

Abstract
The secretin receptor is a prototypic member of family B G protein-coupled receptors that binds and responds to a linear 27-residue peptide natural ligand. The carboxyl-terminal region of this peptide assumes a helical conformation that occupies the peptide-binding cleft within the structurally complex disulphide-bonded amino-terminal domain of this receptor. The amino terminus of secretin is directed toward the core helical bundle domain of this receptor that seems to be structurally distinct from the analogous region of family A G protein-coupled receptors. This amino-terminal region of secretin is critical for its biological activity, to stimulate Gs coupling and the agonist-induced cAMP response. While the natural peptide ligand is known to span the two key receptor domains, with multiple residue-residue approximation constraints well established, the orientation of the receptor amino terminus relative to the receptor core helical bundle domain is still unclear. Fluorescence studies have established that the mid-region and carboxyl-terminal end of secretin are protected by the receptor peptide-binding cleft and the amino terminus of secretin is most exposed to the aqueous milieu as it is directed toward the receptor core, with the mid-region of the peptide becoming more exposed upon receptor activation. Like other family B peptide hormone receptors, the secretin receptor is constitutively present in a structurally specific homo-dimeric complex built around the lipid-exposed face of transmembrane segment four. This complex is important for facilitating G protein association and achieving the high affinity state of this receptor.
LINKED ARTICLES
This article is part of a themed section on Secretin Family (Class B) G Protein-Coupled Receptors. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.166.issue-1
Keywords: secretin receptor, family B GPCR, receptor dimerization, negative cooperativity, RAMPs, photoaffinity labelling
Secretin and its receptor hold special places in history, with secretin representing the first hormone and giving rise to the field of endocrinology (Bayliss and Starling, 1902), and with the secretin receptor representing the first member of the B family of guanine nucleotide-binding protein (G protein)-coupled receptors (GPCRs) to be cloned (Ishihara et al., 1991). This family includes receptors for vasoactive intestinal polypeptide (VIP), pituitary adenylate cyclase-activating peptide (PACAP), glucagon, glucagon-like peptide (GLP), glucose-dependent insulinotropic polypeptide (GIP), calcitonin, calcitonin gene-related peptide, parathyroid hormone (PTH), corticotrophin-releasing factor (CRF) and growth hormone releasing hormone (GHRH) (Ulrich et al., 1998; Mayo et al., 2003), many of which represent potentially very important drug targets. Potential applications span a broad spectrum that includes therapeutics for diabetes mellitus, bone disease, pain, inflammation, migraine, anxiety, depression, short bowel syndrome and even neoplastic disease.
All of the natural agonist ligands for receptors in this family are moderate length peptide hormones that have a diffuse pharmacophoric domain, with critical determinants spread throughout the length of the peptide (Ulrich et al., 1998). While family B GPCRs share the predicted structural motif including transmembrane heptahelical bundle conformation and G protein coupling as the proximal effector event for all other GPCRs, they are structurally distinct, not sharing any of the signature sequences typical of family A GPCRs and being predicted to have a structurally distinct helical bundle conformation (Frimurer and Bywater, 1999; Fredriksson et al., 2003; Foord et al., 2005). The themes that have been established for secretin and its receptor have been consistent for other natural ligands and peptide hormone receptors in this family (Gardella et al., 1994; Holtmann et al., 1995; 1996; Di Paolo et al., 1998; 1999; Solano et al., 2001; Al-Sabah and Donnelly, 2003; Mann et al., 2007). These will be developed in this review.
Secretin family peptides
The natural ligands for the peptide hormone-binding group of family B GPCRs are peptides with 25 or more residues that seem to have substantial tendency to form helical conformations, particularly at their carboxyl-terminal regions (Dong and Miller, 2002). Structure-activity series including alanine-replacement strategies have supported the importance of residues spread throughout the length of these peptides, with many residues at the amino terminus exhibiting structural specificity for high affinity binding and biological activity and with residues spaced every three or four positions in the mid-region and carboxyl terminus exhibiting importance for binding affinity (Figure 1) (Adelhorst et al., 1994; Nicole et al., 2000; Igarashi et al., 2002a,b). This has contributed to the impression that the carboxyl-terminal region of these peptides contributes mainly to binding, with one face of a helical conformation being most important, while the amino-terminal region contributes to both binding and biological activity (Dong et al., 2011). Of note, helix N-capping motifs have been identified in many of these peptides (represented by residues six, seven and ten of many of the peptides, with phenylalanine, threonine and tyrosine in those positions in many of the natural hormonal ligands), presumably stabilizing the extended helical conformations in the mid-region and carboxyl-terminal region of these ligands (Neumann et al., 2008). In calcitonin, a disulphide bond between cysteine residues in positions one and seven seems to play a similar role to the helix N-capping motif (Neumann et al., 2008). Other interesting experimental approaches have stabilized the helical region using constraints such as lactams, effectively improving ligand binding affinities (Ahn et al., 2001a; Taylor et al., 2002; Willick et al., 2004; Murage et al., 2008).
Figure 1.
