

Abstract
The dopamine D3 receptor (D3R) has been implicated in substance abuse and other neuropsychiatric disorders. The high sequence homology between the D3R and D2R, especially within the orthosteric binding site (OBS) that binds dopamine, has made the development of D3R-selective compounds challenging. Here, we deconstruct into pharmacophoric elements a series of D3R-selective substituted-4-phenylpiperazine compounds, and use computational simulations and binding and activation studies to dissect the structural bases for D3R selectivity and efficacy. We find that selectivity arises from divergent interactions within a second binding pocket (SBP) separate from the OBS, whereas efficacy depends on the binding mode in the OBS. Our findings reveal structural features of the receptor that are critical to selectivity and efficacy that can be used to design highly D3R-selective ligands with targeted efficacies. These findings are generalizable to other GPCRs in which the SBP can be targeted by bitopic or allosteric ligands.
The neurotransmitter dopamine is synthesized in and released from dopaminergic neurons to stimulate G-protein coupled receptors, thereby affecting movement, cognition and emotion. Dopamine receptors are classified into two subfamilies, D1-like and D2-like, based on sequence, G-protein coupling and pharmacology. The D1-like subfamily is comprised of the D1 and D5 receptors that are coupled to stimulatory G-protein α subunits (Gs/olf) and activate adenylyl cyclase. In contrast, the D2, D3 and D4 receptors (D2R, D3R, and D4R, respectively), members of the D2-like subfamily, are coupled to inhibitory G-protein α subunits (Gi/o), thus inhibiting adenylyl cyclase as well as modulating other effector pathways.1–3
The high degree of sequence identity within the transmembrane (TM) segments of the D2R-like receptors and the near-identity of the residues that form the binding site in these receptors4 have made it challenging to create subtype-selective agents that possess physicochemical properties suitable for in vivo characterization of the physiological roles of these receptor subtypes. Nevertheless, significant progress has been made for D2-subfamily receptor-selective ligands, including the discovery of selective D2R, D3R and D4R antagonists and partial agonists.1, 5, 6 The selective distribution of D3R in limbic regions of the brain, especially the nucleus accumbens7 has fortified interest in the D3R as a potential target for drug discovery, especially for drug addiction.5 Notably, several D3R-selective agents have been evaluated in animal models of addiction and other neuropsychiatric disorders,5, 8 and clinical trials have been initiated for smoking cessation with one of these agents (GSK598809, compound 1 in Figure 1; see ref9).
Figure 1. D3R-selective compounds.

Although structural templates differ across laboratories, structure-activity relationships (SAR) that have been developed at D3R for antagonists and partial agonists have traditionally focused on a functionalized tertiary amine (e.g. 2,3-diCl-4-phenylpiperazine in Figure 1) separated from an extended arylamide terminus by a requisite 4-atom (n-butyl) linking chain.5, 8 Functionalizing the linker with a 2,3-trans olefin or a 3-substituent (e.g. OH or F) further increases D3R selectivity.10–12 Whereas the 4-phenylpiperazine has been characterized as the primary recognition element, binding selectivity between D2R, D3R and D4R is dependent on the length of the linker that separates this primary pharmacophore from the aryl terminus.5, 13
Despite these successful medicinal chemistry efforts leading to compounds such as R22 (2) with <E100-fold selectivity for D3R over D2R10 (Figure 1), the structural basis for this binding specificity has not been elucidated. The recent availability of a high-resolution structure of the D3R has made structure-based drug design a possibility, but the very high degree of homology between D3R and D2R continues to pose a major challenge. The crystal structure of D3R4 revealed a binding site located in the upper-half of the transmembrane domain, in which the D2-like receptor antagonist eticlopride (3) is bound. This orthosteric binding site (OBS), in which dopamine is also thought to bind, is enclosed by TMs 3, 5, 6, and 7, consistent with our accumulated understanding of aminergic GPCRs based on structure-function studies.14 Of the 18 eticlopride contact residues in the D3R structure, 17 are identical in the D2R and one is similar.4 A predicted binding pose of compound 2 suggested that the 2,3-diCl-4-phenylpiperazine binds in the OBS in close alignment with eticlopride, with its butylamide linking chain extending the 2-indole terminus into a second binding pocket (SBP).4
The discovery of novel D2R/D3R ligands has been reported recently using computational screening of large compound libraries in both a D3R homology model and the D3R crystal structure.15 It is important to note, however, that the two critical features of drug design – specificity and efficacy – were not addressed in that study. More importantly, any screening effort targeted exclusively to the OBS of such similar receptors as the D2R and D3R will almost certainly fail to identify highly-selective compounds. Similar challenges exist for other GPCRs that share a common orthosteric agonist, such as the muscarinic acetylcholine receptors, where much emphasis has gone instead into the development of allosteric agents that target subtype-specific sites (reviewed in ref16). It is interesting to note that the region exploited in the development of these allosteric compounds is thought to include EL2, position 3.28, and the extracellular portion of TM7,16, 17 all parts of the SBP.
Using a strategy that combines synthetic chemistry, binding and functional assays, and computational modeling and simulations, herein we identify components critical to the binding specificity and efficacy of D3R-targeted compounds.
RESULTS AND DISCUSSION
The current study was directed toward understanding the structural bases of the specificity of existing compounds that would facilitate rational drug design as well as provide general principles that extend beyond this particular target. In order to develop an understanding of drug efficacy in addition to specificity, we chose to consider not only the inactive structure represented by the D3R crystal structure but also the active D3R OBS conformation by taking advantage of recent breakthrough high-resolution active-state GPCR structures.18, 19 Thus, by systematically deconstructing the full-length D3R-selective compounds 2, JJC 7-065 (4) and JJC 7-082 (5, Figure 1), examining both binding and efficacy, and using these data to validate computational models, we sought to elucidate structural features responsible both for D3R versus D2R selectivity and for D3R/D2R efficacy.
Binding characterization of compound 2 and two cognate full-length compounds
To investigate the contribution of each component of 2 to D3R binding and selectivity, we synthesized compound 4,20, 21 an analogue of 2 that lacks the 3-OH group on the linking chain. Compound 4 and 2 bind D3R with similar high affinity (Ki=1.4 vs. 1.1 nM), whereas compound 4 has somewhat higher affinity than 2 at D2R (Ki=103 vs. 433 nM, Table 1). Thus, as previously reported, the 3-OH substituent significantly improves D3R selectivity over D2R by reducing affinity at D2R rather than by improving affinity at D3R.10, 11 We also synthesized compound 510, 22 (Figure 1) by replacing the 2,3-diCl substitution of the pendant phenyl ring of compound 4 with a 2-OCH3 group, which is found in many D3R ligands. As for compound 4, the D3R affinity of compound 5 is high (Ki=0.32 nM), whereas D2R affinity is ~100-fold lower (Table 1).
Table 1.
