the tyrosine-kinase domain, which is replaced by a short histidine-rich sequence. FGFR5 is therefore regarded as a decoy receptor, which can inhibit signalling by binding and sequestering FGFs [24].
3. Aberrant FGF Signalling in Urothelial Malignancies
3.1. FGFR3 Alterations in Urothelial Tumours
3.1.1. Activating Mutations
Somatic activating mutations of FGFR3 were first described in UC over ten years ago [25, 26]. Subsequent larger studies established that FGFR3 mutations occur in around 50% of both lower and upper urinary tract tumours and these cluster in three distinct hotspots in exons 7, 10, and 15 [27–32] (Figure 1). The most common mutations in exon 7 and 10 are S249C (~61%), Y375C (~19%), R248C (~8%), and G372C (~6%), with others occurring at very low frequencies (<2%). Mutations in exon 7 and 10 create a cysteine or glutamic acid residue in the proximal extracellular region of the receptor. The abnormal residues form either disulfide or hydrogen bonds between adjacent monomer receptors, favouring ligand-independent dimerization, transactivation, and signalling [33–35]. Mutations in exon 15 are rarer, with a frequency of around 2%, and they all involve the lysine residue at position 652, which is mutated to glutamic acid, glutamine, threonine or methionine. They are thought to induce a conformational change in the kinase domain resulting in ligand-independent receptor activation and signalling [36]. They have also been shown to alter FGFR3 cellular localization, inducing aberrant signalling from the endoplasmic reticulum [37].
Figure 1.
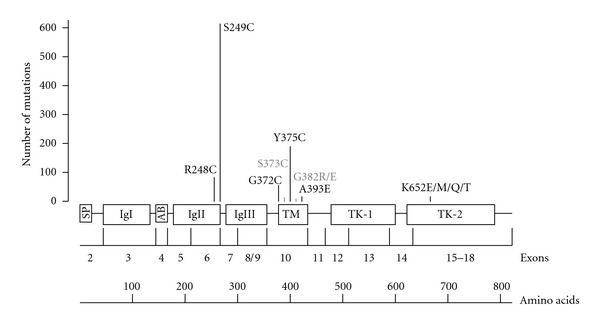
Schematic representation of human FGFR3 protein and corresponding FGFR3 coding exons. Exon numbering based on Tomlinson et al. [38]. Type and total number of reported mutations are based on data pooled from 11 studies [25–29, 31, 32, 39–42], including a total of 1898 bladder tumours. SP: signal peptide; IgI–III: immunoglobulin-like domain; AB: acid box; TM: transmembrane domain; TK; tyrosine-kinase domain.
FGFR3 mutations are frequent in benign skin tumours [43] and have been reported at low frequency in cervical carcinoma [25] and multiple myeloma [44], but are absent in other solid cancers [45, 46], suggesting a tissue-specific role. Interestingly, the relative frequency of different FGFR3 mutations is dependent on the tumour type, with multiple myeloma mostly showing changes in the tyrosine-kinase domain [44], and bladder and cervical tumours mainly exhibiting mutations of the extracellular region. Furthermore, while S249C is by far the most frequent mutation in bladder (Figure 1) and cervical tumours [25], mutation of the adjacent codon (R248C) is the commonest change found in benign skin tumours [43]. It is currently unclear whether the spectrum, frequency and tissue specificity of FGFR3 mutations is determined by exposure to specific carcinogens or by their functional significance. The role of smoking and occupational exposure to polycyclic aromatic hydrocarbons in determining the frequency or the type of FGFR3 mutations in UC has been excluded [47, 48]. However, the limited range of hotspot codons in the receptor makes this a difficult target to study from the epidemiological viewpoint and the possibility of small influences of these exposures cannot be excluded without much larger studies. We have recently shown a correlation between the level of ligand independence, signalling activation, and phenotypic consequences of different FGFR3 mutations expressed in normal urothelial cells and their frequency in UC, suggesting that the spectrum of FGFR3 mutations in bladder tumours may relate to selection for their potency [49]. We also highlighted cell-type-dependent phenotypic and signalling consequences of specific FGFR3 mutations which may explain the differences in the relative frequencies between tumour types [49].
During urothelial transformation, FGFR3 mutations are thought to occur early, as they are reported in flat urothelial hyperplasia, a preneoplastic lesion [50]. Furthermore, FGFR3 mutations are extremely common in the most benign bladder lesions (low malignant potential neoplasms and urothelial papillomas) and low-grade and -stage tumours (PUNLMP; TaG1), reaching frequencies over 80% in these subgroups [27, 28, 39]. This evidence points to an overall “benign” effect of FGFR3 mutation in the bladder. Interestingly, all somatic mutations reported so far in UC have been previously described as germline mutations in skeletal dysplasia syndromes, due to the important role of FGFR3 in regulating chondrocyte proliferation and differentiation [51].
