

Abstract
Disproportionate short stature refers to a heterogeneous group of hereditary disorders that are classified according to their mode of inheritance, clinical skeletal and nonskeletal manifestations, and radiological characteristics. In the present study, we report on an autosomal-recessive osteocutaneous disorder that we termed SOFT (short stature, onychodysplasia, facial dysmorphism, and hypotrichosis) syndrome. We employed homozygosity mapping to locate the disease-causing mutation to region 3p21.1-3p21.31. Using whole-exome-sequencing analysis complemented with Sanger direct sequencing of poorly covered regions, we identified a homozygous point mutation (c.512T>C [p.Leu171Pro]) in POC1A (centriolar protein homolog A). This mutation was found to cosegregate with the disease phenotype in two families. The p.Leu171Pro substitution affects a highly conserved amino acid residue and is predicted to interfere with protein function. Poc1, a POC1A ortholog, was previously found to have a role in centrosome stability in unicellular organisms. Accordingly, although centrosome structure was preserved, the number of centrosomes and their distribution were abnormal in affected cells. In addition, the Golgi apparatus presented a dispersed morphology, cholera-toxin trafficking from the plasma membrane to the Golgi was aberrant, and large vesicles accumulated in the cytosol. Collectively, our data underscore the importance of POC1A for proper bone, hair, and nail formation and highlight the importance of normal centrosomes in Golgi assembly and trafficking from the plasma membrane to the Golgi apparatus.
Main Text
Mutations in a large number of genes have been shown over the past years to be associated with combined defects in skin and bone development in humans and animals. Most of these genes code for proteins associated with the synthesis of components of the extracellular matrix.1 Salient examples include Buschke-Ollendorff syndrome (MIM 166700),2 several subtypes of Ehlers-Danlos syndrome (MIM 130050),3 cutis laxa (MIM 219100),4 gerodermia osteodysplastica (MIM 231070),5 and the recently described macrocephaly, alopecia, cutis laxa, and scoliosis syndrome (MACS [MIM 613075]).6 This association is in line with what is known of the overlapping role played by these molecules in the proper formation of the dermis and in bone osteogenesis.5 In addition, the number of genetic defects shown to cause both skeletal anomalies and ectodermal defects is steadily increasing. Although some of these conditions affect epidermal development per se, as seen in primary autosomal-recessive hypertrophic osteoarthropathy7 (MIM 259100), many of them seem to preferentially target the growth and differentiation of epidermal appendages (rather than the epidermis itself), as in cartilage-hair hypoplasia (MIM 250250)8 or trichorhinophalangeal syndrome (MIM 190350).9 Several osteocutaneous syndromes, such as trichodentoosseous syndrome (MIM 190320),10 have been shown to affect more than two ectodermal appendages and, as such, should be considered as ectodermal dysplasias.11
In the present study, we delineate the molecular basis for a syndrome featuring severely short long bones, peculiar faces associated with paucity of hair, and nail anomalies.12 Specifically, we studied two highly consanguineous Arab Muslim families comprising nine individuals affected by a distinctive form of abnormal skeletal development. The disorder was inherited in both families in an autosomal-recessive manner (Figure 1). Growth retardation was evident on prenatal ultrasound as early as during the second trimester of pregnancy. Affected individuals reached a final stature consistent with a height age of 6–8 years (Figure 2A). Initially, head circumference was elevated during early childhood but became markedly low by adulthood. Psychomotor development was normal. Facial dysmorphism included a long triangular face with a prominent nose (Figure 2B). The affected family members featured an unusual high-pitched voice, small ears, clinodactyly of the fifth finger, brachydactyly, and hypoplastic distal phalanges and fingernails (Figure 2C) associated with postpubertal sparse and short hair (Figure 2D). Typical skeletal findings included short and thick long bones with mild irregular metaphyseal changes. Femoral necks were short. The pelvis and sacrum were hypoplastic. All long bones of hands were short, showing a major delay of carpal ossification, and cone-shaped epiphyses were noted. Vertebral body ossification was also delayed, as recently reported in more detail elsewhere.12
Figure 1.
