

Abstract
Schizophrenia is a severe psychiatric disorder with strong heritability and marked heterogeneity in symptoms, course, and treatment response. There is strong interest in identifying genetic risk factors that can help to elucidate the pathophysiology and that might result in the development of improved treatments. Linkage and genome-wide association studies (GWASs) suggest that the genetic basis of schizophrenia is heterogeneous. However, it remains unclear whether the underlying genetic variants are mostly moderately rare and can be identified by the genotyping of variants observed in sequenced cases in large follow-up cohorts or whether they will typically be much rarer and therefore more effectively identified by gene-based methods that seek to combine candidate variants. Here, we consider 166 persons who have schizophrenia or schizoaffective disorder and who have had either their genomes or their exomes sequenced to high coverage. From these data, we selected 5,155 variants that were further evaluated in an independent cohort of 2,617 cases and 1,800 controls. No single variant showed a study-wide significant association in the initial or follow-up cohorts. However, we identified a number of case-specific variants, some of which might be real risk factors for schizophrenia, and these can be readily interrogated in other data sets. Our results indicate that schizophrenia risk is unlikely to be predominantly influenced by variants just outside the range detectable by GWASs. Rather, multiple rarer genetic variants must contribute substantially to the predisposition to schizophrenia, suggesting that both very large sample sizes and gene-based association tests will be required for securely identifying genetic risk factors.
Introduction
Schizophrenia (MIM 181500) is characterized by positive symptoms (e.g., delusions, hallucinations, and disorganized thinking), negative symptoms (e.g., flat affect, loss of spontaneity, diminished initiative and capacity for pleasure, and impaired volition), numerous cognitive dysfunctions of varying severity, mood disturbances, and suicidality. Antipsychotic drugs are usually effective in treating positive symptoms, and to a much lesser extent, cognitive impairment. Clozapine, the prototypical atypical antipsychotic drug, has also been shown to uniquely reduce the risk of suicide in schizophrenic individuals.1,2 Cognitive impairment is considered the major cause of functioning deficits,3 but the other components of the illness contribute as well by collectively interfering substantially with the quality of life and constituting significant burdens on the families of individuals with schizophrenia. Genetic studies that implicate variants in specific genes as risk factors for the syndrome might help to elucidate the pathophysiology of the syndrome and the identification of novel treatment targets.
Despite many years of study, the genetic basis of schizophrenia remains largely unknown. Many complex diseases, including neuropsychiatric disorders such as epilepsy and Alzheimer disease (MIM 104300),4,5 have been shown to have Mendelian forms; however, no single gene mutations of large effect have been conclusively identified in schizophrenia. Moreover, genome-wide association studies (GWASs) have identified only variants associated with extremely small effects on risk. For example, GWAS meta-analyses on 8,000 cases and 19,000 controls identified several high-frequency associations with very small odd ratios (1.1–1.3), effectively ruling out the possibility that risk of schizophrenia is determined primarily by a modest number of common variants (or even hundreds of common variants).6–8 On the other hand, studies of rare copy-number variants (CNVs) have shown that a modest proportion of schizophrenia cases can be attributed to a heterogeneous collection of rare CNVs with high but incomplete penetrance (the estimated odds ratios range from 2.7 to 25).9–12 Because of their rarity and the multiple genes involved in many of the CNV regions, these CNVs represent daunting targets for drug development unless they lead to more generalized downstream effects that affect a much higher percentage of people with schizophrenia.
Given that very little of the heritability of schizophrenia is explained by nongenetic causes and that there is good evidence for a role for rare variants, there is intense interest in using next-generation sequencing (NGS) for the identification of additional rare variants associated with schizophrenia. These, in turn, might help researchers identify pathways that could inform and motivate novel drug-development efforts. Two recent studies explored the role of highly penetrant individual sequence variants in schizophrenia by examining the number and function of de novo variants in apparent sporadic schizophrenia (in 14 and 53 cases, respectively).13,14 The authors of both studies concluded that an excess of de novo variants was seen in the schizophrenia cases and, additionally, that more of these than expected were damaging, suggesting that at least some schizophrenia cases are caused by highly penetrant de novo variants. However, the high heritability of schizophrenia is not compatible with the hypothesis that most cases are the result of de novo mutations.
The genetic explanation of the majority of schizophrenia cases therefore remains unresolved. One possibility not excluded by current evidence is that variants only slightly below the detection threshold for GWASs have appreciable effects on risk (for example, variants with frequencies approaching 0.5% and relative risks of 2 or slightly more). If there were many such variants, some could be readily detected by the sequencing of case genomes and the genotyping of identified variants in a large cohort of additional cases. This is the design we follow here. Another possibility is that most pathogenic mutations have frequencies well below the GWAS detection threshold. For such variants, the most efficient design will be to employ screens on the basis of the combination of variants across particular genes or regions. There is a clear analogy here with CNVs. Although each individual mutation in a schizophrenia-associated CNV region appears to have arisen either de novo or very recently and thus has an extremely low frequency, they collectively reach frequencies that are significantly different in cases than in controls.9,10,15–19 Whether this will be the requisite paradigm for identifying sequence variants remains to be resolved.
Here, we examined the first possibility—whether a substantial proportion of schizophrenia cases can be explained by individual, moderately rare variants with strong effects. We took a set of 5,155 rare variants identified in 166 sequenced schizophrenia genomes and exomes, and we genotyped them in an additional unrelated 2,617 cases and 1,800 controls to determine whether rare variants overrepresented in unrelated cases relative to controls could be detected in samples of this size.
Subjects and Methods
Study Participants
Discovery cases were 166 individuals with a diagnosis of schizophrenia or schizoaffective disorder and included treatment-resistant cases and/or cases with a strong family history. The original sequenced cases comprised 47 Finnish schizophrenic individuals defined as resistant to treatment (i.e., they qualified for treatment with clozapine), 87 United States individuals with treatment-resistant schizophrenia or schizoaffective disorder (all were whole-exome sequenced), and 32 United States individuals with a diagnosis of schizophrenia and a family history of schizophrenia or other severe neuropsychiatric disorders (these individuals were whole-genome sequenced). The sample was approximately 10% African ancestry, 89% European ancestry, and 1% other (Native American or Hispanic). The institutional review boards (IRBs) of Duke University Medical Center and collaborating institutions approved all procedures. Follow-up cases (n = 2,756) were 544 United States individuals with a family history of schizophrenia or other severe neuropsychiatric disorders (20 of these individuals overlapped with the whole-genome-sequenced subjects for quality-control purposes), 364 United States samples including 168 treatment-resistant individuals (79 overlapped with the exome-sequenced individuals), 360 Italian samples, and 1,567 samples obtained from the Rutgers repository. Follow-up cases were 45% African ancestry, 54% European ancestry, and 1% other (Hispanic, Asian, or Middle Eastern). Informed consent was obtained from all participants or their legal guardians.