D3R and D2R binding data for the D3R-selective compounds and corresponding synthons
| 2,3-diCl | 2-OCH3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Structure | D2 Ki (nM) | D3 Ki (nM) | D2/D3 | Compound | Structure | D2 Ki (nM) | D3 Ki (nM) | D2/D3 |
| 2a |  |
433±29.5 | 1.12±0.21 | 394 | |||||
| 4b |  |
103±20.8 | 1.4±0.4 | 73 | 5c |  |
37.4±6.22 | 0.32±0.12 | 117 |
| 8 |  |
137.3±10.0 | 197.4±36.1 | 0.69 | 9 | 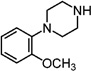 |
679.6±121.3 | 2262±468.9 | 0.30 |
| 10d |  |
45.9±9.7 | 54.6±4.5 | 0.84 | 11e |  |
265.0±19.1 | 589.9±137.6 | 0.45 |
| 12 |  |
63.7±14.4 | 36.3±3.3 | 1.75 | |||||
| 13 |  |
5.2±0.1 | 6.3±1.4 | 0.82 | |||||
| 14 |  |
3.4±0.4 | 1.9±0.2 | 1.79 | 15f |  |
20.6±4.8 | 23.9±5.8 | 0.86 |
| 16g |  |
15.6±2.3 | 4.8±1.2 | 3.25 | 17g |  |
84.0±7.9 | 38.6±7.2 | 2.18 |
| 18 |  |
13570±2102 | 21542±5561 | 0.63 | |||||
Based on SAR and previous modeling studies,10, 13 we hypothesized that the 2,3-diCl- or 2-OCH3-substituted-4-phenylpiperazine termini, defined as the primary pharmacophore (PP), would bind within the OBS of both D3R and D2R.4 This orientation of the ligand within the binding site positions the arylamide terminus, or the secondary pharmacophore (SP), in a second binding pocket (SBP) at the interface of TMs 1, 2, 3, and 7 and the first and second extracellular loops (EL1 and EL2), which can accommodate the SP in several poses. While the OBS is completely conserved between D3R and D2R, the SBP differs substantially, suggesting that the varied interactions of the ligands with the SBP may be responsible for D3R over D2R selectivity.4 To evaluate this hypothesis, we carried out induced-fit docking (IFD), and subsequent molecular dynamics (MD) simulations to predict the binding poses for 2, 4 and 5 in D3R-structure-based models of the D3R and D2R.
In both models, the PP moiety makes extensive interactions with conserved residues from TMs 3, 5, 6, and 7 of the OBS and forms the signature salt bridge between the protonated piperazine amine and Asp3.32 (Ballesteros-Weinstein numbering23) (Figure 2a). In D3R, our results show convergence in the binding of the SP moiety at the interface of TMs 2, 3, and EL1 and EL2 (Figure 2b and c). For all three ligands, the indole ring of the SP packs against a hydrophobic pocket formed by residues Val862.61, Leu892.64, Gly94 (EL1), Phe1063.28, and Cys181(EL2), which interestingly overlaps with the positions of residues identified previously as responsible for D4R over D2R selectivity.13, 24 We termed this portion of the SBP Ptm23 to indicate the TMs that form contacts with the SP in this pose. By contrast, the SP of 2, 4 and 5 did not interact with Ptm23 in any of the binding poses observed in D2R, and our simulations suggest that the binding mode of the SP in this receptor is less well defined and more widely distributed at the interface of TMs 1, 2, and 7 of D2R (Figure 2d).
Figure 2. Predicted binding modes of the full-length compounds 2, 4, and 5 in D3R and D2R.

(a) The binding mode of 2 (orange stick) in D3R shows the interaction of the SP with a hydrophobic pocket at the interface of TMs 2, 3, and EL1 and EL2 (Ptm23). A cone drawn from the C1 atom of the butyl-linker to the indole ring represents the orientation of the SP. (b) The binding poses for 2 (orange), 4 (yellow), and 5 (cyan) in D3R are similar and converged. Snapshots of ligands from the equilibrated stage of MD simulations are superimposed by the OBS of the receptor. (c,d) The ensembles of poses for 2, 4, and 5 in D3R (c) and D2R (d) are shown by cones representing the orientation of the SP. The SP packs against the Ptm23 pocket in D3R (defined by residues Val862.61, Leu892.64, Phe1063.28, Gly94EL1 and Cys181EL2), while it is further away from Ptm23 and more diffusely distributed in D2R. High energy (>2.5 kcal/mol) hydration sites in the SBP of D3R and D2R, as determined using WaterMap, are shown as red spheres. While the 2 SP in D3R displaces several higher-energy waters in Ptm23, in D2R the indole ring only partially displaces a single HEW site, consistent with the selectivity of 2 for D3R over D2R.
Using representative snapshots from the D3R-compound 2 and D2R-compound 2 trajectories, we carried out WaterMap calculations25 to characterize the distributions of hydration sites in the SBP. Water molecules in hydrophobically enclosed regions such as the Ptm23 are less stable then bulk water due to less favorable enthalpy (interactions with non-polar atoms) and/or entropy (constriction in mobility), and displacement of these high-energy waters (HEWs) by ligands has been shown to improve affinity26 and affect selectivity.27 The results indicate that several HEWs are consistently positioned in the Ptm23 of both D3R and D2R, while fewer HEWs are found at the interface of TMs1, 2, and 7 (see the red spheres in Figure 2c and d). Thus, in D3R, but not in D2R, the indole moiety of 2 occupies a region from which several HEWs are displaced, consistent with the higher affinity of 2 for D3R.
Design, synthesis, and binding affinities of synthons
To further characterize the binding modes of each component of these D3R-selective ligands, we systematically deconstructed 2, 4 and 5 into pharmacophoric components, which we termed synthons. Specifically, the 2,3-diCl- and 2-OCH3-4-phenylpiperazine cores (8 and 9, respectively) were systematically substituted in methylene group increments to give the N-Me (1028 and 1129), N-ethanol (12), N-n-propyl (13), and N-n-butyl (14 and 1530) analogues (Table 1). We then further extended the butyl chain to the propionamides (1612 and 1712), and we also prepared the arylamide terminus compound 18 with an appended butyl linker in the 2-position of the indole ring system.
All synthons were evaluated for binding to D3R and D2R stably expressed in HEK 293 cells by displacement of [125I]IABN.31 The binding results for the synthons show that the 4-phenylpiperazine end of the D3R-selective molecules have moderate to high binding affinities for both the D3R and D2R with little to no selectivity (Table 1). Notably, as the alkyl linking chain was extended from the N-phenylpiperazine to the N-n-butyl analogues, binding affinities for both D3R and D2R improved from high to low nanomolar levels. Consistent with previous SAR,10–12, 21 this suggests that hydrophobic interactions between the linker and a previously undefined portion of the receptor contribute to the progressive enhancement of affinity.
Although it substantially enhanced affinity at both receptors, increasing the length of the linking chain only very slightly improved D3R selectivity. Similarly, addition of the 3-OH group to the linking chain in the absence of the SP enhanced D3R selectivity only 2-fold. The propionamides compounds 16 and 17 showed the highest D3R selectivities of their respective synthon series, but their selectivity was still only 3-fold compared to the ~100-fold selectivity seen in the full-length compounds. Not surprisingly, the isolated 2-butyl-indole amide terminus (18) demonstrated very low affinities competing for binding of [125I]IABN at both D3R and D2R.
All the 2,3-diCl-synthons showed ~5–13-fold higher affinities than their respective 2-OCH3-analogues for both D3R and D2R. In the 2,3-diCl series, the attachment of the SP contributes to D3R over D2R selectivity by reducing binding affinity for D2R without a significant impact on D3R binding affinity, while for the 2-OCH3-compounds, the addition of the 2-indole amide to the compound with the four-carbon linker worsened D2R affinity but also enhanced D3R affinity.
Prediction of the binding modes of synthons
To put the observed SAR in the context of receptor structure, we predicted the binding modes in the D3R structure using IFD32 for four pairs of synthons from the 2,3-diCl- and 2-OCH3- series: NH-PP (8 and 9), the N-Me-PP (10 and 11), the N-n-butyl-PP (14 and 15), and the propionamides (16 and 17). In our IFD calculations, overall, the piperazine is in the chair conformation in all poses, and the protonated nitrogen consistently forms a salt-bridge with Asp1103.32. However, the modifications on the phenyl ring and on the protonated piperazine nitrogen lead to distinct binding modes of the 4-phenylpiperazine core in the OBS. Thus, the phenyl ring in the OBS can adopt a variety of orientations, with the substituents pointing towards the intra- or extra-cellular side (henceforth referred to as “downward-facing” and “upward-facing” configurations, both of which are largely “perpendicular” to the membrane), or with the ring in the plane of the membrane (“parallel”). Models of the complexes were selected by identifying representative members of low-energy clusters of poses (Figure S1).
