

Abstract
Excessive growth of terminal hair around the elbows (hypertrichosis cubiti) has been reported both in isolation and in association with a variable spectrum of associated phenotypic features. We identified a cohort of six individuals with hypertrichosis cubiti associated with short stature, intellectual disability, and a distinctive facial appearance, consistent with a diagnosis of Wiedemann-Steiner syndrome (WSS). Utilizing a whole-exome sequencing approach, we identified de novo mutations in MLL in five of the six individuals. MLL encodes a histone methyltransferase that regulates chromatin-mediated transcription through the catalysis of methylation of histone H3K4. Each of the five mutations is predicted to result in premature termination of the protein product. Furthermore, we demonstrate that transcripts arising from the mutant alleles are subject to nonsense-mediated decay. These findings define the genetic basis of WSS, provide additional evidence for the role of haploinsufficency of histone-modification enzymes in multiple-congenital-anomaly syndromes, and further illustrate the importance of the regulation of histone modification in development.
Main Text
The non-androgen-related excessive growth of terminal hair (hypertrichosis terminalis) may arise through conversion of vellus hair to terminal hair, alteration of the hair growth cycle, and increase in hair-follicle density.1 However, little is known about the molecular pathways that control these biological processes. A localized form of hypertrichosis terminalis of the extensor surfaces of the distal upper arm and proximal forearm is known as hypertrichosis cubiti and has also been termed the “hairy elbows syndrome” (MIM 139600). Individuals have been described with hypertrichosis cubiti both in isolation and with a variable spectrum of associated phenotypic features (reviewed in Polizzi et al.2 and Visser et al.3), including several (but not all) individuals reported under the clinical classification of Wiedemann-Steiner syndrome4,5 (WSS, MIM 605130).
We sought to investigate the genetic basis of congenital hypertrichosis cubiti using a whole-exome sequencing approach. Given the heterogeneous spectrum of phenotypic features reported to be associated with hypertrichosis cubiti, identification of a distinct disease phenotype within this group was essential for this study. We defined a discrete subgroup who had hypertrichosis cubiti, intellectual disability, a distinctive facial appearance (Figure 1), and short stature, and we postulated that this phenotype represented a single condition. This group is reminiscent of a subset of reported WSS cases in which hypertrichosis cubiti was observed. Six unrelated individuals with features consistent with this discrete phenotype (Table 1) were recruited to our study. None has a family history of hypertrichosis cubiti or intellectual disability. Each family provided full written consent, and ethical approval for this study was obtained from Guy’s and St. Thomas’ National Health Service (NHS) Foundation Trust local research ethics committee (ref.: 08/H0802/84, “Systematic Characterization of Genes in Inherited Disorders”).
Figure 1.
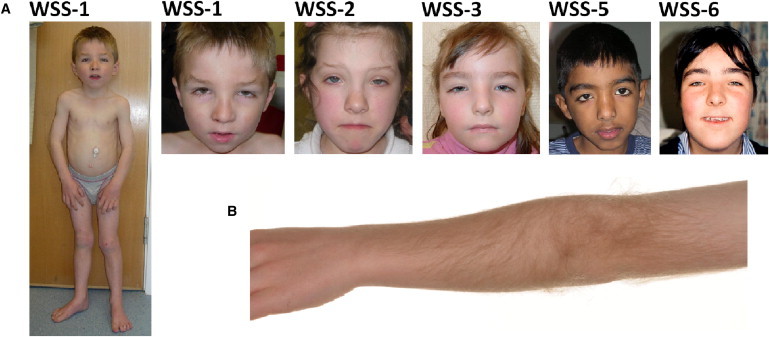
Clinical Images of Individuals in whom Mutations Were Identified in MLL
(A) Slim yet muscular build and the characteristic facial features, including long eyelashes, thick or arched eyebrows with a lateral flare, and downslanting and vertically narrow palpebral fissures. A broad nasal bridge, a wide nasal tip, and a Cupid’s bow contour to the upper lip are also seen.
(B) Hypertrichosis cubiti—excessive hair growth on the extensor surface of the distal upper arm and proximal forearm (individual WSS-1).
Table 1.