Discovery controls (n = 307) and follow-up controls (n = 1,932, including 65 discovery controls for quality-control purposes) either were subjects who were not enriched for (but not specifically screened for) neuropsychiatric disorders, who were enrolled in Duke IRB-approved protocols, and who consented to future unrelated research or were samples received from outside institutions under a Duke IRB exemption. Discovery controls were 6% African ancestry, 92% European ancestry, and 2% other (Hispanic, Native American, or Middle Eastern). Follow-up controls were 37% African ancestry, 59% European ancestry, and 3% other (Hispanic, Asian, or Middle Eastern).
Power calculations were performed with Power for Genetic Association Analyses.20
Targeted Capture and Exome and Genome Sequencing
For whole-exome sequencing, the target regions were captured with the Agilent SureSelect Human All Exon 37 Mb or 50 Mb Kit (Agilent Technologies, Santa Clara, CA) according to the vendor-provided protocols. Sequencing was performed in the Center for Human Genome Variation Genomic Analysis Facility with either Illumina GAII or HiSeq machines. Whole-genome sequencing was performed as previously described.21 Each read was then aligned to the reference genome (National Center for Biotechnology Information human genome assembly build 36; Ensembl core database release 50_36l 1) with the Burrows-Wheeler Alignment tool,22 and single-nucleotide variants (SNVs) and small indels were identified with SAMtools.23 PCR duplicates were removed with Picard software (see Web Resources).
Variant Selection
We focused on variants annotated as functional, which was defined as nonsynonymous, nonsense, or located in the canonical splice sites. The analysis was restricted to variants with a minor allele frequency (MAF) < 0.05 or, for the recessive model, a MAF < 0.3. For each variant tested, only individuals with a minimum coverage of 10× at that site were included (n = 152,511 SNVs for the allelic model and 172,886 for the recessive model).
We first tested for genetic association by using Association Tests for Annotated Variants (ATAV [see Web Resources]) to run Fisher’s exact tests comparing exome-captured regions from cases and controls. We then removed variants that had greater than 50% of the individuals missing as a result of low coverage, variants with a Hardy-Weinberg equilibrium p value < 0.001, variants with a p value < 0.05 but whose frequency was higher in controls, and variants that were clearly driven by differences between the Finnish or African American subjects and the other samples. All other associated (p < 0.05) variants were put forward for scoring in the iSelect follow up (n = 316 allelic, n = 206 recessive).
Because of an expectation of high locus heterogeneity, we recognized that a gene might play a role in only a single case in our sequenced samples, and so we also selected variants that formed genotypes that were present in cases but not in controls. From this set, we removed the following: variants that were present in only one individual (“unique variants”) if that individual had greater than the mean + 1 standard deviation of unique variants (because these might have been poor-quality calls) (n = 485 allelic, n = 5 recessive), unique variants that were present only in the Finnish samples (because we had no Finnish controls) (n = 293 allelic, 12 recessive), and unique variants in a low-coverage sample (23.5–25×; n = 4 samples; n = 33 allelic, n = 1 recessive).
We then included for scoring (1) all variants that were in an essential splice site, destroyed a stop codon, or introduced a new stop codon (n = 1,643 allelic, n = 43 recessive); (2) all variants in a region associated with epilepsy, schizophrenia, autism, or intellectual disability through rare CNVs (n = 863 allelic, 2 recessive), and (3) all nonsynonymous variants that got a PolyPhen224 rating of “probably damaging” (n = 3,736 allelic, n = 51 recessive). These 6,860 variants were submitted to Illumina’s online Assay Design Tool for the prediction of the likelihood of a successful assay. All variants with a score below 0.6 were removed, leaving 5,788 variants.
iSelect Quality Control
The raw data (idats) from the custom iSelect genotyping were brought into the Illumina GenomeStudio software. All variants were clustered with the GenomeStudio default parameters. The call rates were inspected, and any samples with a call rate below 0.95 were removed from analysis. Each variant call was manually inspected for clustering accuracy, and any obvious miscalls were corrected or deleted if correcting was not possible because of irregular clustering. Genders were checked on the basis of X and Y chromosome variants, and mismatched samples were excluded. For a subset of samples, we also had Illumina genome-wide SNP array data, which we used to check the concordance for overlapping SNPs, and discordant samples were excluded from analysis. Of these 5,788 variants, 5,155 variants were successfully genotyped and passed all quality-control checks. We obtained genotypes for these variants in an additional 2,617 cases and 1,800 controls that passed all quality-control checks. Individuals with epilepsy were also genotyped with the iSelect array, and all putative schizophrenia-associated variants were investigated in cases with epilepsy for the evaluation of possible variable expressivity.25
Data Analysis
Because of allele-frequency differences between subjects of African and European descent, we analyzed these two population groups separately and then, where appropriate, combined the p values by using a Fisher’s trend test. Fisher’s allelic and recessive tests were performed with PLINK (see Web Resources). According to PLINK defaults, only females were included in the Fisher’s exact test for X chromosome variants.
Comparison of Validation Rates
To determine whether there were differences between the validation rates among different classes of functional variants, we took all autosomal variants that were seen in only a single sample in the initial sequencing cohort. These were considered to be the most vulnerable calls. We then selected from this group only the variants that were present in an individual who was genotyped with the iSelect array, and we determined for each variant whether it had also been identified by the iSelect genotyping. Those identified were classed as validated, and we compared the percentage of validated variants among different functional groups.