For the NH-PP compound of the 2,3-diCl series (8), the phenyl ring significantly tilts towards the plane of the membrane (Figure 3a). This “parallel” conformation places the chloro-substituents facing TM6 where they interact with Phe6.52, His6.55, and Val6.56 (Figure 3a). In contrast, for the compounds with an alkyl group attached to the protonated nitrogen (10, 14, and 16), the phenyl ring is predominantly positioned perpendicular to the plane of the membrane, and the chloro-substituents are reoriented towards the extracellular part of the binding site (upward-facing) where they interact with the imidazole ring of His6.55 and are in proximity to Val5.39 (Figure 3c, e, and g). However, the phenyl ring of compound 10 still has an intermediate probability of being in a “parallel” orientation (Figure S1).
Figure 3. Predicted binding modes of compounds 8–11 and 14–17 in the OBS of the D3R.

Predicted binding poses, viewed extracellularly, of 8 bound compounds from the 2,3-diCl (8 (a), 10 (c), 14 (e), and 16 (g)), and the 2-OCH3 (9 (b), 11 (d), 15 (f), and 17 (h)) series, based on IFD to the crystal structure of D3R. Side chains of the residues within 3.7 Å of the bound compound are shown. The residues in the OBS are colored in green, while the SBP residues shown in (e–h) are colored in cyan. In (e) and (f), two poses for each compound are shown, with the linker either in an extended form or occupying the Ptm67 (see text). In (i), a zoom-in view of the OBS shows the superimposed binding modes of compounds 8 (yellow) and 14 (orange). The hydrophobic interactions between the linker of compound 14 and Thr7.39 and Tyr7.43, which are absent for compound 8, would prevent the relative rearrangement of the extracellular portions of TM5 and TM7.
All of the 2-OCH3 substituted synthons, from the NH-PP compound to the propionamide (9, 11, 15, and 17), prefer a “perpendicular” orientation. In contrast to the 2,3-diCl substituents in compounds 10, 14, and 16, which are more likely to be “upward-facing”, the predominant binding mode for all the alkyl substituted compounds (9, 11, 15, and 17) is “downward facing” (Figure S1). In this configuration, the 2-OCH3 methyl group makes hydrophobic contacts with Ser5.46 and Phe6.52 (Figure 3b, d, f, and h); in the less frequent “upward-facing” configuration, it interacts with Val5.39 and His6.55.
In both series, the N-methyl group of N-Me-PP (10 and 11) makes hydrophobic contact with Thr7.39 and Tyr7.43 (Figure 3c and d). When the linker length is increased, the hydrophobic and saturated butyl group of N-n-butyl-PP (14 and 15) shows significant flexibility in the docking results. Thus, in addition to being able to adopt extended orientations that point towards the divergent TMs 1, 2, 3, and 7 interface and barely contact individual Ptm23 residues, it can also insert into a hydrophobic pocket formed by Trp6.48, Phe6.51, Thr7.39, Gly7.42 and Tyr7.43, which we termed Ptm67 (Figure 3e and f, Figure S1). The WaterMap calculations of the D3R crystal structure show a HEW in Ptm67 displaced by both compounds 14 and 15 (Figure S2), suggesting that the Ptm67 pocket is the most preferred location of the butyl chain.
The docking results of compounds 16 and 17 show that when the synthons are extended even further (Table 1), the terminal propionamides access Ptm23 (Figure 3g and h). While the indole moiety of the higher-affinity 2, 4, and 5 displaces the HEWs in Ptm23, the shorter propionamide synthons 16 and 17 do not extend as deeply toward EL1 and thus do not displace these HEWs, consistent with their weak D3R-selectivity (Table 1). Interestingly, in both D3R and D2R the addition of the propionamide (16 and 17) prevents the butyl groups from entering the putative high-affinity Ptm67 pocket, thereby reducing affinity ~2–4 fold compared to 14 and 15.
The extensive hydrophobic interactions observed between the longer linkers and TM7 might hinder rearrangements associated with the conformational transition towards the active state18, 19 —specifically, they may prevent the extracellular portions of TM7 and TM5 from moving towards each other (Figure 3i). We predicted, therefore, that there might be a spectrum of efficacy for the synthons, especially within the 2,3-diCl series in which the unsubstituted NH-PP (8) has a significantly different binding mode.
Functional evaluation of the D3R-selective ligands and their synthons
While SAR for D3R binding affinity and selectivity is well defined, elucidating SAR for D3R efficacy has been more challenging.10–12, 33 Since it has been difficult to measure D3R activation reliably using second messenger assays, particularly with Gi-coupled readouts,34, 35 we developed a BRET-based assay that measures activation of GoA by either D3R or D2R to determine the efficacies of 2, 4 and 5. This assay is a modified version of BRET-based assays that have been described previously,36, 37 in which agonist-induced G-protein activation by coexpressed receptor leads to separation of GαoA-91-Rluc8 and complemented mVenus-Gβ1γ2, resulting in a reduction in BRET (Figure 4a). As expected, dopamine and quinpirole (6) acted as full agonists, whereas sulpiride (7) increased the BRET signal through its actions as an inverse agonist (Figure 4b and c). Sulpiride (100 nM) dramatically right-shifted the potency of dopamine at both D3R and D2R, consistent with its expected action as a competitive antagonist (Figure 4b and c).
Figure 4. Evaluation of efficacy for full-length compounds 2, 4 and 5 and their synthons at D3R and D2R.

(a) Schematic of the BRET1-based GoA activation assay in which HEK293T cells transiently express either wild-type D3R or D2R and GαoA-Rluc8, V1-β1 and V2-γ2. (b) Dose-response curves for dopamine (DA), quinpirole (QP) and sulpiride (sulp) are similar for both D3R (left panel) and D2R (right panel). There is a right shift in the DA dose-response curve in the presence of 100 nM sulpiride. Dose-response curves are representative of at least three independent experiments performed with triplicate samples (error bars, s.e.m). (c) 2, 4 and 5 display similar weak partial agonism at D3R and D2R. Whereas compound 9 is similar to the full length compounds, 8 displays substantial partial agonism at both receptors. (d) Whereas all synthons from the 2-OCH3 series (white) display similar weak partial agonism, there is a decreasing trend in efficacy in the 2,3-diCl series (black). Eticlopride (Et) acts as an inverse agonist while compound 18 has no activity. An x on the horizontal axis indicates compounds that have not been synthesized. For c and d, the Emax of a ligand is represented as a percentage of the Emax of DA at the respective receptor. Emax values were determined from dose-response curves ranging from 100 pM to 100 µM, as described in the Experimental Methods section, and are averaged for at least three independent experiments performed with triplicate samples (error bars, s.e.m.).