3.1.2. Overexpression and Alternative Splicing
Overexpression of wild-type FGFR3 due to t(4; 14) translocation, which places FGFR3 in the proximity of the regulatory region of the IgH locus, is common in multiple myeloma [44]. Such rearrangements have not been described in bladder cancer. However several reports have examined FGFR3 protein expression in bladder carcinomas, describing an increase in a high proportion of tumours, particularly in the low-grade and low-stage subgroups [32, 40, 52–54]. Two recent investigations have examined the correlation between mutation status and protein expression, showing that up to 85% of the mutated tumours also have increased protein levels [32, 40]. Overexpression of FGFR3 was also detected in around 40% of wild-type tumours, and this was more common in invasive cases. Overall, around 80% of non-invasive and 54% of invasive UC have dysregulated FGFR3 either through mutation, overexpression or both [32]. Therefore, FGFR3 plays a key role in both superficial and invasive disease. However, while superficial tumours tend to exhibit activating mutations of FGFR3, often accompanied by protein upregulation, invasive tumours more commonly show upregulation of wild-type FGFR3. At this stage, it is not clear whether this difference reflects differential downstream signalling consequences of wild-type and mutant receptors or the different molecular pathways through which these tumours develop. The molecular mechanisms driving FGFR3 protein overexpression in UC are also still largely unknown, although a recent study has shown that FGFR3 expression in urothelial cells is regulated by two microRNAs (miR-99a/100), which are often downregulated in UC, particularly in low-grade and low-stage tumours [55].
Overexpressed FGFR3 could contribute to tumour development by either ligand-dependent or independent mechanisms. FGF levels are often increased in urine and tumour tissue of bladder cancer patients [56, 57]. Parallel overexpression of FGFs and FGFRs could therefore result in upregulated FGF signalling. It is also speculated that overexpression of the wild-type receptor may favour ligand-independent dimerization and signalling due to the close physical proximity of FGFR3 monomers on the cell surface. Overexpression of FGFR3 would be particularly deleterious if accompanied by a switch to alternative isoforms with different FGF affinity profiles, which would allow tumour cells to activate FGF signalling in response to a greater number of substrates. A switch from the epithelial FGFR3b to the mesenchymal FGFR3c isoform, with broader ligand affinity has been described in bladder cancer cell lines [38]. However, as FGFR3c was not detected in a panel of 76 bladder carcinoma [25], the role of FGFR3 isoform switching in UC in vivo is still unclear. The different mechanisms of FGFR3 abnormal activation in bladder cancer are summarized in Figure 2.
Figure 2.
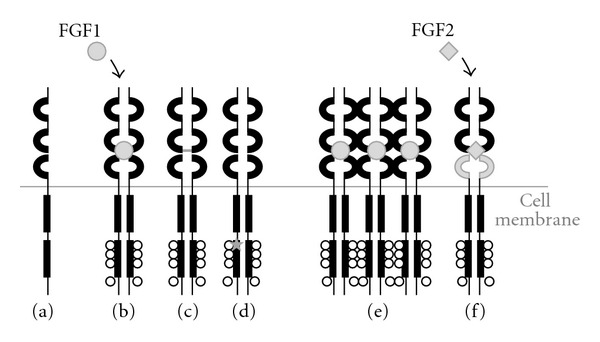
Mechanisms of physiological (a)-(b) and pathological (c)–(f) activation of FGFR3. (a) Monomeric inactive receptor; (b) Ligand-dependent dimerization and activation; (c) Ligand-independent dimerization and activation induced by mutation of the extracellular portion; (d) Ligand-independent activation due to mutations of the tyrosine-kinase domain; (e) Upregulation of signalling due to receptor overexpression; (f) Alteration of splicing favouring isoforms with broader ligand specificity.
3.2. FGFR1 Alterations in Urothelial Tumours
In many malignancies FGFR1 has been implicated as an oncogene whose expression or genetic arrangement is altered compared to normal tissue [58–63]. In mouse models of prostate and breast carcinoma, FGFR1 activation via an inducible regulation system accelerated progression to malignancy [64–67]. Furthermore, FGFR1 signaling was shown to contribute to the survival of a breast cancer cell line, indicating FGFR1 as potential therapeutic target [63]. More, recently it has been shown that FGFR1 is overexpressed in bladder cancer [68]. Interestingly, FGFR1 expression was increased in both noninvasive and invasive tumours. In light of the changes in FGFR3 splicing observed in UC cell lines [38], FGFR1 splicing was examined. This revealed an altered ratio of FGFR1 α and β splice variants, with increased expression of the β isoform, lacking the IgI domain. The increased expression of this splice variant was significantly associated with tumour stage and grade and caused an increased sensitivity to FGF1 and enhanced downstream signalling [69]. Overall these studies demonstrate that FGFR1, via overexpression or altered splicing, may play a key role during bladder tumour development and/or progression.
3.3. Other FGFRs
In contrast to FGFR3 and FGFR1, FGFR2 appears to have a protective or tumour-suppressor role in bladder cancer. Its expression is downregulated in UC and low levels are associated with worse prognosis [70]. Furthermore, FGFR2 re-expression in a UC cell line was associated with reduced proliferation in vitro and diminished tumorigenicity in nude mice [71]. No evidence is available regarding FGFR4 and FGFR5 in UC.
3.4. Phenotypic Consequences of Upregulated FGF Signalling in Urothelial Cells
Few studies have investigated the effects of FGFR dysregulation in normal and malignant urothelial cells. Knockdown or inhibition of FGFR3 signalling in the FGFR3-mutant UC cell lines MGHU3 (Y375C), 97-7 (S249C) and UMUC14 (S249C) is accompanied by diminished cell proliferation and/or anchorage dependent growth in vitro in all, although with different efficacy [72–75]. Tumorigenic potential in vivo is also reduced [72, 74]. Similar effects were seen in UC cell lines RT112 and RT4, which overexpress FGFR3 with no detectable point mutations [74]. These results show that some UC, both FGFR3-mutant and wild-type, have “oncogene addiction” to FGFR3. In contrast, knockdown of FGFR1 in the FGFR1-overexpressing invasive UC cell line UMUC3 did not affect proliferation in vitro despite a clear effect on anchorage independent growth and tumorigenicity in vivo [68].