Results
Association Testing
We performed whole-exome (n = 134) or whole-genome sequencing (n = 32) on 166 schizophrenia cases and 307 controls (n = 256 for exome sequencing, and n = 51 for genome sequencing) and followed this with Fisher’s Exact test to look for association with schizophrenia by using both an allelic and a recessive model after excluding common variants. This resulted in a total of 337,312 variants, for which Bonferroni correction for multiple testing required a p value < 1.5 × 10−7. At this p value threshold in a data set of this size (and with a focus exclusively on rare variants), we would have limited power to detect anything but variants with an extremely high relative risk (Figure 1). As expected, no variant achieved the required level of significance in the initial sequence data. This is not surprising because variants that would show significance in this initial sequence data set would have effect sizes that would be expected to have been identified in linkage analyses.
Figure 1.
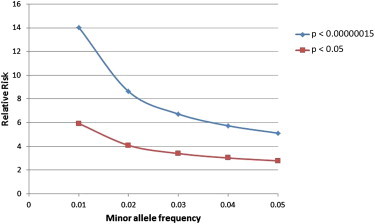
Range of Relative Risks and MAFs that Are Detectable with 99% Power at p < 0.05 and, after Correction for All Included Variants, p < 1.5 × 10−7 in a Cohort of 166 Cases and 307 Controls
If we use a cutoff of p < 0.05 instead, however, we are powered to identify variants over a much broader range of effect sizes and allele frequencies (Figure 1), although the majority of these variants will be false positives. We therefore adopted a two-stage strategy in which (1) we used the discovery cohort to identify a variant set that was likely to be enriched with schizophrenia-associated variants (those with p < 0.05 and selected as described in the Subjects and Methods section; n = 428) and (2) we used a follow-up sample of 2,617 cases and 1,800 controls to look for corroborating evidence of association. We combined the original sequencing data with the iSelect data to produce a final data set of 2,785 cases and 2,120 controls.
Because we didn’t have enough markers to accurately adjust for population stratification, we performed Fisher’s exact tests (for both recessive and allelic models) on individuals of African and European descent separately, and we used a logistic regression with self-described race (including only subjects of European or African ancestry) to look for variants that associated in both ancestral groups. After a Bonferroni correction for all original 337,312 variants, p < 1.5 × 10−7 was required for study-wide significance. The lowest p values in the combined data set were 0.0003 (allelic) and 0.01 (recessive). The most significantly associated variants in the African-American-only analysis had p values of 0.0006 (allelic) and 0.0005 (recessive), and the most significant variants in Europeans were 5.9 × 10−6 (allelic) and 0.01 (recessive). The most strongly associated variant was a nonsynonymous AL589787.16 variant (Human Genome Organization [HUGO]: N/A; rs7098669), which was originally included because it showed association in the recessive model. This variant shows substantial variation in allele frequency across populations, and the association probably reflects population stratification. It is interesting to note, however, that this SNP lies within an open reading frame (C10orf90) found to contain two independent schizophrenia associations in the recent mega-GWAS, although the association did not hold up in the follow-up cohort of that study.26
Evaluation of Genotypes Exclusive to Cases
Most very rare highly penetrant schizophrenia-associated genotypes would not be expected to show a significant association in a data set of the size of our discovery cohort. Such genotypes would be found exclusively in cases but only in very small numbers. We therefore also followed up all genotypes that were present in two cases and no controls (n = 861) and a subset of those present in one case and no controls (n = 4,498; see Subjects and Methods for selection criteria) because this data set is likely to be enriched with very rare variants with high penetrance. Table 1 summarizes the iSelect outcome for the different categories of variants. Of the 4,028 genotypes originally present in only a single case and successfully genotyped in the follow up, 1,588 (39%) were seen in an iSelect control and 1,989 (49%) were seen in only one or no further cases. To further explore the frequency of these variants in controls, we referred to the Exome Variant Server (National Heart, Lung, and Blood Institute [NHLBI] Exome Sequencing Project, Seattle, WA). Tables S1 and S2, available online, show all case-specific variants and case-specific homozygotes, respectively.
Table 1.
Summary of Outcomes for Variants by Original Inclusion Criteria
| Reason for Original Inclusion (n = 5,788) | Failed or Excluded |
Present in Controls, p < 0.05 |
Absent in Controls |
||||||
|---|---|---|---|---|---|---|---|---|---|
| In African Americans Only | In European Ancestry Only | In Both Ancestral Groups | In Neither Ancestral Group | In 0 Additional Cases | In 1 Additional Case | In > 1 Additional Case | |||
| p < 0.05 (n = 428) | allelic (n = 242) | 30 | 3 | 28 | 2 | 173 | 4 | 2 | 0 |
| recessive (n = 186)a | 37 | 5 | 11 | 1 | 131 | 1 | 0 | 0 | |
| In > 1 case and 0 controls (n = 861) | allelic (n = 843) | 94 | 9 | 15 | 0 | 476 | 192 | 34 | 23 |
| recessive (n = 18)a | 2 | 0 | 1 | 0 | 12 | 2 | 1 | 0 | |
| In 1 case and 0 controls (n = 4,498) | allelic (n = 4,280) | 434 | 28 | 9 | 0 | 1,443 | 1,929 | 301 | 136 |
| recessive (n = 218)a | 36 | 2 | 3 | 0 | 103 | 60 | 7 | 7 | |
p values are given for recessive tests; counts are for homozygous genotypes.