Compounds 2, 4 and 5 displayed similar weak partial agonism at D3R and D2R with efficacies ~8–20% that of dopamine (Figure 4c and Table 2). Such similar efficacy phenotypes contrasted with the substantial differences in the binding affinities of these compounds for the two receptors, as well as to the completely different binding modes of the SP within the SBP in D3R and D2R observed in our modeling studies (Figure 2). This suggested that the differential binding of the SP of these compounds does not contribute to their efficacy and further, that the binding of the PP in the OBS is a more likely determinant of activation. To address this more directly, we tested the synthons in the G protein-BRET assay and discovered that the efficacy of 8, the 2,3-diCl-4-phenylpiperazine, was much greater than that of the full-length compounds in both D3R and D2R, reaching 40–60% that of dopamine (Figure 4c and Table 2). In contrast, the 2-OCH3-4-phenylpiperazine (9), displayed only minimal efficacy at both D3R and D2R, similar to its parent compound 5 (Figure 4c and Table 2). This supported our hypothesis that the PP binding in the OBS is critical to determining efficacy but also suggested that different substituents on the 4-phenylpiperazine affect activation, presumably by altering the binding pose within the OBS.
Table 2.
Emax data in the Go BRET assay
| D3R | S.E.M. | D2R | S.E.M. | |
|---|---|---|---|---|
| 2 | −10.31 | 0.61 | ||
| 3 | 20.56 | 5.27 | 16.76 | 1.62 |
| 4 | 16.00 | 1.04 | 16.97 | 5.78 |
| 5 | 8.38 | 3.86 | 13.65 | 1.45 |
| 6 | 79.18 | 1.58 | 87.10 | 0.76 |
| 7 | −6.83 | 3.44 | −6.26 | 3.41 |
| 8 | 57.27 | 4.98 | 40.03 | 4.09 |
| 9 | 6.27 | 0.64 | 14.02 | 1.12 |
| 10 | 37.53 | 4.80 | ||
| 11 | 7.11 | 1.79 | ||
| 12 | 21.08 | 4.36 | ||
| 13 | 17.53 | 3.43 | ||
| 14 | 14.13 | 3.73 | ||
| 15 | 9.40 | 1.78 | ||
| 16 | 22.32 | 5.04 | ||
| 17 | 7.37 | 3.82 | ||
| 18 | −0.65 | 0.65 |
Data are represented as a % of the Emax of dopamine.
These data also established that the linking chain and/or SP reduce efficacy when added to the 2,3-diCl-4-phenylpiperazine core. Functional analysis of the synthons revealed that the SP is not necessary for this effect, as increasing the length of the linker by adding one to three methylenes led to a progressive decrease in efficacy (Figure 4d and Table 2). Compounds with a 3-carbon linker or greater, including the full-length D3R compounds, 2 and 3 had similar efficacies of ~20%. In contrast, all of the 2-OCH3 substituted analogues, including the full-length compound 5, were very weak partial agonists at D3R (~10% efficacy) and thus, in this series, the length of the linker or amide terminus had no effect on efficacy. The SP alone (18) failed to activate D3R, consistent with the critical role of the PP in activation. As an additional control, eticlopride, like sulpiride, acted as an inverse agonist (Figure 4d and Table 2).
Comparing binding modes in the D3R active and inactive conformations
Of note, the D3R high-resolution structure with bound eticlopride represents an inactive conformation. Therefore, to investigate the structural bases of the differences in efficacy between dopamine, synthons 8 and 9, and eticlopride, it was necessary to establish an active conformational state for the OBS. We used the differences in the inactive and active structures of the homologous β1 and β2 adrenergic receptors38–40 to guide the construction of an active conformation of the D3R. Features of the active state near the OBS include the formation of a bulge around Ser5.46 in TM5, tilting of the extracellular segments of TMs 6 and 7 towards TM5, and significantly more H-bonding between the TM5 serines and bound agonist (see Supporting Information). Thus, starting from the D3R inactive conformation, we rotated the side chains of Ser5.42 and Ser5.43 in TM5 to face towards the OBS,41 iteratively docked dopamine in the OBS, and relaxed the system with MD simulations. In the equilibrated D3R model bound with dopamine, there is a tightening of the binding site evidenced by slightly decreased distances from Ser5.42 or Ser5.46 to either Asp3.32 or Thr7.39 compared to the conformation bound with the inverse agonist eticlopride, which parallels the rearrangements observed in the active and inactive conformations of β2 adrenergic receptors (Table S1).
To explore the difference in substituent orientations in the OBS between the 2,3-diCl- and 2-OCH3-series from the perspective of the ligands themselves, electrostatic potential surface calculations were performed on synthons 8 and 9. The 2,3-diCl- and 2-OCH3- substituents result in significantly different charge distributions on the phenyl ring, specifically on the face that interacts with TM5 (Figure 5a). Thus, the polar 3-Cl in compound 8 could form a H-bond with TM5 like dopamine (Figure 5b), while in compound 9, the 2-OCH3 can only form hydrophobic contacts with TM5, like eticlopride (Figure 5c). This finding suggests that the profound difference in efficacy between compounds 8 and 9 is likely due to their different interactions with TM5, which impact transitions of the OBS towards an active conformation.
Figure 5. The interactions of compounds 8, 9, 14, and 15 with TM5 serines compared to those of dopamine and eticlopride.

In (a), the electrostatic potentials (kcal/mol) are superimposed onto a surface of constant electron density (0.001 e/bohr3) with rainbow colors. The most positive and negative potential regions are in deep blue/purple and red, respectively. In (b–g), the binding poses are viewed parallel to the membrane, with a focus on their interactions with TM5 serines. The representative poses of dopamine (b) and eticlopride (c) are taken from the MD trajectories of D3R models, in which the OBS are in active and inactive configurations, respectively. The representative poses of compounds 8 (d) and 9 (e) are taken from the equilibrated MD trajectories that were started from IFD results of these compounds in the D3R model with the OBS in an active configuration (see text), while those of 14 (f) and 15 (g) are the IFD results on the same model — similar to 9, the substituents cannot establish any H-bonding with the TM5 serines. H-bonds are indicated with dotted lines.
To explore this difference further we used IFD to compare the binding modes of the synthons in the active and inactive conformations of the OBS. Interestingly, the overall binding modes of compounds 8 and 9 in the active conformation were similar to their modes in the inactive conformation, but the polar 3-position of 8 is better accommodated in the active conformation due to the rotated TM5 Ser side chains. Indeed, during multiple 30 ns MD relaxations, a H-bond formed between the 3-Cl of compound 8 and Ser5.43 is consistently stable (Figure 5d); in contrast, for compound 9, the serines of TM5 rotate back to their inactive configuration during the MD relaxations, consistent with the observation described above that the 2-OCH3 tilts the 4-phenylpiperazine toward TM5 where it may prevent the formation of the bulge in TM5 that is associated with the active state (Figure 5e). Moreover, when the protonated nitrogen is extended by a linker (10–17), the hydrophobic interactions of the linker formed with TM7 (Thr7.39 and Tyr7.43, see Figure 3) result in an orientation of the 4-phenylpiperazine moiety perpendicular to the membrane, similar to the poses of these compounds docked to the crystal structure of D3R (see above), which also prevents the formation of optimal interactions with the active configuration of the TM5 serines (Figure 5f and g). Similarly, in the inactive eticlopride-bound D3R structure none of the TM5 serines faces the OBS (Figure 5c).