Major Clinical and Genetic Findings in the Six Individuals in the Present Study
|
Individual |
||||||
|---|---|---|---|---|---|---|
| WSS-1 | WSS-2 | WSS-3 | WSS-4 | WSS-5 | WSS-6 | |
| MLL nucleotide alteration | c.8806_8809del | c.8267del | c.6913del | − | c.7144C>T | c.4599dup |
| MLL amino-acid alteration | p.Val2936∗ | p.Leu2756∗ | p.Ser2305Leufs∗2 | − | p.Arg2382∗ | p.Lys1534∗ |
| Gender | male | female | female | male | male | female |
| Age at last examination (years) | 6 | 8 | 12 | 5 | 8 | 24 |
| Growth Parameters | ||||||
| Birth weight (centile) | 75th | 2nd–9th | 0.4th–2nd | 2nd–9th | 9th | 25th–50th |
| Head circumferencea (centile) | 9th | <0.4th | 50th | <0.4th | <2nd | 50th |
| Heighta (centile) | 0.4th–2nd | <0.4th | 9th (GH) | 2nd–9th | 9th | 0.4th–2nd |
| Weighta (centile) | 2nd–9th | <0.4th | 25th | 2nd–9th | 2nd | 25th |
| Facial Features | ||||||
| Eyebrow lateral flare | + | + | − | − | − | − |
| Thick eyebrows | + | − | + | + | + | + |
| Long eyelashes | + | + | + | + | + | + |
| Palpebral fissure, downslanted | + | + | + | − | + | + |
| Palpebral fissure, vertically narrow | + | + | + | − | + | + |
| Wide nasal bridge | + | + | + | − | + | + |
| Broad nasal tip | + | + | + | − | + | + |
| Cupid’s bow, exaggerated | − | + | + | − | + | − |
| Upper vermillion border, thin | + | − | − | + | − | + |
| Musculoskeletal | ||||||
| Broad first digits | + | − | − | − | + | − |
| Tapering fingers | − | + | − | + | + | + |
| 2–3 toe syndactyly | − | + | − | + | + | − |
| Long hallux | − | + | − | − | + | − |
| Slim and muscular build | + | + | − | + | + | − |
| Rib anomalies on X-ray | + | U | + | U | + | − |
| Scoliosis | − | − | − | − | − | + |
| Sacral dimple | + | + | + | − | U | + |
| Gastrointestinal | ||||||
| Constipation | + | + | − | − | + | − |
| Feeding difficulties | + | + | − | + | + | − |
| NG and PEG feeding | + | + | − | − | − | − |
| Hair | ||||||
| Hypertrichosis cubiti | + | + | + | − | + | + |
| Hypertrichosis, back | + | + | + | + | + | + |
| Hypertrichosis, lower limbs | − | − | + | − | − | + |
| Development and Behavior | ||||||
| Developmental delay | moderate | moderate | moderate | mild | mild | mild |
| Autism | + | + | − | − | − | − |
| Aggressive behavior | + | − | + | − | + | − |
| Poor sleep | + | + | − | − | − | − |
| Hyperactivity | − | − | − | + | − | − |
| Cardiac | ||||||
| Cardiac anomaly | − | −b | − | ASD | −b | PDA |
| Investigations | ||||||
| Full blood count | normal | NA | normal | NA | NA | normal |
+, feature present; −, feature absent; GH, growth hormone; U, unknown; NG, nasogastric; PEG, percutaneous endoscopic gastrostomy; ASD, atrial septal defect; PDA, patent ductus arteriosus; NA, not assessed
At last examination.
Normal clinical cardiac examination (has not had echocardiogram).