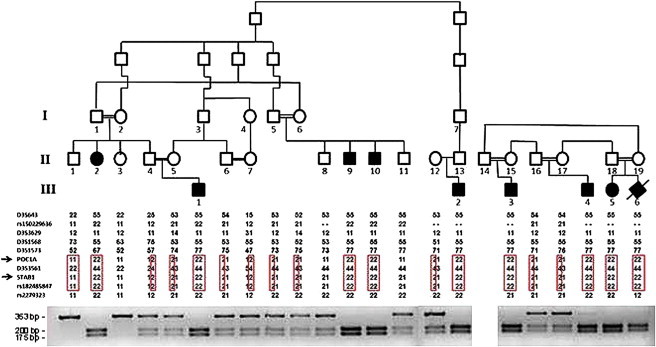
Linkage and PCR-RFLP Analyses of Two Families Affected by SOFT Syndrome
The family trees appear in the upper panel (black symbols denote affected individuals carrying mutation c.512T>C in POC1A). Haplotype analysis with polymorphic markers in chromosomal region 3q21 is depicted in the middle panel and reveals a homozygous haplotype shared by all affected individuals (boxed in red). The lower panel depicts the PCR-RFLP (restriction fragment-length polymorphism) analysis: the c.512T>C substitution generates a recognition site for BtgI. Thus, unaffected individuals display a single fragment of 353 bp, individuals affected with SOFT syndrome show two fragments of 200 and 175 bp, and all three fragments are found in heterozygous carriers of the mutation.
Figure 2.
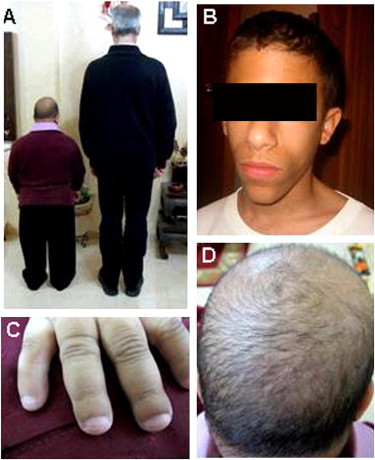
Clinical Features of SOFT Syndrome
(A) Affected individuals show a final height consistent with a height age of 6–8 years.
(B) Affected individuals display typical facial features, including a long triangular face with a prominent nose and small ears.
(C) Clinodactyly of the fifth finger, brachydactyly, and hypoplastic fingernails are noted.
(D) Note sparse and short hair.
All affected and healthy family members or their legal guardians provided written and informed consent according to a protocol approved by our institutional review board and by the Israel National Committee for Human Genetic Studies in adherence to the Helsinki guidelines. DNA was extracted from peripheral-blood lymphocytes. Given the common origin of the two families, the apparent rarity of the syndrome under study, and the fact that the two families were characterized by a high degree of consanguinity, we hypothesized that a founder homozygous mutation underlies the disease in both families. We therefore scrutinized the whole genome for homozygosity regions shared by all affected individuals by using the Illumina HumanLinkage-12 BeadChip (Illumina), which contains ∼6,000 tagged SNPs across the genome. DNA (200 ng) was hybridized according to the Infinium II assay (Illumina) and scanned with an Illumina BeadArray reader. The scanned images were imported into BeadStudio 3.1.3.0 (Illumina) for extraction and quality control, and there was an average call rate of 99.9%. The data were then scanned for homozygous regions with the use of a MatLab (MathWorks)-based software script. We identified two chromosomal regions of homozygosity larger than 2 Mb, one in 4q11-4q11 (3.7 Mb; three consecutive SNPs) and another in 3p21.1-3p21.31 (7.3 Mb; 14 consecutive SNPs). Polymorphic microsatellite or SNP markers spanning those two areas were selected from the GeneLoc site. As previously described,13 we established genotypes by PCR amplification of genomic DNA by using the BigDye terminator system on an automated sequencer (ABI PRISM 3100 Genetic Analyzer; Applied Biosystems, Foster City, CA, USA), and we determined allele sizes with GeneMapper v.4.0 software. Fine mapping of the disease-associated interval with the use of microsatellite typing suggested linkage to 3p21.2-3p21.31 (Figure 1) and excluded the other candidate locus on chromosome 4. Using the Superlink software,14 we obtained a maximal LOD score of 3.91 at marker D3S3561 (Figure S1, available online). Haplotype analysis revealed critical recombinations in individuals III-3 and II-2, setting the disease boundaries at markers D3S1573 and rs2279323 (Figure 1). The 2.156 Mb interval was found to contain 68 genes. Direct sequencing of 19 prominent candidate genes (Table S1) did not disclose any pathogenic change.