Some variants remained absent in all controls and were found to be present in several additional cases (Table 2). The variant present in the most cases (n = 5) is a nonsynonymous mutation (g.1882C>T [Arg628Cys]; ENST00000380099) in KL (MIM 604824), a gene that acts mainly in the renal and cardiovascular systems.27 However, KL has also been implicated in the regulation of vitamin D metabolism. Klotho-deficient mice, for example, show degeneration of mesencephalic dopamine neurons28 and other aging phenotypes, such as hearing loss,29 which can be rescued with a vitamin-D-deficient diet. Although KL has not been previously associated with schizophrenia to our knowledge, substantial evidence implicates alterations in vitamin D levels, especially deficiency, as a schizophrenia risk factor that might also help to explain certain epidemiological findings, such as season of birth and latitude-gradient effects on geographic differences in incidence and prevalence.30–32 In addition, prenatal vitamin D deficiency has been shown to cause lasting changes in NMDA-mediated brain function in adult rats.33 Other genes of interest with case-only variants include EPB41L1 (MIM 602879), SLC1A2 (MIM 600300), STX4 (MIM 186591), HYDIN (MIM 610812), PCLO (MIM 604918), and ZNF804B. EPB41L1 encodes the erythrocyte membrane protein band 4.1-like N,34 which colocalizes with AMPA receptors at excitatory synapses and is thought to mediate the interaction of the AMPA receptors with the cytoskeleton.35 It has also been shown to be necessary for the formation of calcium waves in the mediation of neurite formation.36 A recent study of synaptic protein sequencing reported a de novo functional missense mutation in EPB41L1 in nonsyndromic intellectual disability.37 SLC1A2 (aka, EAAT2 and GLT1) encodes a glial high-affinity glutamate transporter and is the major transporter in the forebrain.38 Mice lacking Slc1a2 have spontaneous lethal seizures and are vulnerable to glutamate neurotoxicity after forebrain trauma.39 STX4 directs membrane fusion at excitatory glutamatergic synapses and is essential for normal dendritic spine morphology, retrograde synaptic signaling, and long-term potentiation at hippocampal synapses.40 A frameshift mutation in Hydin causes recessively transmitted hydrocephalus in the mouse.41 Additionally, a paralog of HYDIN is included in 1q21.1 microdeletions and microduplications, which have been associated with microcephaly, macrocephaly, and neuropsychiatric disorders, including schizophrenia.16 PCLO encodes Picollo, which acts alongside Bassoon, Syntaxin, SNAP-25, and N-Cadherin in the presynaptic active zone, a specialized region where synaptic vesicles dock and fuse.42 Piccolo is upregulated in the nucleus accumbens in response to methamphetamine, and antisense suppression of Piccolo increases the behavioral response to amphetamine and causes synaptic accumulation of dopamine.43 ZNF804B is a paralog of ZNF804A (MIM 612282), which has shown to be associated with schizophrenia in GWASs.44 Some variants were present in both cases and controls but were homozygous only in cases (see Table S2). Two variants were homozygous in three or more cases and absent in all controls, but they were both predicted to be benign.
Table 2.
SNVs that Were Seen in Two or More Follow-Up Schizophrenia Cases and that Were Absent in All Study Controls and Also Invariant in the NHLBI Exome-Sequenced Cohort
| Variant (chr_hg18 position_variant allele)a | Gene | MIM Number | Transcript (Ensembl 50_36l) | RefSeq mRNA | Position of Sequence and Amino Acid Change | Annotated Function | Total Cases | European American Case Counts (Hom/Het/Ref) | European American Control Counts (Hom/Het/Ref) | African American Case Counts (Hom/Het/Ref) | African American Control Counts (Hom/Het/Ref) | Other Case Counts (Hom/Het/Ref) | Other Control Counts (Hom/Het/Ref) | Total Samples (NHLBI Cohort) | European American Samples (NHLBI Cohort) | African American Samples (NHLBI Cohort) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13_32533098_T | KL | 604824 | ENST00000380099 | NM_004795.3 | c.1882C>T (p.Arg628Cys) | NS | 5 | 0/1/1,564 | 0/0/1,359 | 0/4/1,186 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 19_2785443_A | ZNF554 | NA | ENST00000317243 | NM_001102651.1 | c.1210G>A (p.Gly404Arg) | NS | 4 | 0/3/1,561 | 0/0/1,359 | 0/1/1,189 | 0/0/679 | 0/0/30 | 0/0/82 | 5,351 | 3,508 | 1,843 |
| 20_34245584_G | EPB41L1 | 602879 | ENST00000344237 | − | c.59A>G (p.His20Arg) | NS | 4 | 0/4/1,560 | 0/0/1,359 | 0/0/1,187 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 2_238150673_A | RAB17 | 602206 | ENST00000264601 | NM_022449.3 | c.401C>T (p.Thr134Met) | NS | 4 | 0/0/1,565 | 0/0/1,359 | 0/4/1,186 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 21_44639800_T | TRPM2 | 603749 | ENST00000397928 | NM_003307.3 | c.1870G>T (p.Asp624Tyr) | NS | 4 | 0/3/1,561 | 0/0/1,359 | 0/1/1,189 | 0/0/679 | 0/0/30 | 0/0/82 | 5,376 | 3,508 | 1,868 |
| 1_55290595_T | PCSK9 | 607786 | ENST00000302118 | NM_174936.3 | c.580C>T (p.Arg194Trp) | NS | 3 | 0/0/1,565 | 0/0/1,358 | 0/3/1,187 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 11_111555378_T | BCDO2 | 611740 | ENST00000393032 | NM_001037290.2 | c.154C>T (p.Arg52∗) | X | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 11_35290538_T | SLC1A2 | 600300 | ENST00000395750 | NM_001195728.2 | c.317G>A (p.Arg106His) | NS | 3 | 0/2/1,563 | 0/0/1,359 | 0/1/1,189 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 12_12952740_A | GPRC5A | 604138 | ENST00000014914 | NM_003979.3 | c.290G>A (p.Arg97His) | NS | 3 | 0/0/1,565 | 0/0/1,359 | 0/3/1,187 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 15_41875227_T | SERINC4 | 614550 | ENST00000299969 | − | c.918-1C>T | splice | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 16_1083724_C | C1QTNF8 | 614147 | ENST00000328449 | NM_207419.3 | c.537C>G (p.Tyr179∗) | X | 3 | 0/3/1,557 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,368 | 3,500 | 1,868 |
| 16_30958421_A | STX4 | 186591 | ENST00000313843 | NM_004604.3 | c.761G>A (p.Arg254His) | NS | 3 | 0/1/1,564 | 0/0/1,359 | 0/2/1,188 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 16_69543869_T | HYDIN | 610812 | ENST00000316490 | − | c.6340G>A (p.Gly2114Arg) | NS | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 4,650 | 3,215 | 1,435 |
| 17_7769756_A | KCNAB3 | 604111 | ENST00000303790 | NM_004732.3 | c.508C>T (p.Arg170∗) | X | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 19_19506937_A | AC011448.5 | NA | ENST00000397179 | NM_198537.3 | c.413G>A (p.Arg138Gln) | NS | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,167 | 3,422 | 1,745 |
| 19_60041100_T | KIR3DL3 | 610095 | ENST00000391729 | − | c.328C>T (p.Gln110∗) | X | 3 | 0/0/1,559 | 0/0/1,355 | 0/3/1,186 | 0/0/679 | 0/0/30 | 0/0/82 | 5,264 | 3,418 | 1,846 |
| 2_32578230_C | BIRC6 | 605638 | ENST00000261359 | − | c.8497G>C (p.Glu2833Gln) | NS | 3 | 0/0/1,542 | 0/0/1,337 | 0/3/1,187 | 0/0/679 | 0/0/29 | 0/0/75 | 5,379 | 3,510 | 1,869 |