Our group has recently begun to elucidate the specific phenotypic differences between FGFR1 and FGFR3 activation in urothelial cells. When mutant FGFR3 was overexpressed in normal urothelial cells, subtle phenotypic changes were observed. The cells had a higher proliferative rate and reduced apoptosis only in confluent cultures, suggesting that activation of FGFR3 signalling may assist premalignant urothelial cells in overcoming cell-cell contact inhibition and favour the formation of hyperplastic bladder lesions [49]. These phenotypes are compatible with the hypothesis that FGFR3 mutation contributes early in the process of tumour development. FGFR1 overexpression and activation, in contrast, has a more profound effect on proliferation and survival of normal urothelial cells, even in subconfluent culture conditions [68]. As expected, neither mutant FGFR3 or upregulated FGFR1 was sufficient alone to confer on normal urothelial cells a fully transformed phenotype, such as anchorage-independent growth or the ability to form tumours in nude mice [49, 68].
Our recent data, however, show that activation of overexpressed FGFR1 in bladder cancer cell lines is sufficient to induce an epithelial mesenchymal transition (EMT) [76]. EMT developed over a period of 72 hours. Initially a rapid increase in actin stress fibres occurred, followed by an increase in cell size, altered morphology and increased migration and invasion. By using site-directed mutagenesis and small molecule inhibitors, it was shown that combined activation of the mitogen-activated protein kinase (MAPK) and phospholipase C gamma (PLCγ) pathways regulated this EMT. Expression array analysis identified COX-2 as a major upregulated transcript following FGFR1 activation and this led to increased intracellular prostaglandin E2 levels, which promoted migration. This suggests that the timing and cellular context of FGFR1 dysregulation may be crucial in determining its phenotypic consequences and may influence the development of either superficial or invasive bladder tumours.
Interestingly, despite driving different phenotypes, the signalling pathways activated by FGFR1 and FGFR3 in both normal and malignant cells are similar and involve FRS2, PLCγ1 and ERK1/2 [49, 68]. These observations imply that context-specific downstream effectors of these signalling pathways and interaction with other molecular events need to be elucidated to fully understand the observed phenotypic differences.
4. Clinical Applications
The potential applications of FGFRs in the early diagnosis and clinical management of bladder cancer are summarized in Figure 3.
Figure 3.
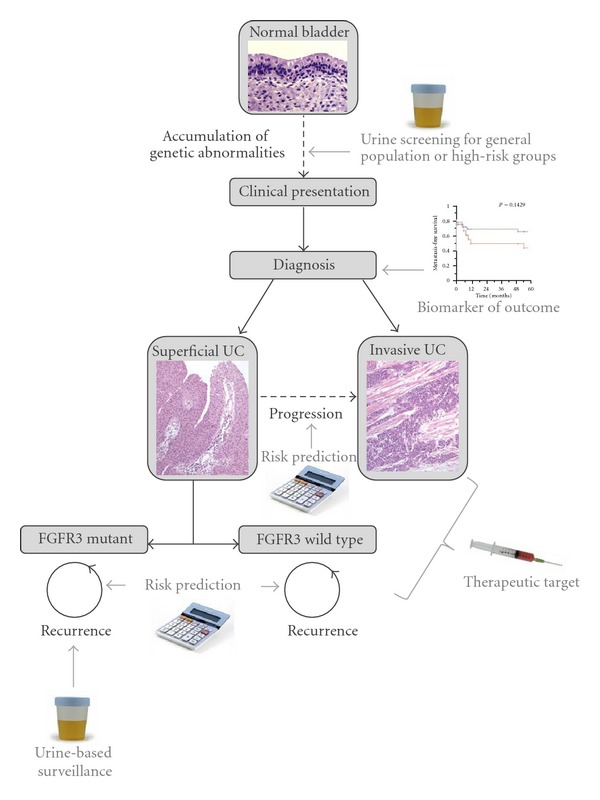
Potential applications of FGFRs in the early detection and clinical management of bladder tumours.
4.1. Surveillance and Screening
As mutations of FGFR3 are found in up to 80% of primary Ta tumours, which are characterized by a high recurrence rate, detection of FGFR3 mutations in urine is currently under study as a noninvasive and inexpensive method for the surveillance of superficial FGFR3 mutation-positive bladder tumours. A test has been developed to detect eleven common FGFR3 mutations by multiplex polymerase chain reaction amplification of the three hotspot regions followed by SNaPshot mutation analysis [77]. When applied to urine samples pooled within a 24-hr period, this test is able to detect all mutant tumours irrespective of their size [78]. However, overall sensitivity is around 80%, as it is limited by the fact that around one-fifth of patients with an FGFR3-mutant primary tumour have FGFR3 wild-type recurrences [78, 79].
Detection of FGFR3 mutations in urine could also be employed for general population screening aimed at early detection of primary tumours. Preliminary results show that a combined test for mutation of FGFR3, PIK3CA and RAS could potentially detect 75% of primary tumours, including 88% of the pTa-T1G1-2 tumours but only 36% of the high-grade and -stage malignancies [79]. Addition of other markers is being considered to improve detection of invasive tumours [80, 81].