DNA of individual III-1 was then used for whole-exome capture and NextGen sequencing (see Table S2 for exome-sequencing details). To this end, genomic DNA was subjected to exome capture and sequencing (Otogenetics, Norcross, GA, USA). After fragmentation (Covaris, Woburn, MA, USA), DNA was tested for size distribution and concentration with an Agilent Bioanalyzer 2100 and Nanodrop. Illumina libraries were made from qualified fragmented gDNA with NEBNext reagents (New England Biolabs, Ipswich, MA, USA; catalog number E6040), and the resulting libraries were subjected to exome enrichment with NimbleGen SeqCap EZ Human Exome Library v.2.0 (Roche NimbleGen, Madison, WI, USA; catalog number 05860482001). Enriched libraries were tested for enrichment by qPCR, as well as for size distribution and concentration, by an Agilent Bioanalyzer 2100. The samples were then sequenced on an Illumina HiSeq2000. The platform provided by DNAnexus (DNAnexus, Mountain View, CA, USA) was used for uploading and analyzing data for quality, exome coverage, and exome-wide SNPs and indels.
Two mutations segregating with the disease phenotype in both families were identified in the disease interval: (1) c.694-3C>A in STAB1 (MIM 608560; RefSeq accession number NM_015136.2) was predicted by Splice Site Prediction by Neural Network to disrupt the intron 7 donor splice site; and (2) c.512T>C in POC1A (RefSeq NM_015426.4) (Figures 1 and 3A) was predicted to result in amino acid substitution p.Leu171Pro. We then screened a panel of 300 population-matched healthy individuals. The c.694-3C>A mutation in STAB1 was identified in three individuals. In addition, we were unable to demonstrate aberrant splicing of STAB1 in both peripheral leukocytes and skin fibroblasts obtained from affected individual III-1 (not shown), suggesting that the SOFT (short stature, onychodysplasia, facial dysmorphism, and hypotrichosis) syndrome phenotype results from the c.512T>C mutation in POC1A. A number of facts further point to the pathogenicity of the c.512T>C mutation: (1) c.512T>C in POC1A was not found in a series of 300 healthy individuals of Arab Muslim origin; (2) PolyPhen-2, SIFT, and align-GVGD predicted that the mutation would result in the substitution of a highly conserved amino acid residue (Figure 3B and Figure S2) (ConSeq score = 7/9) and would have a deleterious effect on protein (Table S3); and (3) a comparative study of transcription profiles of fibroblasts obtained from affected and healthy individuals with the use of the HumanHT-12 v.3 Expression BeadChip Kit (Illumina) revealed strong enrichment of biological networks and functions that are associated with the cell cycle, also known to be affected by defective POC1A function,15 as well as connective tissue and dermatology disorders (p values < 0.0005; Figure S3 and Table S4).
Figure 3.