CONCLUSIONS
Given the conservation of the D3R and D2R residues in the OBS that bind the PP and the linker, it is not surprising that the synthons lacking the SP have similar affinities (little or no selectivity) for these two receptor subtypes. Nonetheless, structure-based SAR of the synthons from the 2,3-diCl and 2-OCH3 series indicates that substitutions on the terminal phenyl ring, in combination with the linker, can impact the orientation of the PP in the OBS, which consequently influences the exact orientation of the SP. Consistent with this notion, in the 2-OCH3 series, D3R affinity for the full length compound 5 increases by >7000-fold compared to its PP (9), whereas D2R affinity improves by <20-fold; in the 2,3-diCl series, D3R affinities of the full length compounds 2 and 4 increase ~140-fold at D3R, but D2R affinity is not appreciably different from the core PP (8). Thus, the structure of the PP and linker, although intrinsically lacking receptor selectivity, is critical for optimal binding of the SP, which on its own binds with extremely low affinity at both D3R and D2R (Table 1). On the other hand, in both the 2,3-diCl and 2-OCH3 series, the similar or higher affinities of the full-length compounds (2, 4 and 5) for D3R compared to their PP-plus-linker synthons 14 and 15 suggest that the SP is well tolerated in the SBP of D3R. In contrast, decreased affinities for D2R compared to D3R for the full-length compounds suggest that the SP cannot bind in the favored Ptm23 pocket within the D2R SBP. The similar D3R affinities between compounds 4 and 14 of the 2,3-diCl series, however, is likely a result of an offsetting effect in which the contribution of the SP in Ptm23 to binding affinity is countered by the SP restraining the butyl linker from binding within its favored Ptm67 pocket. Interestingly, it has been noted in developing multivalent ligands for muscarinic acetylcholine receptors that the hydrophobic linkers might be in different orientations with or without the attached moiety.42
In our examination of the correlation between the binding mode and efficacy, we found that the PP was the critical component in receptor activation. Specifically in the 2,3-diCl series, there is a progressive decrease in efficacy with the incremental addition of methylenes to the NH-PP (8). In contrast, in the 2-OCH3 series, all synthons are very weak partial agonists. These data correspond to our modeling study, which demonstrates that the binding mode of the 2,3-diCl compound 8 in the OBS is distinct from the binding mode in the 2-OCH3 synthon series. Moreover, as linker length increases in the 2,3-diCl series, the orientation of the phenylpiperazine moiety becomes more similar to that in the 2-OCH3 series (Figure S1).
Further analysis using a D3R model with the OBS in an active conformation revealed that like dopamine, compound 8 forms a H-bond with TM5, which is not present for the inverse agonist eticlopride, is less favorable in the 2,3-diCl synthons with longer linker length, and cannot form in the 2-OCH3 synthons. In addition, compound 8’s size and its binding mode in the OBS tend to stabilize a more contracted OBS (Figure 3i). Thus it is likely that interactions with these residues and corresponding conformational rearrangements are critical for the observed efficacy of the D3R/D2R partial agonists, in agreement with studies indicating that the serines in TM5 are critical for activation in other aminergic receptors.14, 43, 44
In summary, the molecular identification of the SBP, which differentiates D3R and D2R, and the role of their nearly identical OBS in efficacy provide insight into the molecular basis for both D3R selectivity as well as experimentally-validated computational models to guide the design of compounds with targeted efficacies. The fact that residues of the SBP have been shown to play a role in D4R-selectivity24 as well as in the binding of allosteric ligands16 suggest that this pocket has been targeted by drugs in multiple GPCRs, for both bitopic ligands and allosteric modulators, but can now be exploited more rationally in engineering ligand specificity.
EXPERIMENTAL METHODS
Molecular docking and the selection of the preferred binding mode
Selection of the preferred binding mode for each compound was based on molecular docking calculations and the following analysis with the crystal structure of the D3R (PDB: 3PBL).4 Briefly, the structure was prepared with the Protein Preparation Wizard in Maestro (version 9.2, Schröinger, LLC, New York, NY, 2011) using default options. Ligand docking calculations were performed with Induced Fit Docking.32 Ligands were docked in the protonated form. Poses were clustered into upward, downward, parallel conformations by determining the angle of the phenyl ring with the membrane normal. The PP of the alkylated compounds was able to adopt a large variety of orientations, as revealed by a large spread of the angle distribution for all compounds. To obtain reliable predictions, we selected only low energy solutions that occurred multiple times in the IFD ensemble. The ensemble of poses within 1 kcal/mol of the lowest energy state for each of the compounds was considered in selecting the final models, and the representative members of the largest low-energy clusters were selected as the final prediction (see Figure S1). The ensemble of poses that survived this protocol is shown in Figure 3.
Binding modes of the full-length compounds were predicted using core-constrained IFD. In this customized protocol, initial poses are obtained by a separate Glide calculation with softened van der Waals radii on both protein and ligand (using a scaling factor of 0.50 for non-polar atoms). At this stage, core-constraints were used to keep the PP largely in the orientation shown in Figure 3 (RMSD of the core kept below 2.0 Å). An ensemble of 20 poses was subsequently submitted to the second stage of regular IFD calculations, comprised of a Prime side-chain optimization, a minimization and a Glide docking stage using a hard van der Waals potential. The final docking stage was carried out in the absence of core constraints.
D3R and D2R homology models and molecular dynamics
Starting from our D3R and D2R models built previously using the crystal structure of D3R (PDB:3PBL) as the template,4 we extended the extracellular end of TM1 with Modeller 9v245 to better conform to the lipid bilayer dimension. In the new models, TM1 of the D3R model starts at residue 23 and of the D2R starts at residue 29. The new models, bound with eticlopride, were reinserted into the MD simulation systems and were relaxed with MD for at least 90 ns using Desmond Molecular Dynamics System (version 3.0, D. E. Shaw Research, New York, NY, 2011).
The resulting models were used as the starting points for the MD simulations of the complexes of D3R/D2R and 4-phenylpiperazine compounds – to characterize further the binding modes of selected compounds with MD, their binding poses from the docking study with the D3R crystal structure described above were transferred to the equilibrated D3R and D2R models.
The MD simulation system includes a lipid bilayer comprised of ~180 palmitoyoleoylphosphatidylcholine molecules and a water phase with Na+ and Cl− ions corresponding to a concentration of 150 mM NaCl. The entire system totally has ~65,000 atoms. We used the all-atom OPLS_AA force field46 throughout and semi-isotropic pressure treatment in the isothermal-isobaric ensemble (NPT) stages. When the receptor-compound complex was inserted back into the equilibrated system, a relaxation protocol was applied first, which included two energy minimization stages, first with and then without restraints on protein heavy atoms, a heating stage to elevate the system temperature from 10 to 310 K in 60 ps, and a protein relaxation stage to gradually reduce the restraints. In the subsequent production runs, a constant surface tension of 4000 bar·Å was applied.
Watermap calculations
Characterization of solvent thermodynamics in the D3R and D2R binding sites was performed using the WaterMap program,25 using default settings, as described previously for the A2A receptor.26 WaterMap calculations were performed on the crystal structure of D3R (Figure S2) and on representative frames from the D3R- and D2R-2 MD trajectories (Figure 2c and d).
Conformational search and electrostatic potential surface calculation
The conformational search of selected compounds was performed using MacroModel (version 9.9, Schröinger, LLC, New York, NY, 2011), with a CHCl3 solvent model. The resulting conformers were optimized at the quantum mechanical level using B3LYP-Density Functional Theory and a 6–31G* Basis set, as implemented in Jaguar (version 7.8, Schröinger, LLC, New York, NY, 2011), and then ranked by quantum mechanical Relative Energies. The electrostatic potential was calculated for the lowest-energy conformer of each compound using Jaguar and was mapped on the electrostatic density surfaces shown in Figure 5.