4.2. Prognosis
FGFR3 mutation status has been investigated as a prognostic marker for recurrence, progression, and survival. A small study including 53 pTaG1-2 tumours showed that wild-type FGFR3 is predictive of disease recurrence [41]. In contrast, in a subsequent larger study of 764 superficial tumours FGFR3 mutation was predictive of a higher rate of recurrence in TaG1 but not TaG3 or T1 tumours [28]. In this investigation, TaG2 tumours showed a trend towards a higher recurrence rate but did not reach statistical significance. Whilst an association between FGFR3 mutation and risk of progression was not detected in this cohort, where progression rate was small [28], other studies suggested a negative correlation [30, 31, 82]. An international prospective study including 221 superficial tumours indicated that FGFR3 status is not associated with recurrence but is predictive of disease progression in some subgroups (pT1 and high-grade malignancies) [30]. Inverse correlation between FGFR3 mutation and progression in pT1 tumours was also confirmed in a subsequent investigation [83]. A multicentre study comprising 230 superficial tumours suggested that adding FGFR3 mutation status and Ki-67 positivity to current histopathological criteria improved prediction of progression in about 7% of patients [84]. Furthermore, better survival rates were suggested for patients with muscle invasive tumours harbouring an FGFR3 mutation [31]. In a recent study, FGFR3 mutation status was found to be predictive of progression, recurrence, and outcome only when combined with 9p22 LOH status [85], but this was in a relatively small sample set, including only 29 FGFR3-mutant tumours. Overall, further research is needed to confirm the utility of FGFR3-mutation status as molecular marker for patient stratification alongside current prognostic criteria.
4.3. FGFR-Targeted Therapy in Bladder Cancer
As FGFR3 and FGFR1 are altered in the majority of superficial tumours and in a good proportion of invasive tumours, they represent very inviting therapeutic targets. As discussed in paragraph 3.4, in vitro and in vivo studies using siRNA or shRNA knockdown or specific antibodies to block FGFRs activity have shown that some UC cell lines are FGFR3-dependent.
A number of FGFR-targeted therapeutic agents have been tested in bladder cancer cell lines in vitro and in vivo. PD173074 is a selective FGFR-inhibitor, which functions by competing with ATP binding and inhibiting autophosphorylation [86]. TKI258 and SU5402 are broader-profile inhibitors which target both FGFRs and VEGFR [87, 88]. All three compounds were found to be cytotoxic and/or cytostatic on a range of FGFR3- or FGFR1-dependent bladder cancer cell lines in vitro and to reduce FGFR phosphorylation and downstream signalling, with PD173074 and TKI258 showing the greatest effect [89, 90]. PD173074 had no toxicity on normal bladder cells, suggesting the existence of a useful therapeutic window, while TKI258 had some detrimental effects, perhaps due to its broader target range [90]. PD173074 was also tested in vivo on xenografts obtained from UMUC14, MGHU3, RT112, and SW780 cells and was shown to inhibit growth and induce tumour regression, although growth resumed following drug withdrawal [89, 90]. Notably, response to PD173074 appeared to be related to the level of FGFR expression and dependence, rather than to mutation status. Some FGFR3-mutant cell lines (J82, 94-10) were less sensitive than FGFR3-overexpressing cell lines (RT112, RT4, SW780) and other FGFR3-mutant cell lines (UMUC14, MGHU3, 97-7). Similarly, in FGFR1-overexpressing cell lines, treatment with PD173074 was effective in JMSU1 but not UMUC3, despite similar levels of FGFR1 expression [68]. This may be attributable to the fact that UMUC3 cells also have a KRAS2 mutation, which activates the same downstream pathways as FGFR signalling. Alternatively, it is possible that FGFR-dependence may confer an initial survival advantage on bladder cancer cells, which may later be replaced by other oncogenic events. There may therefore be an early “susceptibility window” during which tumours are treatable with FGFR inhibitors as single agents. Overall, the in vivo and in vitro studies confirm that FGFR inhibitors may be of clinical relevance in the treatment of bladder cancer but also raise some crucial issues, particularly the requirement for biomarkers of FGFR dependence to predict response to treatment and the need for combination therapy with other agents due to the likely recurrence and/or resistance after treatment withdrawal. Clinical studies currently underway with several FGFR inhibitors [91] are hoped to shed light on some of these issues.
FGFR-blocking antibodies represent an alternative approach to the use of small molecule inhibitors. Humanized and fully human synthetic antibodies have recently become available for therapeutic purposes and present several advantages including low toxicity, high target specificity, easy tissue penetration, and the possibility to be combined with immunotoxins or radionucleotides for specific targeting to malignant cells [92]. Results so far are promising. For example, a single-chain Fv against FGFR3 conjugated to the gelonin toxin was shown to block proliferation and induce apoptosis of the FGFR3-overexpressing cell lines RT112 and RT4 both in vivo and in vitro [93].
The elucidation of downstream targets of FGFR signalling in bladder tumours could also open up new avenues for therapeutic intervention. For example, the discovery that FGFR1 may drive EMT through COX-2 activation [76] suggests that COX-2 inhibition may be particularly beneficial in FGFR1-dependent invasive tumours. A clinical trial utilising a COX-2 inhibitor is currently in progress and it would be interesting to see whether the clinical outcomes correlate with FGFR1 expression and activation levels.