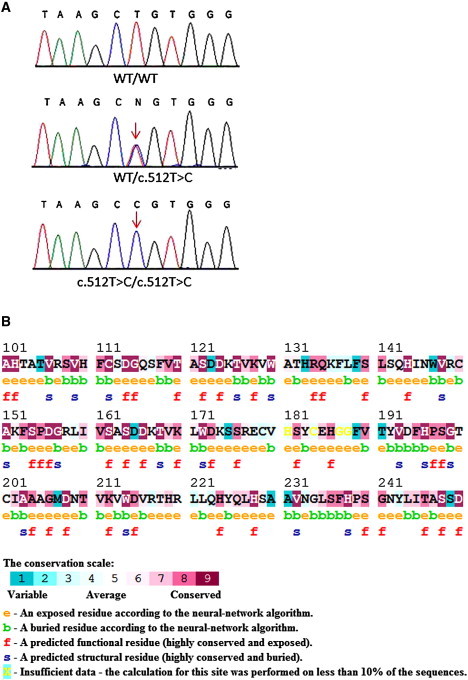
Mutation Analysis
(A) Sequence analysis of POC1A reveals a homozygous base substitution at cDNA position 512 (red arrow) in all affected individuals (lower panel). Healthy family members were found to be heterozygous carriers of the mutation (middle panel). The wild-type sequence is given for comparison (upper panel).
(B) The mutation in POC1A is predicted to alter a highly conserved amino acid residue as indicated by conservation analysis made with the ConSurf Server. The L171 residue is indicated by a black arrow.
Two POC1 proteins, POC1A and POC1B, have been described in humans, whereas in lower organisms, only one Poc1 protein is present.15 POC1 proteins are well conserved evolutionarily.16 They all contain an N-terminal WD40 domain, which forms a seven-bladed β-propeller, as well as a C-terminal coiled coil domain (Figure S2). They are part of centrosomes and are required for basal body and cilia formation. Some studies have suggested additional roles for POC1 proteins because they have been identified in association with mitochondria in human cells.15 Poc1 in lower organisms seems to play a dual role by regulating centriole formation and being essential for proper ciliogenesis.17
Given these data, we examined centrosome distribution in skin fibroblasts derived from affected individuals. Staining with γ-tubulin antibodies revealed that in the mutation-carrying fibroblasts, there was a significantly higher number of cells with an increased number of centrosomes (Figures 4A and 4B). Using electron microscopy (Figure 4C), we found that compared with normal cells, mutant cells had normal centrosome ultrastructure. Of note is the fact that microtubule organization tested by staining with β-tubulin did not present a gross change (Figure S4).
Figure 4.
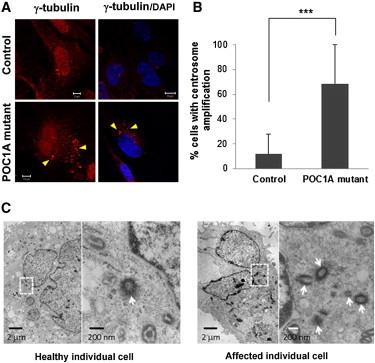
Centrosome Amplification
(A) Affected and healthy fibroblasts were fixed, stained for γ-tubulin (centrosomes) (Santa Cruz Biotechnology, Santa Cruz, CA, USA or Sigma-Aldrich, Saint Louis, MO, USA), and visualized by confocal microscopy. Nuclei were stained with DAPI. POC1A-mutant cells show significant amplification of centrosomes (>2/cell; arrows), which also appear to be in various shapes (sand like). Scale bars represent 5 μm and 10 μm.
(B) The number of cells displaying an abnormal centrosome number (>2/cell) and morphology was determined by the examination of 130–250 randomly chosen cells stained with γ-tubulin antibodies. Results are provided as a percentage of abnormal cells out of the total number of cells counted ± standard error of the mean (SEM). Asterisks indicate statistical significance (∗∗∗p < 0.0001) as analyzed by the Student's t test.
(C) Electron microscopic examination does not reveal significant differences between the ultrastructural appearances of centrioles (arrows) of affected and control cells. Scale bars represent 2 μm and 200 nm.