| 3_114481460_A | BOC | 608708 | ENST00000355385 | NM_033254.2 | c.2120G>A (p.Arg707His) | NS | 3 | 0/2/1,563 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/1/29 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 3_42714733_A | HHATL | 614071 | ENST00000341477 | − | c.325C>T (p.Arg109Cys) | NS | 3 | 0/1/1,564 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/2/28 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 6_117745049_A | ROS1 | 165020 | ENST00000368507 | − | c.6067A>T (p.Met2023Leu) | NS | 3 | 0/0/1,565 | 0/0/1,359 | 0/3/1,187 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 7_65077123_A | GUSB | 611499 | ENST00000345660 | − | c.916C>T (p.Arg305∗) | X | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 7_82419591_T | PCLO | 604918 | ENST00000333891 | − | c.8407G>A (p.Val2803Ile) | NS | 3 | 0/0/1,512 | 0/0/1,312 | 0/3/1,186 | 0/0/679 | 0/0/29 | 0/0/80 | 5,032 | 3,389 | 1,643 |
| 7_86891201_A | ABCB4 | 171060 | ENST00000394680 | − | c.2168G>T (p.Gly723Val) | NS | 3 | 0/0/1,565 | 0/0/1,359 | 0/3/1,187 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
| 7_88803894_A | ZNF804B | NA | ENST00000333190 | NM_181646.2 | c.3662C>A (p.Ala1221Asp) | NS | 3 | 0/3/1,562 | 0/0/1,359 | 0/0/1,190 | 0/0/679 | 0/0/30 | 0/0/82 | 5,379 | 3,510 | 1,869 |
A full list of variants, including those that were absent in our study but that were either seen in NHLBI controls or not covered in the NHLBI exome-sequenced cohort, can be seen in Table S1. The following abbreviations are used: NS, nonsynonymous; X, nonsense; and NA, not available.
No variant had an rs number.
Interestingly, it was recently shown that loss-of-function mutations have a lower validation rate than other classes of variants.45 This is expected because deleterious variants are likely to be less common than those with moderate or no function, and thus, all else being equal, there is a lower prior that deleterious variants are real. We searched for a similar effect in our data, although we expected our validation rate in general to be higher because (1) we required high stringency for variants to be carried to follow up and (2) we looked almost exclusively at rare variants predicted to have strong functional effects, so the magnitude of the difference among them would be expected to be reduced. Nevertheless, we do see an attenuated effect as described by MacArthur et al.,45 such that if we compare validation rates (as defined by a variant being observed on the iSelect array) among types of variants, the rates for very rare nonsense (76%, n = 388) and essential splice variants (73%, n = 221) are lower than those for nonsynonymous variants (86%, n = 2826).
Discussion
In this large-scale report of sequenced schizophrenia genomes, we have looked for evidence of association of individual, rare, highly penetrant sequence variants in a discovery cohort of 166 cases and 307 controls and in a follow-up cohort of 2,756 cases and 1,932 controls. We found no significantly associated variants after Bonferroni correction for multiple hypothesis testing.
Having found no significantly associated SNVs, we focused on variants that were present in more than one case and absent in controls. At least some of these might ultimately prove to be statistically associated with schizophrenia, even though each was individually too rare to reach study-wide significance in this data set. We found a small number of variants that remain good candidates as individual schizophrenia-associated variants and that are high priority for follow-up studies. However, because of the low frequency of these variants, large collaborative studies will be required for providing statistically significant evidence for their association with schizophrenia. As an illustrative example, we can consider the top hit, a missense KL mutation that was present in 5 of 2,780 cases and absent in 7,417 controls. Assuming that the frequency in cases remains at 0.18% and that we have equal numbers of cases and controls, we would need to see about 23 more schizophrenia cases with this variant, which would require approximately 13,000 schizophrenia cases and controls for this individual variant to obtain study-wide significance (p < 1.5 × 10−7).
This study had 99% power to detect moderately rare (1%–5%) variants with relative risks between 2 and 6. Because some variants in this frequency range would be poorly represented on GWAS chips46 and because consistently detecting effect sizes in part of this range would be difficult with linkage studies,47 variants with these properties have not previously been systematically investigated on a genome-wide scale in relation to schizophrenia. On the basis of our findings and those of previous studies, some possible genetic architectures for schizophrenia risk appear increasingly unlikely and include (1) a small number of highly penetrant loci explaining the majority of cases (linkage studies), (2) a moderate number (less than several hundred) of common variants with low relative risks underlying most cases (GWASs), and (3) so-called “goldilocks alleles,”48 or moderately rare variants that have moderate relative risks and that explain most cases (present study). This finding suggests that the majority of schizophrenia-associated variants will be of very low frequency and that their association with schizophrenia will most readily be confirmed by their collective presence in genes associated with schizophrenia with the use of collapsing methods and related approaches49–52 rather than with variant-specific frequency differences between cases and controls.
These findings have a number of implications. First, one outcome of the 1000 Genomes Project is novel GWAS arrays that extend to variants in the 1%–5% frequency range. Our results suggest that the use of these arrays in relation to schizophrenia is unlikely to reveal significant associations for relative risks above 2.
Second, our data strongly suggest that genetic risk factors for schizophrenia are outside the range of what is easily detected in sample sizes of the sort used here. For example, although our power to detect variants with frequencies below 1% is low, we should have detected some of them if there were many risk factors with modest effect sizes near this frequency cutoff. Thus, at least in terms of the relatively rare and high impact variants contributing to schizophrenia risk, the genetic architecture is one characterized by high locus heterogeneity, high allelic heterogeneity, or, more likely, both. Beyond locus and allelic heterogeneity, the results could also be consistent with oligogenic, polygenic, or epistatic models in which effect sizes for individual variants are very modest and the genotypes are reasonably penetrant only in combination. We did not explicitly test for such models given the current sample sizes. Considering the number of tests required for open-ended screens of interacting variants (even those limited to interacting pairs and restricting analyses to common variants), we suspect that such interactions will need to be identified secondarily to the identification of a main effect, however small, for a single variant or through testing of specific hypotheses analyzing small sets of variants in defined biological pathways.