An active configuration of the OBS
Based on our analysis of the available active and inactive adrenergic receptors (see Supplementary Results), to establish an active configuration of the OBS we first rotated the sidechains of Ser5.42 and Ser5.43 of our D3R model to face towards the OBS41 and then carried out three rounds of iterative IFD and MD relaxation — the IFD of each subsequent round used a representative MD snapshot from the previous round as the input receptor conformation. Multiple MD trajectories were collected for each MD round, with a total of ~50 ns each for the first two rounds, and 120 ns for the third round. In the first round of IFD/MD, the meta-OH of dopamine was more intracellular relative to the para-OH, whereas in the second and third rounds, the meta-OH was consistently more extracellular than the para-OH, consistent with the topology of the -OH substituents of isoprenaline bound in an β1 adrenergic receptor crystal structure.40
Radioligand Binding Assays
A filtration binding assay was used to characterize the binding properties of the compounds. For human D2R (D2long isoform) and D3R stably expressed in HEK 293 cells, membrane homogenates (50 µL) were suspended in 50 mM Tris-HCl/150 mM NaCl/10 mM EDTA buffer pH 7.5 and incubated with 50 µL of [125I]IABN at 37 °C for 60 min. Nonspecific binding was determined using 15 µM (+)-butaclamol. For competition experiments, the radioligand concentration was generally equal to 0.5 times the Kd value, and the concentration of the competitive inhibitor ranged over 5 orders of magnitude. Binding was terminated by the addition of cold wash buffer (10 mM Tris-HCl/150 mM NaCl, pH 7.5) and filtration over glass-fiber filters (Schleicher and Schuell No. 32). Filters were washed with 10 mL of cold buffer, and the radioactivity was measured using a Packard Cobra gamma counter. Estimates of the equilibrium dissociation constant and maximum number of binding sites were obtained using unweighted nonlinear regression analysis of data modeled according to the equation describing mass action binding. Data from competitive inhibition experiments were modeled using nonlinear regression analysis to determine the concentration of inhibitor that displaced 50% of the specific binding of the radioligand. Competition curves were modeled for a single site, and the IC50 values were converted to equilibrium dissociation constants (Ki values) using the Cheng-Prusoff correction. Mean Ki values ± s.e.m. are reported for at least three independent experiments.
Constructs for expression vectors and transfection
The human D2shortR construct was previously reported.47 cDNA for human D3R and GαoA-RLuc8 were kindly provided by D. Sibley (NINDS) and C. Gales (INSERM), respectively. The following human G-protein constructs were cloned into the pcDNA3.1 expression vector (Invitrogen): Gβ1 fused with V1 (N-terminal split of mVenus; residues 1–155) at its N-terminus, and Gγ2 fused with V2 (C-terminal split of mVenus, residues 156–240) at its N-terminus. The split mVenus probes were linked to their respective G-protein with an amino acid linker consisting of GGSAGT. All the constructs were confirmed by sequencing analysis.
Plasmid cDNA was transfected into HEK-293T cells using polyethylenimine (Polysciences Inc.) in a 1:3 ratio in 10-cm dishes (BD Falcon). Amounts of receptor and the GαoA, Gβ1 and Gγ2 sensors transfected were optimized by testing various ratios of plasmids. Cells were maintained in culture with DMEM (GIBCO) supplemented with 10% FBS, and transfection media was replaced with fresh media after 24 h. Experiments were performed ~48 h after transfection.
BRET1-based GoA activation assay
Cells were washed, harvested and resuspended in PBS containing 0.1% (w/v) glucose at room temperature. Cells (40 µg of protein per well measured by BCA protein assay kit, ThermoScientific) were distributed into a 96-well microplate (Wallac, PerkinElmer Life and Analytical Sciences). After incubation with coelenterazine h (5 µM) (Dalton Pharma Services) for 8 minutes, different ligands were injected and incubated for 2 minutes. Using the Pherastar FS (BMG Labtech), BRET1 signal was determined by quantifying and calculating the ratio of the light emitted by mVenus (510–540 nm) over that emitted by RLuc8 (485 nm). Data were normalized to vehicle (0%) and dopamine (100%) and nonlinear regression analysis was performed using the sigmoidal dose-response function in GraphPad Prism 5.1.
Synthesis
1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer. Chemical shifts are reported and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26; DMSO-d6, 2.50), 13C spectra (CDCl3, 77.2), 19F spectra (CFCl3, 0). Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and agrees within 0.4% of calculated values. Melting point determinations were conducted using a Thomas-Hoover melting point apparatus and are uncorrected. Anhydrous solvents were purchased from Aldrich were used without further purification, except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Aldrich Chemical Co., Combi-Blocks, TCI, America., Matrix Scientific; Lancaster Synthesis, Inc. (Alfa Aesar) and AK Scientific, Inc. Final compounds (free base) were purified by column chromatography (EMD Chemicals, Inc.; 230–400 mess, 60 Å) or preparative thin layer chromatography (silica gel, Analtech, 1000 µm). The final products were analyzed as free bases unless otherwise stated. Yields and reaction conditions are not optimized. Generally, yields and spectroscopic data refer to the free base. On the basis of NMR, GC-MS (where obtainable), and combustion analysis data, all final compounds are >95% pure.
N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-1H-indole-2-carboxamide (4)
This compound20 was prepared using the previously described method21 from 2,3-dichlorophenylpiperazine linked to the N-phthalimido-protected 1-butylamine using standard N-alkylation conditions followed by deprotection with hydrazine and then coupling with 1H-indole-2-carboxylic acid using CDI in THF. The yield of the amidation step was 65%. The free base was converted to the HCl salt and recrystallized from hot MeOH/i-PrOH; Mp: 250–251 °C ; 1H NMR (400 MHz, CDCl3), δ: 1.65–1.73 (m, 4H), 2.47–2.50 (m, 2H), 2.64 (m, 4H), 3.07 (m, 4H), 3.51–3.54 (m, 2H), 6.83 (m, 1H), 6.91–6.93 (m, 1H), 7.10–7.16 (m, 2H), 7.26–7.31 (m, 2H), 7.43–7.45 (m, 1H), 7.63–7.65 (m, 1H); 13C NMR (100 MHz, CDCl3), δ: 24.3, 27.6, 39.4, 51.2, 53.3, 57.9, 101.7, 103.2, 111.8, 118.6, 120.7, 121.8, 124.5, 124.6, 127.4, 127.5, 127.6, 130.7, 134.0, 151.1, 167.8; Anal. (C23H26Cl2·HCl·0.25H2O), C, H, N.
1-(2,3-Dichlorophenyl)-4-methylpiperazine (10)
This compound was previously reported28 but synthesized using a different method. Compound 8 (Aqchem, USA; 694 mg, 3 mmol) and formaldehyde 37 % in H2O (1 mL) were mixed in 1,2-dichloroethane (10 mL) and then treated with sodium triacetoxyborohydride (1.3 g, 2 ee). The mixture was stirred at room temperature (RT) for 12 h and quenched by adding 1N NaOH (20 mL). The product was extracted with ethyl acetate (EtOAc). The organic extract was washed with brine, dried over sodium sulphate (Na2SO4) and concentrated in vacuo. The crude product was purified by column chromatography using EtOAc/CHCl3/MeOH (5:5:1) to afford compound 10 (596 mg) in 81% yield. 1H NMR (400 MHz, CDCl3), δ: 2.36 (b s, 3H), 2.61 (br s, 4H), 3.07 (br s, 4H), 6.96 (dd, J = 6.4, 3.2 Hz, 1H), 7.13–7.15 (m, 2H); 13C NMR (100 MHz, CDCl3), δ: 46.20, 51.38, 55.29, 118.74, 124.66, 127.54, 127.60, 134.11, 151.35; GC/MS RT(RIMS): 14.56 min (244 [M]+ m/z); Anal. (C11H14Cl2N2) C, H, N.