It has been recently shown that centrosomal microtubules are necessary for proper Golgi assembly.18 On the other hand, a large number of osteocutaneous syndromes, including autosomal-recessive cutis laxa type IIa (MIM 219200),19 gerodermia osteodysplastica,20 and MACS syndrome,6 have been shown to result from abnormal function of Golgi-related biological activities. Given that we noted amplification of centrosome number accompanied by centrosome disorganization in the mutant cells, we wondered whether this could affect Golgi structure, as well as centrosomal-microtubule-associated trafficking to the Golgi. A significant fraction of POC1A-mutant fibroblasts presented dispersed cisternae stacks of Golgi, as visualized by staining with the Golgi marker GM130 (Figure 5A). Although 78% of SOFT syndrome cells had a dispersed abnormal Golgi, only 10% of control fibroblasts showed altered Golgi morphology (Figure 5B).
Figure 5.
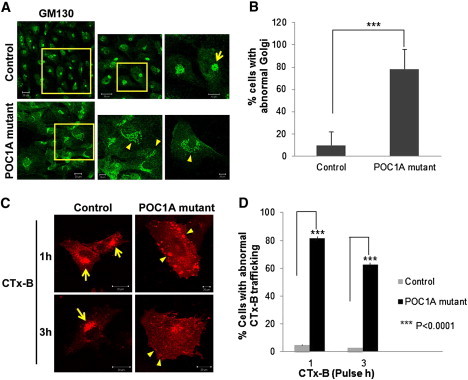
Altered Golgi Morphology and Aberrant Trafficking to the Golgi in SOFT Syndrome Fibroblasts
(A) SOFT syndrome fibroblasts and control fibroblasts were fixed and stained with GM130 antibodies (Sigma-Aldrich, Saint Louis, MO, USA). Arrows point to dispersed Golgi. Scale bars represent 10 μm and 20 μm.
(B) Cells displaying a dispersed or typical perinuclear Golgi pattern were counted (150–250 randomly chosen cells stained with GM130 antibodies were counted for each experiment). Results are provided as a percentage of abnormal cells out of the total number of cells counted ± SEM. Asterisks indicate statistical significance (∗∗∗p < 0.0001) as analyzed by the Student's t test.
(C) Cells were labeled with AlexaFluor 555-conjugated subunit B of cholera toxin (CTx-B; Invitrogen/Molecular Probes, Eugene, OR, USA) for the indicated times, fixed, and visualized by confocal microscopy. Arrows indicate normal transport of CTx-B to the Golgi, and arrowheads show atypical peripheral large vesicles representing abnormal CTx-B transport. The scale bar represents 20 μm.
(D) Abnormal CTx-B trafficking was quantified after 1 hr and 3 hr pulse in 40–85 randomly chosen cells. Results are provided as a percentage of cells displaying abnormal transport out of the total number of cells counted ± SEM. Asterisks indicate statistical significance (∗∗∗p < 0.0001) as analyzed by the Student's t test.
We next examined the functional efficacy of cellular trafficking from the plasma membrane to the Golgi because such trafficking depends on centrosome-derived microtubules. For this purpose, we followed the transport of subunit B of cholera toxin (CTx-B). The uptake of CTx-B, which binds GM1 on the plasma membrane, into mammalian cells can be mediated by several endocytic mechanisms.21,22 It does not recycle but reaches the Golgi, from which it traffics to the cytoplasm through the endoplasmic reticulum. To follow intracellular trafficking, we cultured cells on coverslips coated with fibronectin and labeled them with 0.5 μg/ml AlexaFluor 555-conjugated CTx-B at 37°C for either 1 hr or 3 hr. We then fixed and mounted them for confocal microscopy. The results (Figures 5C and 5D) showed that trafficking of the CTx-B from the plasma membrane to the Golgi apparatus was significantly altered in POC1A-mutant cells. Cholera toxin reached the Golgi after 1 hr of internalization in normal fibroblasts, whereas in POC1A-mutant cells, it accumulated in large peripheral vesicles in the cytosol. After 3 hr of pulse, CTx-B partially reached the Golgi in mutant cells; however, positive-CTx-B vesicles were still visible in their cytosol.