5. Conclusions
In the last decade, it has become clear that FGFRs play a key role in the development of UC and hold promise as therapeutic targets, screening tools, and diagnostic, and prognostic biomarkers. Future challenges include detailed elucidation of downstream signalling, refining FGFR-based screening and prognostic tests, identification of markers to select patients most likely to benefit from FGFR-targeted therapies and development of strategies to overcome recurrence after treatment withdrawal or development of resistance. There is great hope that in the near future the results of research on the role of FGFRs in UC will be translated into the clinical management of these tumours.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Eble JNSG, Epstein JI, Sesterhenn IA, editors. EditorWorld Health Organization. Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon, France: IARC Press; 2004. [Google Scholar]
- 3.Sobin LH, Gospodarowicz MK, Wittekind C. TNM Classification of Malignant Tumours. Oxford, UK: Wiley-Blackwell; 2010. [Google Scholar]
- 4.Mostofi FK, Davies CJ, Sesterhenn I. Histological Typing of Urinary Bladder Tumours. New York, NY, USA: Springer; 1999. [Google Scholar]
- 5.Brausi M, Witjes JA, Lamm D, Persad R, Palou J, et al. A review of current guidelines and best practice recommendations for the management of nonmuscle invasive bladder cancer by the international bladder cancer group. The Journal of Urology. 2011;186(6):2158–2167. doi: 10.1016/j.juro.2011.07.076. [DOI] [PubMed] [Google Scholar]
- 6.Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R. The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics. 2003;21(18):1315–1330. doi: 10.1007/BF03262330. [DOI] [PubMed] [Google Scholar]
- 7.Sangar VK, Ragavan N, Matanhelia SS, Watson MW, Blades RA. The economic consequences of prostate and bladder cancer in the UK. BJU International. 2005;95(1):59–63. doi: 10.1111/j.1464-410X.2005.05249.x. [DOI] [PubMed] [Google Scholar]
- 8.Pashos CL, Botteman MF, Laskin BL, Redaelli A. Bladder cancer: epidemiology, diagnosis, and management. Cancer Practice. 2002;10(6):311–322. doi: 10.1046/j.1523-5394.2002.106011.x. [DOI] [PubMed] [Google Scholar]
- 9.Sylvester RJ, van der Meijden APM, Oosterlinck W, et al. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: a combined analysis of 2596 patients from seven EORTC trials. European Urology. 2006;49(3):466–475. doi: 10.1016/j.eururo.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Witjes JA, Kiemeney LALM, Schaafsma HE, Debruyene FMJ. The influence of review pathology on study outcome of a randomized multicentre bladder cancer trial. British Journal of Urology. 1994;73(2):172–176. doi: 10.1111/j.1464-410x.1994.tb07487.x. [DOI] [PubMed] [Google Scholar]
- 11.Tosoni I, Wagner U, Sauter G, et al. Clinical significance of interobserver differences in the staging and grading of superficial bladder cancer. BJU International. 2000;85(1):48–53. doi: 10.1046/j.1464-410x.2000.00356.x. [DOI] [PubMed] [Google Scholar]
- 12.Böttcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocrine Reviews. 2005;26(1):63–77. doi: 10.1210/er.2003-0040. [DOI] [PubMed] [Google Scholar]
- 13.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocrine-Related Cancer. 2000;7(3):165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 14.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nature Reviews Drug Discovery. 2009;8(3):235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsen SK, Ibrahimi OA, Raucci A, et al. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(4):935–940. doi: 10.1073/pnas.0307287101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klint P, Claesson-Welsh L. Signal transduction by fibroblast growth factor receptors. Frontiers in Bioscience. 1999;4:D165–D177. doi: 10.2741/klint. [DOI] [PubMed] [Google Scholar]
- 17.Ornitz DM, Xu J, Colvin JS, et al. Receptor specificity of the fibroblast growth factor family. The Journal of Biological Chemistry. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family: the complete mammalian FGF family. The Journal of Biological Chemistry. 2006;281(23):15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chellaiah AT, McEwen DG, Werner S, Xu J, Ornitz DM. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. The Journal of Biological Chemistry. 1994;269(15):11620–11627. [PubMed] [Google Scholar]
- 20.Wang F, Kan M, Yan G, Xu J, McKeehan WL. Alternately spliced NH2-terminal immunoglobulin-like loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. The Journal of Biological Chemistry. 1995;270(17):10231–10235. doi: 10.1074/jbc.270.17.10231. [DOI] [PubMed] [Google Scholar]
- 21.Root LL, Shipley GD. Normal human fibroblasts produce membrane-bound and soluble isoforms of FGFR-1. Molecular Cell Biology Research Communications. 2000;3(2):87–97. doi: 10.1006/mcbr.2000.0199. [DOI] [PubMed] [Google Scholar]
- 22.Terada M, Shimizu A, Sato N, Miyakaze SI, Katayama H, Kurokawa-Seo M. Fibroblast growth factor receptor 3 lacking the Ig IIIb and transmembrane domains secreted from human squamous cell carcinoma DJM-1 binds to FGFs. Molecular Cell Biology Research Communications. 2001;4(6):365–373. doi: 10.1006/mcbr.2001.0306. [DOI] [PubMed] [Google Scholar]
- 23.Sleeman M, Fraser J, McDonald M, et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271(2):171–182. doi: 10.1016/s0378-1119(01)00518-2. [DOI] [PubMed] [Google Scholar]