1-(2-Methoxyphenyl)-4-methylpiperazine (11)
This compound29 was prepared by a different method as described for compound 10, employing compound 9 (Aldrich, USA; 576 mg, 3 mmol), formaldehyde 37% in H2O (1 mL) and sodium triacetoxyborohydride (1.3 g, 6 mmol), to afford 11 (531 mg) in 86% yield. 1H NMR (400 MHz, CDCl3), δ: 2.36 (s, 3H), 2.63 (br s, 4H), 3.10 (br s, 4H), 3.86 (s, 3H), 6.86 (dd, J = 7.6, 1.2 Hz, 1H), 6.89–7.02 (m, 3H); 13C NMR (100 MHz, CDCl3), δ: 46.32, 50.76, 55.51, 111.28, 118.38, 121.10, 123.04, 141.44, 152.39; GC/MS RT(RIMS): 9.93 min (206 [M]+ m/z); Anal. (C12H18N2O) C, H, N.
2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethanol (12)
This compound was prepared by the method described for compound 13, employing compound 8 (694 mg, 3 mmol), 2-bromoethanol (375 mg, 3 mmol) and K2CO3 (1.24 g, 9 mmol), to afford compound 12 (478 mg) in 58% yield.1H NMR (400 MHz, CDCl3), δ: 2.61–2.65 (m, 2H), 2.71 (br s, 4H), 3.07 (br s, 4H), 3.66 (t, J = 5.2 Hz, 2H), 6.95 (dd, J = 6.4, 3.2 Hz, 1H), 7.14–7.16 (m, 2H); 13C NMR (100 MHz, CDCl3), δ: 51.47, 53.08, 57.85, 59.39, 118.70, 124.75, 127.57, 134.14, 151.23; GC/MS RT(RIMS): 14.01 min (274 [M]+ m/z); Anal. (C12H16Cl2N2O.0.1H2O) C, H, N.
1-(2,3-Dichlorophenyl)-4-propylpiperazine (13)
A suspension of compound 8 (694 mg, 3 mmol), 1-bromopropane (370 mg, 1 eq) and K2CO3 (1.24 g, 3 eq) in acetone was stirred at reflux overnight. Acetone was removed under reduced pressure, and the residue was diluted with ethyl acetate, washed with H2O and brine, dried over Na2SO4, concentrated, and purified by column chromatography (ethyl acetate/hexane, 1:1) to afford compound 13 (755 mg) in 92% yield.1H NMR (400 MHz, CDCl3), δ: 0.93 (t, J = 7.6 Hz, 3H), 1.50–1.60 (m, 2H), 2.36–2.40 (m, 2H), 2.63 (br s, 4H), 3.07 (br s, 4H), 6.96 (dd, J = 6.4, 3.2 Hz, 1H), 7.13–7.15 (m, 2H); 13C NMR (100 MHz, CDCl3), δ: 12.13, 20.21, 51.48, 53.46, 60.81, 118.70, 124.62, 127.56, 127.61, 134.12, 151.49; GC/MS RT(RIMS): 12.84 min (272 [M]+ m/z); Anal. (C13H18Cl2N2) C, H, N.
1-Butyl-4-(2,3-dichlorophenyl)piperazine (14)
This compound was prepared by the method described for compound 13, employing compound 8 (694 mg, 3 mmol), 1-bromobutane (412 mg, 3 mmol) and K2CO3 (1.24 g, 9 mmol), to afford compound 14 (820 mg) in 95% yield.1H NMR (400 MHz, CDCl3), δ: 0.94 (t, J = 7.6 Hz, 3H), 1.30–1.40 (m, 2H), 1.47–1.55 (m, 2H), 2.39–2.43 (m, 2H), 2.63 (br s, 4H), 3.07 (br s, 4H), 6.96 (dd, J = 6.4, 3.2 Hz, 1H), 7.13–7.16 (m, 2H); 13C NMR (100 MHz, CDCl3), δ: 14.20, 20.91, 29.18, 51.44, 53.46, 58.59, 118.68, 124.59, 127.54, 127.57, 134.09, 151.45; GC/MS RT(RIMS): 13.53 min (286 [M]+ m/z); Anal. (C14H20Cl2N2) C, H, N.
1-Butyl-4-(2-methoxyphenyl)piperazine (15)
This compound30 was prepared by the method described for compound 13, employing compound 9 (576 mg, 3 mmol), 1-bromobutane (412 mg, 3 mmol) and K2CO3 (1.24 g, 9 mmol), to afford compound 15 (690 mg) in 95% yield. 1H NMR (400 MHz, CDCl3), δ: 0.93 (t, J = 7.6 Hz, 3H), 1.30–1.39 (m, 2H), 1.47–1.56 (m, 2H), 2.38–2.42 (m, 2H), 2.65 (br s, 4H), 3.11 (br s, 4H), 3.86 (s, 3H), 6.85 (dd, J = 7.6, 1.2 Hz, 1H), 6.89–7.02 (m, 3H); 13C NMR (100 MHz, CDCl3), δ: 14.24, 20.98, 29.25, 50.82, 53.67, 55.45, 58.77, 111.22, 118.31, 121.09, 122.96, 141.55, 152.39; GC/MS RT(RIMS): 12.03 min (248 [M]+ m/z); Anal. (C15H24N2O) C, H, N.
N-Butyl-1H-indole-2-carboxamide (18)
This compound was prepared employing 1H-indole-2-carboxylic acid (322 mg, 2 mmol), CDI (324 mg, 2 mmol) and 1-butylamine (146 mg, 2 mmol), to afford compound 18 (393 mg) in 91% yield. Mp: 171–173 °C; 1H NMR (400 MHz, CDCl3), δ: 0.98 (t, J = 7.2 Hz, 3H), 1.39–1.48 (m, 2H), 1.59–1.67 (m, 2H), 3.47–3.53 (m, 2H), 6.17 (br s, 1H), 6.81–6.82 (m, 1H), 7.12–7.16 (m, 1H), 7.26–7.31 (m, 1H), 7.45 (dd, J = 8.0, 0.8 Hz, 1H), 7.65 (dd, J = 8.0, 0.8 Hz, 1H), 9.42 (br s, 1H); 13C NMR (100 MHz, CDCl3), δ: 13.93, 20.28, 31.95, 39.65, 101.77, 112.23, 120.68, 121.93, 124.46, 127.74, 131.01, 136.54, 161.93; GC/MS RT(RIMS): 13.60 min (216 [M]+ m/z); Anal. (C13H16N2O) C, H, N.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to R. Abel, W. Sherman, D. Lupyan, C. Galès, and R. Stevens for helpful discussion, and R. Mach for the precursor used for the iodination of [125I]IABN. This work was supported in part by NIDA Intramural Research Program (AHN), DA022413 and MH54137 (JAJ), DA23957 and DA13584 (RRL), and DA023694 (LS). Computations were performed on the Ranger at the Texas Advanced Computing Center (TG-MCB090022).
Abbreviations
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- OBS
orthosteric binding site
- SBP
second binding pocket
- Gs/olf
stimulatory G-protein α subunits
- Gi/o
inhibitory G-protein α subunits
- TM
transmembrane
- PP
primary pharmacophore
- SP
secondary pharmacophore
- EL
extracellular loop
- IFD
induced-fit docking
- MD
molecular dynamics
- HEW
high energy waters
- BRET
bioluminescence resonance energy transfer
- RMSD
root-mean-square deviation
- NPT
isothermal-isobaric ensemble
- OPLS_AA
optimized potentials for liquid simulations all-atom force field
Footnotes
Supporting Information Available: This material is free via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Lober S, Hubner H, Tschammer N, Gmeiner P. Recent advances in the search for D(3)- and D(4)-selective drugs: probes, models and candidates. Trends Pharmacol Sci. 2011;32:148–157. doi: 10.1016/j.tips.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 3.Levant B. The D3 dopamine receptor: neurobiology and potential clinical relevance. Pharmacol Rev. 1997;49:231–252. [PubMed] [Google Scholar]
- 4.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heidbreder CA, Newman AH. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci. 2010;1187:4–34. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vangveravong S, Zhang Z, Taylor M, Bearden M, Xu J, Cui J, Wang W, Luedtke RR, Mach RH. Synthesis and characterization of selective dopamine D receptor ligands using aripiprazole as the lead compound. Bioorg Med Chem. 2011;19:3502–3511. doi: 10.1016/j.bmc.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokoloff P, Giros B, Martres MP, Andrieux M, Besancon R, Pilon C, Bouthenet ML, Souil E, Schwartz JC. Localization and function of the D3 dopamine receptor. Arzneimittelforschung. 1992;42:224–230. [PubMed] [Google Scholar]
- 8.Micheli F. Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem. 2011;6:1152–1162. doi: 10.1002/cmdc.201000538. [DOI] [PubMed] [Google Scholar]
- 9. [last accessed 5/3/2012]; http://clinicaltrials.gov/ct2/results?term=GSK598809.