DNA was extracted from blood and the role of large-scale chromosomal abnormalities was excluded in each of the six cases (WSS-1–WSS-6) via comparative genomic hybridization (Agilent Human Genome CGH Microarray 44K: WSS-1, WSS-2, WSS-3, and WSS-4; Agilent Human Genome CGH Microarray 60K: WSS-5 and WSS-6; data not shown). We therefore sought to identify disease-causing alleles with the use of a whole-exome sequencing strategy. Three micrograms of genomic DNA from four of the six cases was sheared with the use of focused acoustic technology (Covaris), which yielded a mean fragment size of 150 bp. Fragments were end repaired and sequencing adaptors were ligated. The sequence library was hybridized for 24 hr with biotinylated 120 bp RNA probes designed against coding regions of the genome (Agilent). Streptavidin-coated magnetic beads were utilized to retain DNA bound to the RNA probes, and unbound DNA was washed off. The exome-enriched pool of DNA was then eluted and amplified with a low-cycle PCR. Finally, the enriched DNA fragments were sequenced with 100 bp paired-end reads on the Illumina HiSeq2000. The resulting reads were aligned to the reference human genome (hg19, National Center for Biotechnology Information build 37) with Novoalign (Novocraft Technologies). Over 7.0 Gb of sequence was generated for each subject, such that > 85% of the coding bases of the GENCODE-defined exome were represented by at least 20 reads (Table S1 available online). Duplicate reads, resulting from PCR clonality or optical duplicates, and reads mapping to multiple locations were excluded from the downstream analysis. The depth and breadth of sequence coverage was calculated with custom scripts and the BedTools package.6 Single-nucleotide substitutions and small insertion-deletion variants were identified with SAMtools7 and annotated with respect to coding genes with the ANNOVAR software package8 (Table S2).
We interrogated the exome variant profiles of the four affected cases under the assumption that each of the causative alleles was dominant and had arisen de novo, highlighting candidate genes harboring at least one previously unobserved heterozygous nonsynonymous or splice-site substitution or a coding insertion or deletion in the same gene in all four individuals. This process did not reveal a single gene matching the criteria in all four individuals (Table S3). However, evaluation of these data with a prior expectation of genetic heterogeneity highlighted MLL (MIM 159555) as the only candidate gene harboring previously unobserved heterozygous variants in three of the four subjects (Table S3): a four-nucleotide deletion (WSS-1: c.8806_8809del [p.Val2936∗]) and two single-nucleotide deletions (WSS-2: c.8267del [p.Leu2756∗], WSS-3: c.6913del [p.Ser2305Leufs∗2]) (Figure 2, Table 1).
Figure 2.
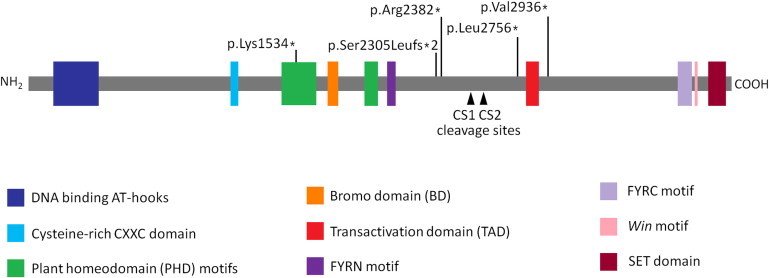
Domain Structure of MLL and the Locations of the Observed Mutations
The identified mutations are each predicted to cause premature termination of the protein product before the translation of the C-terminal region containing the FRYC motif, the Win motif, and the SET domain.