- 24.Trueb B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cellular and Molecular Life Sciences. 2011;68(6):951–964. doi: 10.1007/s00018-010-0576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nature Genetics. 1999;23(1):18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 26.Sibley K, Cuthbert-Heavens D, Knowles MA. Loss of heterozygosity at 4p16.3 and mutation of FGFR3 in transitional cell carcinoma. Oncogene. 2001;20(6):686–691. doi: 10.1038/sj.onc.1204110. [DOI] [PubMed] [Google Scholar]
- 27.van Rhijn BWG, Montironi R, Zwarthoff EC, Jöbsis AC, van der Kwast TH. Frequent FGFR3 mutations in urothelial papilloma. The Journal of Pathology. 2002;198(2):245–251. doi: 10.1002/path.1202. [DOI] [PubMed] [Google Scholar]
- 28.Hernández S, López-Knowles E, Lloreta J, et al. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. Journal of Clinical Oncology. 2006;24(22):3664–3671. doi: 10.1200/JCO.2005.05.1771. [DOI] [PubMed] [Google Scholar]
- 29.Billerey C, Chopin D, Aubriot-Lorton MH, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. American The Journal of Pathology. 2001;158(6):1955–1959. doi: 10.1016/S0002-9440(10)64665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burger M, van der Aa MNM, van Oers JMM, et al. Prediction of progression of non-muscle-invasive bladder cancer by WHO 1973 and 2004 grading and by FGFR3 mutation status: a prospective study. European Urology. 2008;54(4):835–844. doi: 10.1016/j.eururo.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 31.van Oers JMM, Zwarthoff EC, Rehman I, et al. FGFR3 mutations indicate better survival in invasive upper urinary tract and bladder tumours. European Urology. 2009;55(3):650–658. doi: 10.1016/j.eururo.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Tomlinson DC, Baldo O, Hamden P, Knowles MA. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. The Journal of Pathology. 2007;213(1):91–98. doi: 10.1002/path.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. Journal of Bone and Mineral Research. 2002;17(5):860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- 34.D’Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth and Differentiation. 1998;9(1):71–78. [PubMed] [Google Scholar]
- 35.Chen F, Degnin C, Laederich M, Horton WA, Hristova K. The A391E mutation enhances FGFR3 activation in the absence of ligand. Biochimica et Biophysica Acta. 2011;1808(8):2045–2050. doi: 10.1016/j.bbamem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster MK, D’Avis PY, Robertson SC, Donoghue DJ. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Molecular and Cellular Biology. 1996;16(8):4081–4087. doi: 10.1128/mcb.16.8.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lievens PMJ, Roncador A, Liboi E. K644E/M FGFR3 mutants activate Erk1/2 from the endoplasmic reticulum through FRS2α and PLCγ-independent pathways. Journal of Molecular Biology. 2006;357(3):783–792. doi: 10.1016/j.jmb.2006.01.058. [DOI] [PubMed] [Google Scholar]
- 38.Tomlinson DC, L’Hôte CG, Kennedy W, Pitt E, Knowles MA. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Research. 2005;65(22):10441–10449. doi: 10.1158/0008-5472.CAN-05-1718. [DOI] [PubMed] [Google Scholar]
- 39.Kimura T, Suzuki H, Ohashi T, Asano K, Kiyota H, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92(10):2555–2561. doi: 10.1002/1097-0142(20011115)92:10<2555::aid-cncr1607>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 40.Bodoor K, Ghabkari A, Jaradat Z, et al. FGFR3 mutational status and protein expression in patients with bladder cancer in a Jordanian population. Cancer Epidemiology. 2010;34(6):724–732. doi: 10.1016/j.canep.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 41.van Rhijn BWG, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Research. 2001;61(4):1265–1268. [PubMed] [Google Scholar]
- 42.van Rhijn B, van Tilborg A, Lurkin I, et al. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. European Journal of Human Genetics. 2002;10(12):819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- 43.Hafner C, Van Oers JMM, Hartmann A, et al. High frequency of FGFR3 mutations in adenoid seborrheic keratoses. Journal of Investigative Dermatology. 2006;126(11):2404–2407. doi: 10.1038/sj.jid.5700422. [DOI] [PubMed] [Google Scholar]
- 44.Chesi M, Nardini E, Brents LA, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nature Genetics. 1997;16(3):260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karoui M, Hofmann-Radvanyi H, Zimmermann U, et al. No evidence of somatic FGFR3 mutation in various types of carcinoma. Oncogene. 2001;20(36):5059–5061. doi: 10.1038/sj.onc.1204651. [DOI] [PubMed] [Google Scholar]
- 46.Sibley K, Stern P, Knowles MA. Frequency of fibroblast growth factor receptor 3 mutations in sporadic tumours. Oncogene. 2001;20(32):4416–4418. doi: 10.1038/sj.onc.1204543. [DOI] [PubMed] [Google Scholar]
- 47.Wallerand H, Bakkar AA, de Medina SGD, et al. Mutations in TP53, but not FGFR3, in urothelial cell carcinoma of the bladder are influenced by smoking: contribution of exogenous versus endogenous carcinogens. Carcinogenesis. 2005;26(1):177–184. doi: 10.1093/carcin/bgh275. [DOI] [PubMed] [Google Scholar]
- 48.Bakkar AA, Allory Y, Iwatsubo Y, et al. Occupational exposure to polycyclic aromatic hydrocarbons influenced neither the frequency nor the spectrum of FGFR3 mutations in bladder urothelial carcinoma. Molecular Carcinogenesis. 2010;49(1):25–31. doi: 10.1002/mc.20573. [DOI] [PubMed] [Google Scholar]