- 10.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem. 2009;52:2559–2270. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundt P, Prevatt KM, Cao J, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J Med Chem. 2007;50:4135–4146. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 12.Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem. 2011;54:3581–3594. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrlich K, Gotz A, Bollinger S, Tschammer N, Bettinetti L, Harterich S, Hubner H, Lanig H, Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52:4923–4935. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 14.Shi L, Javitch JA. The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annu Rev Pharmacol Toxicol. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- 15.Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D(3) receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohr K, Trankle C, Kostenis E, Barocelli E, De Amici M, Holzgrabe U. Rational design of dualsteric GPCR ligands: quests and promise. Br J Pharmacol. 2010;159:997–1008. doi: 10.1111/j.1476-5381.2009.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leach K, Davey AE, Felder CC, Sexton PM, Christopoulos A. The role of transmembrane domain 3 in the actions of orthosteric, allosteric, and atypical agonists of the M4 muscarinic acetylcholine receptor. Mol Pharmacol. 2011;79:855–865. doi: 10.1124/mol.111.070938. [DOI] [PubMed] [Google Scholar]
- 18.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 20.Leopoldo M, Berardi F, Colabufo NA, De Giorgio P, Lacivita E, Perrone R, Tortorella V. Structure-affinity relationship study on N-[4-(4-arylpiperazin-1-yl)butyl]arylcarboxamides as potent and selective dopamine D(3) receptor ligands. J Med Chem. 2002;45:5727–5735. doi: 10.1021/jm020952a. [DOI] [PubMed] [Google Scholar]
- 21.Newman AH, Cao J, Bennett CJ, Robarge MJ, Freeman RA, Luedtke RR. N-(4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl, butenyl and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg Med Chem Lett. 2003;13:2179–2183. doi: 10.1016/s0960-894x(03)00389-5. [DOI] [PubMed] [Google Scholar]
- 22.Campiani G, Butini S, Trotta F, Fattorusso C, Catalanotti B, Aiello F, Gemma S, Nacci V, Novellino E, Stark JA, Cagnotto A, Fumagalli E, Carnovali F, Cervo L, Mennini T. Synthesis and pharmacological evaluation of potent and highly selective D3 receptor ligands: inhibition of cocaine-seeking behavior and the role of dopamine D3/D2 receptors. J Med Chem. 2003;46:3822–3839. doi: 10.1021/jm0211220. [DOI] [PubMed] [Google Scholar]
- 23.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods in Neurosciences. 1995;25:366–428. [Google Scholar]
- 24.Simpson MM, Ballesteros JA, Chiappa V, Chen J, Suehiro M, Hartman DS, Godel T, Snyder LA, Sakmar TP, Javitch JA. Dopamine D4/D2 receptor selectivity is determined by A divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol Pharmacol. 1999;56:1116–1126. doi: 10.1124/mol.56.6.1116. [DOI] [PubMed] [Google Scholar]
- 25.Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc. 2008;130:2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higgs C, Beuming T, Sherman W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Medicinal Chemistry Letters. 2010;1:160–164. doi: 10.1021/ml100008s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beuming T, Farid R, Sherman W. High-energy water sites determine peptide binding affinity and specificity of PDZ domains. Protein Sci. 2009;18:1609–1619. doi: 10.1002/pro.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morita S, Kitano K, Matsubara J, Ohtani T, Kawano Y, Otsubo K, Uchida M. Practical application of the palladium-catalyzed amination in phenylpiperazine synthesis: An efficient synthesis of a metabolite of the antipsychotic agent aripiprazole. Tetrahedron. 1998;54:4811–4818. [Google Scholar]
- 29.ten Hoeve W, Kruse CG, Luteyn JM, Thiecke JRG, Wynberg H. Direct substitution of aromatic ethers by lithium amides. A new aromatic amination reaction. The Journal of Organic Chemistry. 1993;58:5101–5106. [Google Scholar]
- 30.Mokrosz JL, Paluchowska MH, Chojnacka-Wojcik E, Filip M, Charakchieva-Minol S, Deren-Wesolek A, Mokrosz MJ. Structure-activity relationship studies of central nervous system agents. 13. 4-[3-(Benzotriazol-1-yl)propyl]-1-(2-methoxyphenyl)piperazine, a new putative 5-HT1A receptor antagonist, and its analogs. J Med Chem. 1994;37:2754–2760. doi: 10.1021/jm00043a014. [DOI] [PubMed] [Google Scholar]
- 31.Luedtke RR, Freeman RA, Boundy VA, Martin MW, Huang Y, Mach RH. Characterization of (125)I-IABN, a novel azabicyclononane benzamide selective for D2-like dopamine receptors. Synapse. 2000;38:438–449. doi: 10.1002/1098-2396(20001215)38:4<438::AID-SYN9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 33.Taylor M, Grundt P, Griffin SA, Newman AH, Luedtke RR. Dopamine D3 receptor selective ligands with varying intrinsic efficacies at adenylyl cyclase inhibition and mitogenic signaling pathways. Synapse. 2010;64:251–266. doi: 10.1002/syn.20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaworski PG, Alberts GL, Pregenzer JF, Im WB, Slightom JL, Gill GS. Efficient functional coupling of the human D3 dopamine receptor to G(o) subtype of G proteins in SH-SY5Y cells. Br J Pharmacol. 1999;128:1181–1188. doi: 10.1038/sj.bjp.0702905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane JR, Powney B, Wise A, Rees S, Milligan G. G protein coupling and ligand selectivity of the D2L and D3 dopamine receptors. J Pharmacol Exp Ther. 2008;325:319–330. doi: 10.1124/jpet.107.134296. [DOI] [PubMed] [Google Scholar]
- 36.Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE, Bouvier M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- 37.Urizar E, Yano H, Kolster R, Gales C, Lambert N, Javitch JA. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7:624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J Med Chem. 2008;51:4978–4985. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- 42.Steinfeld T, Mammen M, Smith JA, Wilson RD, Jasper JR. A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol Pharmacol. 2007;72:291–302. doi: 10.1124/mol.106.033746. [DOI] [PubMed] [Google Scholar]
- 43.Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. The forgotten serine. A critical role for Ser-203(5.42) in ligand binding to and activation of the beta 2-adrenergic receptor. J Biol Chem. 2000;275:37779–37788. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- 44.Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. Journal of Biological Chemistry. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 45.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 46.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptidesâ€. The Journal of Physical Chemistry B. 2001;105:6474–6487. [Google Scholar]
- 47.Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, Javitch JA. Dopamine D2 receptors form higher order oligomers at physiological expression levels. Embo J. 2008;27:2293–2304. doi: 10.1038/emboj.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.