It is valuable to consider these observations in light of the example of CNVs. CNVs were the first type of rare variants that could be detected on a genome-wide scale. When all similar (but not necessarily identical by descent) CNVs in a region were considered together, moderately rare CNVs with relative risks from 2 to 25 were immediately and definitively associated with schizophrenia risk across different cohorts and populations.9,10,17 Because other types of rare genetic variation, such as SNVs and indels, were not available for comparison, it was not possible to determine whether these properties were specific to large structural variants or were representative of rare schizophrenia-associated variants in general. Here, we were able to systematically search for associated SNVs that have similar relative risks and that explain as high a proportion of cases as some of the CNV regions, and we failed to identify any.
It is essential to appreciate that the schizophrenia-associated CNVs were detectable because recurrent mutation greatly increases the number of affected cases. Any given mutation event leading to a CNV appears to be responsible only for a few cases among closely related individuals.10 If SNVs prove analogous, we would expect certain individual genes or gene pathways to associate with schizophrenia, although individual variants in those genes would not be frequent enough to reach statistical significance by themselves in the sample sizes used in our follow-up genotyping experiment. The clear implication is that collapsing methods that combine qualifying variants in individual genes and/or much larger sample sizes will be required for finding secure evidence of risk-conferring genes. Such a conclusion should not necessarily discourage the prospects for gene discovery in schizophrenia; rather, it suggests that the clearest path to discovery should focus on both significantly expanded samples sizes and sophisticated methods for appropriately combining qualifying variants affecting the same genes.49–52 Thus, we propose two possible strategies: one that sequences a sufficiently large discovery cohort to prioritize genes in terms of their load of qualifying mutations and that further sequences those genes of interest in additional samples and/or a second strategy that generates complete sequence data on 10,000 or more case genomes. Significant findings from either approach will implicate particular genes as risk factors for schizophrenia, and these genes will hopefully converge on a limited number of pathways and will thus provide valuable information about the genetic etiology of schizophrenia. Either way, only very rarely will an individual variant be determined to be causal in an individual schizophrenia case, thus limiting the scope for genetics in individual risk prediction.
The conclusions of this study must be tempered by a number of considerations. First, we analyzed only coding regions. It therefore remains possible that moderately rare regulatory variants with relatively strong effect sizes explain the majority of cases of schizophrenia. This would seem to be an unlikely model on the basis of the prominent role of coding variants in Mendelian disorders, but it will be tested in the near future as variants identified from whole-genome sequencing of individuals with schizophrenia are investigated in larger data sets. One example of a regulatory region that was not represented in this study was that around the miRNA MIR137 (miR-137), which was recently associated with schizophrenia through a “mega-GWAS.”26 It is also likely that some of the variants that we included as likely to be functional are in fact errors of reference-based mapping and that other functional variants (e.g., nonsense mutations) were excluded from the analysis as a result of mismapping.45
Second, our initial analysis included cases from multiple populations, including Finns and African Americans. If rare schizophrenia-associated variants are population specific, power would be reduced by the lack of focus on a single ethnic or racial group. We know from previous studies that different racial and ethnic groups share rare CNVs,17 but this could be because they are in hypermutable regions. Thus, the same structural changes arise multiple independent times, which is much less likely to be the case for SNVs. Although we performed separate analyses for samples of self-reported European and African ancestry, we were not able to control for population structure within these groups. Stratification usually acts to inflate test statistics, but the effect on power was expected to be modest. Additionally, existing methods are not effective for the type of structure caused by rare variants.53 We therefore employed the approach that was maximally inclusive in this exploratory study and intended that any variants that were supported in the second stage would be further investigated in cohorts that are robust to the effects of population stratification (e.g., in family-based studies, as suggested in Mathieson et al.53). Finally, the discovery sample comprised mostly people with treatment-resistant schizophrenia; these individuals are a special population representing about 30% of people who meet the DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition) criteria for schizophrenia.54 The replication sample had a much smaller percentage of treatment-resistant cases, so there remains a possibility that real associated variants were not supported in the follow-up sample because they increase the risk for treatment-resistant schizophrenia specifically, but not for schizophrenia more generally.
Our findings will aid in the interpretation of future small-scale sequencing studies. Here, we have investigated almost all obviously functional variants in a set of 166 schizophrenia cases and have found no study-wide significant associations. Notably, the majority of nonprivate variants that were exclusive to cases in the discovery cohort were either seen in controls in the follow-up sample (60%) or not seen in any further cases (23%). Thus, associations based on small-scale sequencing studies, however biologically enticing, must be interpreted with caution pending validation in larger follow-up studies.
If the majority of variants occur at a very low frequency, there are a number of potential investigative strategies. The most obvious and immediate strategy would be to use gene-based NGS approaches for comprehensively investigating candidate genes (e.g., from CNV studies or GWASs) in large data sets, as described above. This approach would allow us to accurately gauge how much of the heritability of schizophrenia is explained by genes that have already been identified. We can also take advantage of the relatively small early sequencing data sets to identify a small number of promising candidate rare variants, such as those that continue to be absent in controls in the present study, for genotyping in larger data sets. Although the majority of these candidates will turn out to be false positives, potential insight into schizophrenia etiology is worth the cost if even one is statistically proven. Finally, rare variants can be investigated via family studies through either segregation analyses in multiplex families or the search for de novo or recessive variants in sporadic cases. Although both of these approaches are likely to produce multiple candidate variants per family, with adequate control data sets and by the comparison of multiple families in collaborative efforts, it should be possible to identify either recurring variants or repeatedly disrupted genes that will ultimately prove to contribute to schizophrenia risk.