- 49.Di Martino E, L'Hote CG, Kennedy W, Tomlinson DC, Knowles MA. Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type-and mutation-specific manner. Oncogene. 2009;28(48):4306–4316. doi: 10.1038/onc.2009.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Oers JMM, Adam C, Denzinger S, et al. Chromosome 9 deletions are more frequent than FGFR3 mutations in flat urothelial hyperplasias of the bladder. International Journal of Cancer. 2006;119(5):1212–1215. doi: 10.1002/ijc.21958. [DOI] [PubMed] [Google Scholar]
- 51.Wilkie AOM. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine and Growth Factor Reviews. 2005;16(2):187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Matsumoto M, Ohtsuki Y, Ochii K, et al. Fibroblast growth factor receptor 3 protein expression in urothelial carcinoma of the urinary bladder, exhibiting no association with low-grade and/or non-invasive lesions. Oncology Reports. 2004;12(5):967–971. [PubMed] [Google Scholar]
- 53.Gómez-Román JJ, Saenz P, González JC, et al. Fibroblast growth factor receptor 3 is overexpressed in urinary tract carcinomas and modulates the neoplastic cell growth. Clinical Cancer Research. 2005;11(2):459–465. [PubMed] [Google Scholar]
- 54.Mhawech-Fauceglia P, Cheney RT, Schwaller J. Genetic alterations in urothelial bladder carcinoma: an updated review. Cancer. 2006;106(6):1205–1216. doi: 10.1002/cncr.21743. [DOI] [PubMed] [Google Scholar]
- 55.Catto JWF, Miah S, Owen HC, et al. Distinct microRNA alterations characterize high- and low-grade bladder cancer. Cancer Research. 2009;69(21):8472–8481. doi: 10.1158/0008-5472.CAN-09-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ravery V, Jouanneau J, Gil Diez S, et al. Immunohistochemical detection of acidic fibroblast growth factor in bladder transitional cell carcinoma. Urological Research. 1992;20(3):211–214. doi: 10.1007/BF00299719. [DOI] [PubMed] [Google Scholar]
- 57.Chopin DK, Caruelle JP, Colombel M, et al. Increased immunodetection of acidic fibroblast growth factor in bladder cancer, detectable in urine. Journal of Urology. 1993;150(4):1126–1130. doi: 10.1016/s0022-5347(17)35705-1. [DOI] [PubMed] [Google Scholar]
- 58.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clinical Cancer Research. 1999;5(5):1063–1071. [PubMed] [Google Scholar]
- 59.Penault-Llorca F, Bertucci F, Adelaide J, et al. Expression of FGF and FGF receptor genes in human breast cancer. International Journal of Cancer. 1995;61(2):170–176. doi: 10.1002/ijc.2910610205. [DOI] [PubMed] [Google Scholar]
- 60.Yamaguchi F, Saya H, Bruner JM, Morrison RS. Differential expression of two fibroblast growth factor-receptor genes is associated with malignant progression in human astrocytomas. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(2):484–488. doi: 10.1073/pnas.91.2.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Research. 2010;70(5):2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cross NCP, Reiter A. Tyrosine kinase fusion genes in chronic myeloproliferative diseases. Leukemia. 2002;16(7):1207–1212. doi: 10.1038/sj.leu.2402556. [DOI] [PubMed] [Google Scholar]
- 63.Reis-Filho JS, Simpson PT, Turner NC, et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clinical Cancer Research. 2006;12(22):6652–6662. doi: 10.1158/1078-0432.CCR-06-1164. [DOI] [PubMed] [Google Scholar]
- 64.Freeman KW, Welm BE, Gangula RD, et al. Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Research. 2003;63(23):8256–8263. [PubMed] [Google Scholar]
- 65.Freeman KW, Gangula RD, Welm BE, et al. Conditional activation of fibroblast growth factor receptor (FGFR) 1, but not FGFR2, in prostate cancer cells leads to increased osteopontin induction, extracellular signal-regulated kinase activation, and in vivo proliferation. Cancer Research. 2003;63(19):6237–6243. [PubMed] [Google Scholar]
- 66.Welm BE, Freeman KW, Chen M, Contreras A, Spencer DM, Rosen JM. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. Journal of Cell Biology. 2002;157(4):703–714. doi: 10.1083/jcb.200107119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Acevedo VD, Gangula RD, Freeman KW, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12(6):559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 68.Tomlinson DC, Lamont FR, Shnyder SD, Knowles MA. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Research. 2009;69(11):4613–4620. doi: 10.1158/0008-5472.CAN-08-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomlinson DC, Knowles MA. Altered splicing of FGFR1 is associated with high tumor grade and stage and leads to increased sensitivity to FGF1 in bladder cancer. American The Journal of Pathology. 2010;177(5):2379–2386. doi: 10.2353/ajpath.2010.100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Medina SGD, Chopin D, El Marjou A, et al. Decreased expression of keratinocyte growth factor receptor in a subset of human transitional cell bladder carcinomas. Oncogene. 1997;14(3):323–330. doi: 10.1038/sj.onc.1200830. [DOI] [PubMed] [Google Scholar]
- 71.Ricol D, Cappellen D, El Marjou A, et al. Tumour suppressive properties of fibroblast growth factor receptor 2-IIIb in human bladder cancer. Oncogene. 1999;18(51):7234–7243. doi: 10.1038/sj.onc.1203186. [DOI] [PubMed] [Google Scholar]
- 72.Bernard-Pierrot I, Brams A, Dunois-Lardé C, et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis. 2006;27(4):740–747. doi: 10.1093/carcin/bgi290. [DOI] [PubMed] [Google Scholar]