The three observed variants in MLL are all located within exon 27 of the 36-exon gene, which is located at chromosome 11q23 and encodes a 16.6 kb transcript (NM_001197104.1). Each is predicted to lead to the disruption of the reading frame, resulting in a predicted premature termination of the protein product with a high likelihood of functional impact (Figure 2). None of the observed mutations are present in dbSNP, have been observed by the 1000 Genomes Project, or were detected in 600 unrelated control exome profiles. The variants were confirmed with Sanger sequencing in the respective probands. Moreover, none of the mutations were present in unaffected parental DNA from leukocytes, consistent with the notion that the disease-causing alleles have arisen de novo. We sought to identify mutations in MLL in the remaining two individuals who met our clinical criteria. Sequencing of the coding regions and associated splice sites of MLL revealed a heterozygous variant in each subject; each is predicted to lead to premature termination of the protein product (WSS-5: c.7144C>T [p.Arg2382∗], WSS-6: c.4599dup [p.Lys1534∗]). Neither mutation was observed in the respective unaffected parental DNA from leukocytes. In total, we investigated six individuals who met our clinical criteria and identified mutations in five of them. Following a review of the findings, it was evident on clinical examination that hypertrichosis cubiti was observed in conjunction with a homogeneous group of associated features in the five individuals in whom we had identified MLL mutations (Table 1): distinct, consistent facial characteristics including long eyelashes, thick or arched eyebrows with a lateral flare, and downslanting and vertically narrow palpebral fissures (Figure 1); mild to moderate intellectual disability; and behavioral difficulties. In addition, the distribution of hair on the lower upper arm and upper forearm was observed in combination with excessive hair on the back with a whorl-like distribution. All five individuals were < 10th centile for height, and a sacral dimple was observed in four of the five. The affected individual (WSS-4), in whom exome sequencing did not reveal any potentially pathogenic variation in MLL, and high-resolution comparative genomic hybridization did not reveal genomic abnormalities (affecting MLL or elsewhere in the genome), presented some subtle phenotypic differences. The hair on the arms was at the upper limit of normal and was more diffuse than the hypertrichosis cubiti observed in the five individuals with mutant MLL alleles. Hair growth on the back was notably longer than that of the other five cases (extending up to 12 cm in length). His facial features also differed; downslanting palpebral fissures were absent. Although this case may be explained by an allele comprising a noncoding or undetected coding variant in MLL, in light of the observed phenotypic differences, it is reasonable to speculate that this individual most likely represents a phenotype within the spectrum of WSS for which the molecular genetic basis is distinct from the five cases in which mutations have been identified in MLL.
MLL encodes a histone methyltransferase that has been demonstrated to regulate chromatin-mediated transcription, including the transcription of multiple Hox and Wnt genes, through the catalysis of mono-, di-, and trimethylation of histone H3K4.9 The large (3,969 aa), multidomain MLL protein comprises three DNA binding AT-hooks at the N terminus, followed by a cysteine-rich CXXC domain, plant homeodomain finger motifs, a bromo domain, a transactivation domain, a nuclear receptor interaction motif, a WDR5 interaction (Win) motif, and a C-terminal SET domain, which is responsible for the protein’s histone-methyltransferase activity.10 The mature MLL is proteolytically processed at two independent sites (CS1, aa 2666–2667 and CS2, aa 2718–2719; Figure 2), and the cleaved C- and N-terminal fragments form a stable complex that localized to a subnuclear compartment.11 The identified mutations were each predicted to cause premature termination of the protein product, leading to a mature protein product lacking the SET domain and, therefore, histone-methyltransferase activity (Figure 2). To establish whether the mutant MLL alleles are subject to nonsense-mediated decay, we used a quantitative real-time PCR approach (Kapa Biosystems) performed on a 7900HT Fast Real-Time PCR System (Life Technologies) to assess the total abundance of MLL transcripts in primary skin fibroblast cells derived from individual WSS-3, heterozygous for the c.6913del allele. We observed that the level of MLL transcript is reduced in comparison to unrelated healthy controls (Figure 3), consistent with the notion that transcripts arising from the mutant MLL alleles are subject to nonsense-mediated decay.
Figure 3.
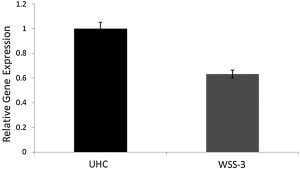
Total Abundance of MLL Transcript in Primary Skin Fibroblasts from WSS-3 and the Average of Two Unrelated Healthy Controls
MLL expression normalized to endogenous GAPDH and RPLO expression levels. Error bars represent SEM of three independent experiments. UHC, unrelated healthy controls.