In summary, the present report delineates the molecular basis of a disproportionate-short-stature syndrome that we termed SOFT syndrome.12 We demonstrate that a missense mutation in POC1A is associated with arrested growth of bone and ectodermal tissues and results in a severe form of dwarfism together with facial dysmorphism, hypoplastic nails, and hypotrichosis. The substitution in centrosome-associated protein POC1A leads to centrosome disorganization and an increase in centrosome number, which is associated with abnormal trafficking from the plasma membrane to the Golgi. This abnormal transport most likely reflects the aberrant distribution of centrosome-associated microtubules, which are important for proper assembly of the Golgi stacks, as well as for normal trafficking to the Golgi.18 These functional defects are in line with a steadily expanding set of data suggesting an association between various defects in Golgi-associated proteins and syndromes featuring common bone and cutaneous abnormalities.6,19,23–25
Thus, through the study of a rare osteocutaneous syndrome, we confirm in a human context previous data attributing to POC1A an essential role in centrosome function, and we provide evidence for the importance of this molecule in normal bone, hair, and nail formation.
Acknowledgments
We would like to acknowledge the participation of all family members in this study.
Contributor Information
Stavit Shalev, Email: stavit_sh@clalit.org.il.
Eli Sprecher, Email: elisp@tasmc.health.gov.il.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ArrayExpress, http://www.ebi.ac.uk/arrayexpress/
ConSurf Server, http://consurftest.tau.ac.il/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
GeneLoc, http://genecards.weizmann.ac.il/geneloc/index.shtml
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
Superlink, http://bioinfo.cs.technion.ac.il/superlink-online-twoloci/makeped/TwoLociMultiPoint.html
UCSC Genome Browser, http://genome.ucsc.edu/
Accession Numbers
The ArrayExpress accession number for the microarray data presented in the Supplemental Data is E-MEXP-3650.
References
- 1.Bateman J.F., Boot-Handford R.P., Lamandé S.R. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- 2.Hellemans J., Preobrazhenska O., Willaert A., Debeer P., Verdonk P.C., Costa T., Janssens K., Menten B., Van Roy N., Vermeulen S.J. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat. Genet. 2004;36:1213–1218. doi: 10.1038/ng1453. [DOI] [PubMed] [Google Scholar]
- 3.Malfait F., Wenstrup R.J., De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet. Med. 2010;12:597–605. doi: 10.1097/GIM.0b013e3181eed412. [DOI] [PubMed] [Google Scholar]
- 4.Milewicz D.M., Urbán Z., Boyd C. Genetic disorders of the elastic fiber system. Matrix Biol. 2000;19:471–480. doi: 10.1016/s0945-053x(00)00099-8. [DOI] [PubMed] [Google Scholar]
- 5.Mohamed M., Kouwenberg D., Gardeitchik T., Kornak U., Wevers R.A., Morava E. Metabolic cutis laxa syndromes. J. Inherit. Metab. Dis. 2011;34:907–916. doi: 10.1007/s10545-011-9305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basel-Vanagaite L., Sarig O., Hershkovitz D., Fuchs-Telem D., Rapaport D., Gat A., Isman G., Shirazi I., Shohat M., Enk C.D. RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am. J. Hum. Genet. 2009;85:254–263. doi: 10.1016/j.ajhg.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uppal S., Diggle C.P., Carr I.M., Fishwick C.W., Ahmed M., Ibrahim G.H., Helliwell P.S., Latos-Bieleńska A., Phillips S.E., Markham A.F. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat. Genet. 2008;40:789–793. doi: 10.1038/ng.153. [DOI] [PubMed] [Google Scholar]
- 8.Ridanpää M., van Eenennaam H., Pelin K., Chadwick R., Johnson C., Yuan B., vanVenrooij W., Pruijn G., Salmela R., Rockas S. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 2001;104:195–203. doi: 10.1016/s0092-8674(01)00205-7. [DOI] [PubMed] [Google Scholar]