Acknowledgments
We thank the participants who made their DNA available for research. We thank Jordan Silver and Marlyne Silver; doctors and staff at Central Regional Hospital; Robert Millet, Edward Leuth, and other doctors and staff at Carolina Behavioral Care for local patient recruitment; and Latasha Little, Ken Cronin, Melora McCall, and Alex McKenzie for lab support. For the Italian cohort, we acknowledge Catia Scassellati and Cristian Bonvicini for sample handling and Stefano Bignotti, Giuseppe Rossi, and Alessandra Minelli for recruitment. We thank the Murdock study community registry and biorepository pro00011196, D. Daskalakis, R. Brown, A. Holden, E. Behr, W. Lowe, P. Lugar, J. Milner, K. Welsh-Bohmer, D. Valle, J. Hoover-Fong, D. Marchuk, S. Palmer, E. Pras, D. Lancet, and Z. Farfel for control DNA. This work was funded by National Institute of Mental Health (NIMH) grant 3RC2MH089915-01W1 and R01 grant MH071523, National Institute of Neurological Disorders and Stroke grant RC2NS070344, National Institute of Allergy and Infectious Diseases grant UO1AIO67854, Italian Ministry of Health (Ricerca Corrente) and Associazione Fatebenefratelli per la Ricerca grants, Ellison Funding, National Institute on Aging P30 grant AG028377, the Sidney R. Baer, Jr. Foundation, the Essel Foundation, the Carmela and Menachem Abraham Fund, and the Brain and Behavior Research Foundation. This research was performed in collaboration with the International Serious Adverse Events Consortium, whose membership includes Abbott, Amgen, Astra-Zeneca, Cerner, Daiichi-Sankyo, GlaxoSmithKline, Merck, Novartis, Pfizer, Takeda, and the Wellcome Trust. The NIMH Cell Repository at Rutgers University provided schizophrenia follow-up samples. H.Y.M. is a shareholder in SureGene, LLC.
Contributor Information
Anna C. Need, Email: anna.need@duke.edu.
David B. Goldstein, Email: d.goldstein@duke.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
HUGO Gene Nomenclature Committee, http://www.genenames.org
NHLBI Exome Sequencing Project Exome Variant Server, http://snp.gs.washington.edu/EVS
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Picard, http://picard.sourceforge.net
Reactome, http://www.reactome.org
SequenceVariantAnalyzer (SVA), http://www.svaproject.org
References
- 1.Tiihonen J., Lönnqvist J., Wahlbeck K., Klaukka T., Niskanen L., Tanskanen A., Haukka J. 11-year follow-up of mortality in patients with schizophrenia: A population-based cohort study (FIN11 study) Lancet. 2009;374:620–627. doi: 10.1016/S0140-6736(09)60742-X. [DOI] [PubMed] [Google Scholar]
- 2.Meltzer H.Y., Alphs L., Green A.I., Altamura A.C., Anand R., Bertoldi A., Bourgeois M., Chouinard G., Islam M.Z., Kane J., International Suicide Prevention Trial Study Group Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT) Arch. Gen. Psychiatry. 2003;60:82–91. doi: 10.1001/archpsyc.60.1.82. [DOI] [PubMed] [Google Scholar]
- 3.Green M.F., Kern R.S., Braff D.L., Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: Are we measuring the “right stuff”? Schizophr. Bull. 2000;26:119–136. doi: 10.1093/oxfordjournals.schbul.a033430. [DOI] [PubMed] [Google Scholar]
- 4.Blennow K., de Leon M.J., Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 5.Baulac S., Baulac M. Advances on the genetics of Mendelian idiopathic epilepsies. Clin. Lab. Med. 2010;30:911–929. doi: 10.1016/j.cll.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 6.Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietiläinen O.P., Mors O., Mortensen P.B., Genetic Risk and Outcome in Psychosis (GROUP) Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J., Levinson D.F., Duan J., Sanders A.R., Zheng Y., Pe’er I., Dudbridge F., Holmans P.A., Whittemore A.S., Mowry B.J. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O’Donovan M.C., Sullivan P.F., Sklar P., International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vacic V., McCarthy S., Malhotra D., Murray F., Chou H.H., Peoples A., Makarov V., Yoon S., Bhandari A., Corominas R. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bassett A.S., Chow E.W. Schizophrenia and 22q11.2 deletion syndrome. Curr. Psychiatry Rep. 2008;10:148–157. doi: 10.1007/s11920-008-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girard S.L., Gauthier J., Noreau A., Xiong L., Zhou S., Jouan L., Dionne-Laporte A., Spiegelman D., Henrion E., Diallo O. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 14.Xu B., Roos J.L., Dexheimer P., Boone B., Plummer B., Levy S., Gogos J.A., Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rees E., Moskvina V., Owen M.J., O’Donovan M.C., Kirov G. De novo rates and selection of schizophrenia-associated copy number variants. Biol. Psychiatry. 2011;70:1109–1114. doi: 10.1016/j.biopsych.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Need A.C., Ge D., Weale M.E., Maia J., Feng S., Heinzen E.L., Shianna K.V., Yoon W., Kasperaviciūte D., Gennarelli M. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 2009;5:e1000373. doi: 10.1371/journal.pgen.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vrijenhoek T., Buizer-Voskamp J.E., van der Stelt I., Strengman E., Sabatti C., Geurts van Kessel A., Brunner H.G., Ophoff R.A., Veltman J.A., Genetic Risk and Outcome in Psychosis (GROUP) Consortium Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 2008;83:504–510. doi: 10.1016/j.ajhg.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rujescu D., Ingason A., Cichon S., Pietilainen O.P., Barnes M.R., Toulopoulou T., Picchioni M., Vassos E., Ettinger U., Bramon E. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 2008;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menashe I., Rosenberg P.S., Chen B.E. PGA: power calculator for case-control genetic association analyses. BMC Genet. 2008;9:36. doi: 10.1186/1471-2156-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelak K., Shianna K.V., Ge D., Maia J.M., Zhu M., Smith J.P., Cirulli E.T., Fellay J., Dickson S.P., Gumbs C.E. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6:6. doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinzen E.L., Depondt C., Cavalleri G.L., Ruzzo E.K., Walley N.M., Need A.C., Ge D., He M., Cirulli E.T., Zhao Q. Exome sequencing followed by large scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am. J. Hum. Genet. 2012 doi: 10.1016/j.ajhg.2012.06.016. Published online August 2, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ripke S., Sanders A.R., Kendler K.S., Levinson D.F., Sklar P., Holmans P.A., Lin D.Y., Duan J., Ophoff R.A., Andreassen O.A., Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernheim J., Benchetrit S. The potential roles of FGF23 and Klotho in the prognosis of renal and cardiovascular diseases. Nephrol. Dial. Transplant. 2011;26:2433–2438. doi: 10.1093/ndt/gfr208. [DOI] [PubMed] [Google Scholar]