- 73.Tomlinson DC, Hurst CD, Knowles MA. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene. 2007;26(40):5889–5899. doi: 10.1038/sj.onc.1210399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qing J, Du X, Chen Y, et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. The Journal of Clinical Investigation. 2009;119(5):1216–1229. doi: 10.1172/JCI38017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martínez-Torrecuadrada J, Cifuentes G, López-Serra P, Saenz P, Martínez A, Casal JI. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clinical Cancer Research. 2005;11(17):6280–6290. doi: 10.1158/1078-0432.CCR-05-0282. [DOI] [PubMed] [Google Scholar]
- 76.Tomlison DC, Baxter EW, Loadman et al. PM. FGFR1-induced epithelial to mesenchymal transition through MAPK/PLCγ/COX-2-mediated mechanisms. Plos ONE. 2012;7(6) doi: 10.1371/journal.pone.0038972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Oers JMM, Lurkin I, van Exsel AJA, et al. A simple and fast method for the simultaneous detection of nine fibroblast growth factor receptor 3 mutations in bladder cancer and voided urine. Clinical Cancer Research. 2005;11(21):7743–7748. doi: 10.1158/1078-0432.CCR-05-1045. [DOI] [PubMed] [Google Scholar]
- 78.Zuiverloon TCM, Tjin SS, Busstra M, Bangma CH, Boevé ER, Zwarthoff EC. Optimization of nonmuscle invasive bladder cancer recurrence detection using a urine based FGFR3 mutation assay. Journal of Urology. 2011;186(2):707–712. doi: 10.1016/j.juro.2011.03.141. [DOI] [PubMed] [Google Scholar]
- 79.Kompier LC, Lurkin I, van der Aa MNM, van Rhijn BWG, van der Kwast TH, Zwarthoff EC. FGFR3, HRAS, KRAS, NRAS AND PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS ONE. 2010;5(11) doi: 10.1371/journal.pone.0013821.e13821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roobol MJ, Bangma CH, el Bouazzaoui S, Franken-Raab CG, Zwarthoff EC. Feasibility study of screening for bladder cancer with urinary molecular markers (the BLU-P project) Urologic Oncology. 2010;28(6):686–690. doi: 10.1016/j.urolonc.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 81.Serizawa RR, Ralfkiær U, Steven K, et al. Integrated genetic and epigenetic analysis of bladder cancer reveals an additive diagnostic value of FGFR3 mutations and hypermethylation events. International Journal of Cancer. 2011;129(1):78–87. doi: 10.1002/ijc.25651. [DOI] [PubMed] [Google Scholar]
- 82.Kompier LC, van der Aa MNM, Lurkin I, et al. The development of multiple bladder tumour recurrences in relation to the FGFR3 mutation status of the primary tumour. The Journal of Pathology. 2009;218(1):104–112. doi: 10.1002/path.2507. [DOI] [PubMed] [Google Scholar]
- 83.van Rhijn BW, van der Kwast TH, Liu L, Fleshner NE, Bostrom PJ, et al. The FGFR3 mutation is related to favorable pT1 bladder cancer. The Journal of Urology. 2012;187(1):310–314. doi: 10.1016/j.juro.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Van Rhijn BWG, Zuiverloon TCM, Vis AN, et al. Molecular grade (FGFR3/MIB-1) and EORTC risk scores are predictive in primary non-muscle-invasive bladder cancer. European Urology. 2010;58(3):433–441. doi: 10.1016/j.eururo.2010.05.043. [DOI] [PubMed] [Google Scholar]
- 85.Ploussard G, Soliman H, Dubosq F, Meria P, Verine J, et al. The prognostic value of FGFR3 mutational status for disease recurrence and progression depends on allelic losses at 9p22. American Journal of Cancer Research. 2011;1(4):498–507. [PMC free article] [PubMed] [Google Scholar]
- 86.Mohammadi M, Froum S, Hamby JM, et al. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO Journal. 1998;17(20):5896–5904. doi: 10.1093/emboj/17.20.5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sarker D, Molife R, Evans TRJ, et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clinical Cancer Research. 2008;14(7):2075–2081. doi: 10.1158/1078-0432.CCR-07-1466. [DOI] [PubMed] [Google Scholar]
- 88.Mohammadi M, McMahon G, Sun L, et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276(5314):955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- 89.Miyake M, Ishii M, Koyama N, et al. 1-tert-butyl-3-[6-(3,5-dimethoxy-phenyl)-2-(4-diethylamino-butylamino) -pyrido[2,3-d]pyrimidin-7-yl]-urea (PD173074), a selective tyrosine kinase inhibitor of fibroblast growth factor receptor-3 (FGFR3), inhibits cell proliferation of bladder cancer carrying the FGFR3 gene mutation along with up-regulation of p27/Kip1 and G1/G0 arrest. Journal of Pharmacology and Experimental Therapeutics. 2010;332(3):795–802. doi: 10.1124/jpet.109.162768. [DOI] [PubMed] [Google Scholar]
- 90.Lamont FR, Tomlinson DC, Cooper PA, Shnyder SD, Chester JD, Knowles MA. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo . British Journal of Cancer. 2011;104(1):75–82. doi: 10.1038/sj.bjc.6606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Greulich H, Pollock PM. Targeting mutant fibroblast growth factor receptors in cancer. Trends in Molecular Medicine. 2011;17(5):283–292. doi: 10.1016/j.molmed.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Harris M. Monoclonal antibodies as therapeutic agents for cancer. The Lancet Oncology . 2004;5(5):292–302. doi: 10.1016/S1470-2045(04)01467-6. [DOI] [PubMed] [Google Scholar]
- 93.Martínez-Torrecuadrada JL, Cheung LH, López-Serra P, et al. Antitumor activity of fibroblast growth factor receptor 3-specific immunotoxins in a xenograft mouse model of bladder carcinoma is mediated by apoptosis. Molecular Cancer Therapeutics. 2008;7(4):862–873. doi: 10.1158/1535-7163.MCT-07-0394. [DOI] [PubMed] [Google Scholar]