- 9.Momeni P., Glöckner G., Schmidt O., von Holtum D., Albrecht B., Gillessen-Kaesbach G., Hennekam R., Meinecke P., Zabel B., Rosenthal A. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat. Genet. 2000;24:71–74. doi: 10.1038/71717. [DOI] [PubMed] [Google Scholar]
- 10.Price J.A., Bowden D.W., Wright J.T., Pettenati M.J., Hart T.C. Identification of a mutation in DLX3 associated with tricho-dento-osseous (TDO) syndrome. Hum. Mol. Genet. 1998;7:563–569. doi: 10.1093/hmg/7.3.563. [DOI] [PubMed] [Google Scholar]
- 11.Itin P.H., Fistarol S.K. Ectodermal dysplasias. Am. J. Med. Genet. C. Semin. Med. Genet. 2004;131C:45–51. doi: 10.1002/ajmg.c.30033. [DOI] [PubMed] [Google Scholar]
- 12.Shalev-Alon S., Sr., Borochowitz Z.-U. A distinctive autosomal recessive syndrome of severe disproportionate short stature with short long bones, brachydactyly, and hypotrichosis in two consanguineous Arab families. Eur. J. Hum. Genet. 2012;55:256–264. doi: 10.1016/j.ejmg.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000;18:233–234. doi: 10.1038/72708. [DOI] [PubMed] [Google Scholar]
- 14.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 15.Hames R.S., Hames R., Prosser S.L., Euteneuer U., Lopes C.A., Moore W., Woodland H.R., Fry A.M. Pix1 and Pix2 are novel WD40 microtubule-associated proteins that colocalize with mitochondria in Xenopus germ plasm and centrosomes in human cells. Exp. Cell Res. 2008;314:574–589. doi: 10.1016/j.yexcr.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Fourrage C., Chevalier S., Houliston E. A highly conserved Poc1 protein characterized in embryos of the hydrozoan Clytia hemisphaerica: Localization and functional studies. PLoS ONE. 2010;5:e13994. doi: 10.1371/journal.pone.0013994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearson C.G., Osborn D.P., Giddings T.H., Jr., Beales P.L., Winey M. Basal body stability and ciliogenesis requires the conserved component Poc1. J. Cell Biol. 2009;187:905–920. doi: 10.1083/jcb.200908019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vinogradova T., Paul R., Grimaldi A.D., Loncarek J., Miller P.M., Yampolsky D., Magidson V., Khodjakov A., Mogilner A., Kaverina I. Concerted effort of centrosomal and Golgi-derived microtubules is required for proper Golgi complex assembly but not for maintenance. Mol. Biol. Cell. 2012;23:820–833. doi: 10.1091/mbc.E11-06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kornak U., Reynders E., Dimopoulou A., van Reeuwijk J., Fischer B., Rajab A., Budde B., Nürnberg P., Foulquier F., Lefeber D., ARCL Debré-type Study Group Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 20.Hennies H.C., Kornak U., Zhang H., Egerer J., Zhang X., Seifert W., Kühnisch J., Budde B., Nätebus M., Brancati F. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat. Genet. 2008;40:1410–1412. doi: 10.1038/ng.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirkham M., Fujita A., Chadda R., Nixon S.J., Kurzchalia T.V., Sharma D.K., Pagano R.E., Hancock J.F., Mayor S., Parton R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005;168:465–476. doi: 10.1083/jcb.200407078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinnapen D.J., Chinnapen H., Saslowsky D., Lencer W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007;266:129–137. doi: 10.1111/j.1574-6968.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hucthagowder V., Morava E., Kornak U., Lefeber D.J., Fischer B., Dimopoulou A., Aldinger A., Choi J., Davis E.C., Abuelo D.N. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet. 2009;18:2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Syx D., Malfait F., Van Laer L., Hellemans J., Hermanns-Lê T., Willaert A., Benmansour A., De Paepe A., Verloes A. The RIN2 syndrome: A new autosomal recessive connective tissue disorder caused by deficiency of Ras and Rab interactor 2 (RIN2) Hum. Genet. 2010;128:79–88. doi: 10.1007/s00439-010-0829-0. [DOI] [PubMed] [Google Scholar]
- 25.Morava E., Guillard M., Lefeber D.J., Wevers R.A. Autosomal recessive cutis laxa syndrome revisited. Eur. J. Hum. Genet. 2009;17:1099–1110. doi: 10.1038/ejhg.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.