- 28.Kosakai A., Ito D., Nihei Y., Yamashita S., Okada Y., Takahashi K., Suzuki N. Degeneration of mesencephalic dopaminergic neurons in klotho mouse related to vitamin D exposure. Brain Res. 2011;1382:109–117. doi: 10.1016/j.brainres.2011.01.056. [DOI] [PubMed] [Google Scholar]
- 29.Carpinelli M.R., Wise A.K., Burt R.A. Vitamin D-deficient diet rescues hearing loss in Klotho mice. Hear. Res. 2011;275:105–109. doi: 10.1016/j.heares.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 30.McGrath J.J., Burne T.H., Féron F., Mackay-Sim A., Eyles D.W. Developmental vitamin D deficiency and risk of schizophrenia: A 10-year update. Schizophr. Bull. 2010;36:1073–1078. doi: 10.1093/schbul/sbq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGrath J.J., Eyles D.W., Pedersen C.B., Anderson C., Ko P., Burne T.H., Norgaard-Pedersen B., Hougaard D.M., Mortensen P.B. Neonatal vitamin D status and risk of schizophrenia: A population-based case-control study. Arch. Gen. Psychiatry. 2010;67:889–894. doi: 10.1001/archgenpsychiatry.2010.110. [DOI] [PubMed] [Google Scholar]
- 32.Amato R., Pinelli M., Monticelli A., Miele G., Cocozza S. Schizophrenia and vitamin D related genes could have been subject to latitude-driven adaptation. BMC Evol. Biol. 2010;10:351. doi: 10.1186/1471-2148-10-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kesby J.P., O’Loan J.C., Alexander S., Deng C., Huang X.F., McGrath J.J., Eyles D.W., Burne T.H. Developmental vitamin D deficiency alters MK-801-induced behaviours in adult offspring. Psychopharmacology (Berl.) 2012;220:455–463. doi: 10.1007/s00213-011-2492-0. [DOI] [PubMed] [Google Scholar]
- 34.Parra M., Gee S., Chan N., Ryaboy D., Dubchak I., Mohandas N., Gascard P.D., Conboy J.G. Differential domain evolution and complex RNA processing in a family of paralogous EPB41 (protein 4.1) genes facilitate expression of diverse tissue-specific isoforms. Genomics. 2004;84:637–646. doi: 10.1016/j.ygeno.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Shen L., Liang F., Walensky L.D., Huganir R.L. Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J. Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiedler M.J., Nathanson M.H. The type I inositol 1,4,5-trisphosphate receptor interacts with protein 4.1N to mediate neurite formation through intracellular Ca waves. Neurosignals. 2011;19:75–85. doi: 10.1159/000324507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamdan F.F., Gauthier J., Araki Y., Lin D.T., Yoshizawa Y., Higashi K., Park A.R., Spiegelman D., Dobrzeniecka S., Piton A., S2D Group Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benediktsson A.M., Marrs G.S., Tu J.C., Worley P.F., Rothstein J.D., Bergles D.E., Dailey M.E. Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia. 2012;60:175–188. doi: 10.1002/glia.21249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka K., Watase K., Manabe T., Yamada K., Watanabe M., Takahashi K., Iwama H., Nishikawa T., Ichihara N., Kikuchi T. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 40.Kennedy M.J., Davison I.G., Robinson C.G., Ehlers M.D. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davy B.E., Robinson M.L. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003;12:1163–1170. doi: 10.1093/hmg/ddg122. [DOI] [PubMed] [Google Scholar]
- 42.Zhai R.G., Vardinon-Friedman H., Cases-Langhoff C., Becker B., Gundelfinger E.D., Ziv N.E., Garner C.C. Assembling the presynaptic active zone: A characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- 43.Nitta A., Hibi Y., Miyamoto Y., Nabeshima T. [Identification of Piccolo as a regulator of behavioral plasticity and dopamine transporter internalization] Nihon Arukoru Yakubutsu Igakkai Zasshi. 2010;45:525–529. [PubMed] [Google Scholar]
- 44.O’Donovan M.C., Craddock N., Norton N., Williams H., Peirce T., Moskvina V., Nikolov I., Hamshere M., Carroll L., Georgieva L., Molecular Genetics of Schizophrenia Collaboration Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 45.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrett J.C., Cardon L.R. Evaluating coverage of genome-wide association studies. Nat. Genet. 2006;38:659–662. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- 47.Botstein D., Risch N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 48.Antonarakis S.E., Chakravarti A., Cohen J.C., Hardy J. Mendelian disorders and multifactorial traits: The big divide or one for all? Nat. Rev. Genet. 2010;11:380–384. doi: 10.1038/nrg2793. [DOI] [PubMed] [Google Scholar]
- 49.Dering C., Hemmelmann C., Pugh E., Ziegler A. Statistical analysis of rare sequence variants: An overview of collapsing methods. Genet. Epidemiol. 2011;35(Suppl 1):S12–S17. doi: 10.1002/gepi.20643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li, L., Zheng, W., Lee, J.S., Zhang, X., Ferguson, J., Yan, X., and Zhao, H. (2011). Collapsing-based and kernel-based single-gene analyses applied to Genetic Analysis Workshop 17 mini-exome data. BMC proceedings 5 Suppl 9, S117. [DOI] [PMC free article] [PubMed]
- 51.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun Y.V., Sung Y.J., Tintle N., Ziegler A. Identification of genetic association of multiple rare variants using collapsing methods. Genet. Epidemiol. 2011;35(Suppl 1):S101–S106. doi: 10.1002/gepi.20658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mathieson I., McVean G. Differential confounding of rare and common variants in spatially structured populations. Nat. Genet. 2012;44:243–246. doi: 10.1038/ng.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meltzer H.Y. Treatment-resistant schizophrenia—the role of clozapine. Curr. Med. Res. Opin. 1997;14:1–20. doi: 10.1185/03007999709113338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.