MLL is widely expressed in most cell types,12 but it has been most extensively studied in the context of hematological malignancies. The MLL locus has been reported as the site of somatic chromosome translocations in acute myeloid, acute lymphoblastoid, or mixed-lineage leukemia, and more than 50 different fusion partners have been identified. In mice, homozygosity for MLL null alleles is embryonically lethal.13 However, heterozygous mice were demonstrated to be viable; they were small at birth and had retarded growth.13 These observations in the haploinsufficient murine model are consistent with birth weight in the lower range (4/5 individuals, <10th centile, Table 1), subsequent growth retardation, and height in the lower range (5/5 individuals, <10th centile, Table 1) in our cohort of individuals with heterozygous mutations in MLL. The heterozygous mice were also reported to be anemic and have decreased platelets, although the morphology of hematopoietic cells was normal. Immune function has not been fully investigated in our cohort; basic hematological investigations (full blood count), where available, had normal results (Table 1). However, it may be of potential functional importance that individual WSS-1 had a history of recurrent infections in childhood. Given the intellectual disability in all the individuals in whom we identified mutations in MLL (Table 1), it is of note that MLL also has a demonstrated neuronal function and has been shown to be required for neurogenesis in the postnatal brain.14
MLL demonstrates homology to other proteins with histone-methylation activity, including MLL2 (MIM 602113).15 Recently, several studies have reported an allelic series of heterozygous nonsense and frameshift mutations in MLL2 in individuals diagnosed with Kabuki syndrome (MIM 147920), a multiple-anomaly syndrome that is also associated with short stature, feeding difficulties, intellectual disability, and a distinctive facial appearance, including unusual eyebrows and long eyelashes.16–19 Furthermore, de novo truncating and missense mutations in CREBBP (MIM 600140), which encodes a protein with histone-acetyltransferase activity that has also previously been demonstrated to interact with MLL,20 underlie Rubinstein Taybi syndrome,21 and de novo truncating mutations in the gene encoding SRCAP (MIM 611421), a coactivator of CREBBP, are responsible for Floating Harbor syndrome22 (MIM 180849). Both of these multiple-anomaly disorders overlap phenotypically with WSS, including postnatal growth restriction, hypertrichosis, and intellectual disability.
The distinct phenotype observed in individuals in whom we have identified de novo MLL mutations is similar to that of certain individuals reported in the WSS literature. Specifically, the individual reported by Steiner and Marques4 and the individuals reported by Koenig et al.5 all had hypertrichosis, intellectual disability, and similar facial characteristics, including thick or arched eyebrows and downslanting and vertically narrow palpebral fissures. Within the WSS literature, there is substantial phenotypic variation; taken together with the fact that one of our six cases (WSS-4) does not harbor a mutation in MLL, these findings suggest that haploinsufficiency of MLL may underlie a distinct clinical and genetic entity within the WSS spectrum. Further investigation of the pathogenic role of MLL in a wider cohort of individuals with WSS and other congenital forms of hypertrichosis cubiti is needed to fully delineate the phenotypic spectrum associated with mutations in this gene.
In summary, we have identified the causal role of MLL haploinsufficiency in a subset of individuals with hypertrichosis cubiti who have additional phenotypic features that are consistent with a diagnosis of WSS. The features observed in these individuals, including the excessive growth of terminal hair, are probably the result of secondary downstream effects on transcription via disruption of the genomic distribution and the timing of histone-methylation events. Our findings provide additional evidence to the growing body of research that has demonstrated the role of haploinsuficiency of histone-modification enzymes in multiple-congenital-anomaly disorders16,23–25 and reinforce the important role that epigenetic control, and chromatin modification in particular, plays in both human development and disease.
Acknowledgments
The authors express their gratitude to the families for participating in this study. The authors also acknowledge support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s and St. Thomas’ NHS Foundation Trust, in partnership with the King’s College London and King’s College Hospital NHS Foundation Trust. We thank Alistair Calder (Department of Radiology, Great Ormond Street Hospital for Children, Great Ormond Street, London) for his assistance and expertise in reviewing the radiography.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
GENCODE Project, http://www.gencodegenes.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Wendelin D.S., Pope D.N., Mallory S.B. Hypertrichosis. J. Am. Acad. Dermatol. 2003;48:161–179. doi: 10.1067/mjd.2003.100. quiz 180–181. [DOI] [PubMed] [Google Scholar]
- 2.Polizzi A., Pavone P., Ciancio E., La Rosa C., Sorge G., Ruggieri M. Hypertrichosis cubiti (hairy elbow syndrome): a clue to a malformation syndrome. J. Pediatr. Endocrinol. Metab. 2005;18:1019–1025. doi: 10.1515/jpem.2005.18.10.1019. [DOI] [PubMed] [Google Scholar]
- 3.Visser R., Beemer F.A., Veenhoven R.H., De Nef J.J. Hypertrichosis cubiti: two new cases and a review of the literature. Genet. Couns. 2002;13:397–403. [PubMed] [Google Scholar]
- 4.Steiner C.E., Marques A.P. Growth deficiency, mental retardation and unusual facies. Clin. Dysmorphol. 2000;9:155–156. doi: 10.1097/00019605-200009020-00021. [DOI] [PubMed] [Google Scholar]
- 5.Koenig R., Meinecke P., Kuechler A., Schäfer D., Müller D. Wiedemann-Steiner syndrome: three further cases. Am. J. Med. Genet. A. 2010;152A:2372–2375. doi: 10.1002/ajmg.a.33587. [DOI] [PubMed] [Google Scholar]
- 6.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne T.A., Briggs S.D., Brock H.W., Martin M.E., Gibbs D., Allis C.D., Hess J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 10.Cosgrove M.S., Patel A. Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. FEBS J. 2010;277:1832–1842. doi: 10.1111/j.1742-4658.2010.07609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh J.J.D., Ernst P., Erdjument-Bromage H., Tempst P., Korsmeyer S.J. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol. Cell. Biol. 2003;23:186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler L.H., Slany R., Cui X., Cleary M.L., Mason D.Y. The HRX proto-oncogene product is widely expressed in human tissues and localizes to nuclear structures. Blood. 1997;89:3361–3370. [PubMed] [Google Scholar]
- 13.Yu B.D., Hess J.L., Horning S.E., Brown G.A., Korsmeyer S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 14.Lim D.A., Huang Y.C., Swigut T., Mirick A.L., Garcia-Verdugo J.M., Wysocka J., Ernst P., Alvarez-Buylla A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 2009;458:529–533. doi: 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prasad R., Zhadanov A.B., Sedkov Y., Bullrich F., Druck T., Rallapalli R., Yano T., Alder H., Croce C.M., Huebner K. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene. 1997;15:549–560. doi: 10.1038/sj.onc.1201211. [DOI] [PubMed] [Google Scholar]
- 16.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulussen A.D., Stegmann A.P., Blok M.J., Tserpelis D., Posma-Velter C., Detisch Y., Smeets E.E., Wagemans A., Schrander J.J., van den Boogaard M.J. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011;32:E2018–E2025. doi: 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- 18.Li Y., Bögershausen N., Alanay Y., Simsek Kiper P.O., Plume N., Keupp K., Pohl E., Pawlik B., Rachwalski M., Milz E. A mutation screen in patients with Kabuki syndrome. Hum. Genet. 2011;130:715–724. doi: 10.1007/s00439-011-1004-y. [DOI] [PubMed] [Google Scholar]
- 19.Banka S., Veeramachaneni R., Reardon W., Howard E., Bunstone S., Ragge N., Parker M.J., Crow Y.J., Kerr B., Kingston H. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012;20:381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ernst P., Wang J., Huang M., Goodman R.H., Korsmeyer S.J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 2001;21:2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petrij F., Giles R.H., Dauwerse H.G., Saris J.J., Hennekam R.C., Masuno M., Tommerup N., van Ommen G.J., Goodman R.H., Peters D.J. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 22.Hood R.L., Lines M.A., Nikkel S.M., Schwartzentruber J., Beaulieu C., Nowaczyk M.J., Allanson J., Kim C.A., Wieczorek D., Moilanen J.S., FORGE Canada Consortium Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome. Am. J. Hum. Genet. 2012;90:308–313. doi: 10.1016/j.ajhg.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clayton-Smith J., O’Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., Voss A.K., Thomas T., Biesecker L.G., Smith P. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simpson M.A., Deshpande C., Dafou D., Vissers L.E., Woollard W.J., Holder S.E., Gillessen-Kaesbach G., Derks R., White S.M., Cohen-Snuijf R. De novo mutations of the gene encoding the histone acetyltransferase KAT6B cause Genitopatellar syndrome. Am. J. Hum. Genet. 2012;90:290–294. doi: 10.1016/j.ajhg.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lederer D., Grisart B., Digilio M.C., Benoit V., Crespin M., Ghariani S.C., Maystadt I., Dallapiccola B., Verellen-Dumoulin C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.