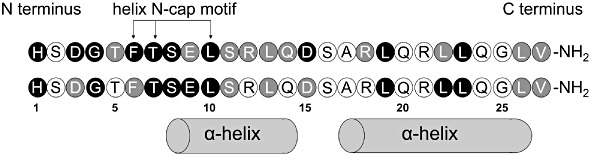
Secretin. Shown is the amino acid sequence of rat secretin from the amino terminus on the left to the carboxyl-terminal amide on the right. The position of the helix N-capping motif and the helical components (in solution) are also labelled. Each of the residues in secretin was replaced with an alanine residue, with the resulting peptide studied for its binding affinity and ability to stimulate a cAMP response from secretin receptor-bearing cells (Dong et al., 2011). Residues in which alanine replacement had major effects [>100-fold reduction in binding affinity (top sequence) and/or biological activity (bottom sequence)] are shown as black-filled circles; those with intermediate effects (>10-fold and <100-fold) are shown as gray-filled circles; those with minimal or no effects (<10-fold) are shown as open circles. This provides a graphic display of the distribution of functionally important residues.
Much less is understood about the conformation of the amino-terminal regions of these peptide ligands than about their helical carboxyl-terminal regions. The amino-terminal regions of these ligands have not been solved in the co-crystallization studies with receptor amino-terminal domains (Parthier et al., 2007; Pioszak et al., 2008; Pioszak and Xu, 2008; Runge et al., 2008; Underwood et al., 2010). They are clearly not constrained or stabilized in those structures. There is one important report in which the transfer Nuclear Overhauser effect nuclear magnetic resonance approach was successfully utilized to determine the receptor-bound conformation of PACAP as bound to its receptor (Inooka et al., 2001). While that approach did not provide insights into the conformation of that receptor or even what portion of that receptor might contribute to peptide ligand docking, it does provide our only clear insight into the conformation of the amino terminus of the receptor-bound ligand. Of interest, this seems to assume a conformation fully consistent with the disulphide-constrained calcitonin and with the presence of a helix N-capping motif.
Consistent with this general theme for the functional importance of different regions of the natural ligands for many members of this family, truncation of the amino terminus of the peptides results in reduced or absent biological activity and can yield receptor antagonists (Robberecht et al., 1976; Turner et al., 1986; Pozvek et al., 1997; Ahn et al., 2001b). Series of studies with modification of peptide and receptor structures, including powerful chimeric constructs, have predicted that the carboxyl-terminal regions of the peptide ligands interact with the receptor amino-terminal domain, while the amino-terminal region of the peptides interact with the receptor core helical bundle domain (Al-Sabah and Donnelly, 2003; Gardella et al., 1994; Holtmann et al., 1996; Di Paolo et al., 1998; Runge et al., 2003; Mann et al., 2007). Further, many other types of studies, including photoaffinity labelling (Bisello et al., 1998; Dong et al., 2004a; Dong et al., 2004b) and co-crystalization of peptide ligands with the soluble receptor amino-terminal domain, support this prediction (Parthier et al., 2007; Pioszak et al., 2008; Pioszak and Xu, 2008; Runge et al., 2008; Underwood et al., 2010).
Structure of family B GPCRs
Primary sequence analysis has clearly identified a series of seven segments of 18 or more relatively hydrophobic residues, thought to represent transmembrane helices contributing to an intramembranous helical bundle. Of note, the signature sequences found in the large and well-characterized family A GPCRs, are completely absent in the family B GPCRs. Theoretical analysis of these segments, noting specifically conserved residues and positions of charged residues, has resulted in predictions of a helical bundle quite distinct from that of the family A GPCRs (Fredriksson et al., 2003; Foord et al., 2005; Frimurer and Bywater, 1999). While we now have several examples of crystal structures of family A GPCRs (Cherezov et al., 2007; Jaakola et al., 2008; Scheerer et al., 2008; Warne et al., 2008; Chien et al., 2010; Wu et al., 2010; Rosenbaum et al., 2011), and we have a relatively clear understanding of that structure, no such information yet exists for family B GPCRs. Using computational predictive techniques, such as ‘cold spot’ analysis, transmembrane helical bundle structures for family B GPCRs have been proposed, recognizing that these represent general structures to be tested.
A major recent advance in our insights into structure of family B GPCRs has come from the ability to directly solve structures of soluble amino-terminal domains of several members of this family that bind peptide hormones (Grace et al., 2004; 2007; Parthier et al., 2007; Pioszak et al., 2008; Koth et al., 2010; Pioszak and Xu, 2008; Sun et al., 2007; Runge et al., 2008; ter Haar et al., 2010; Underwood et al., 2010). This is an extremely important domain of these receptors that contributes critical determinants of ligand binding (Miller et al., 2007; Parthier et al., 2009). It also includes many of the signature sequences for family B GPCRs, including the six conserved cysteine residues predicted to contribute to conserved functionally important intradomain disulphide bonds (Miller et al., 2007; Parthier et al., 2009). This new insight has been accomplished using nuclear magnetic resonance for the CRF2β receptor, the PACAP receptor and the calcitonin receptor-like receptor (CLR)–receptor activity-modifying protein-1 (RAMP-1) complex, as well as more recent crystal structures for the GIP, GLP-1, PTH and CRF1, and the CLR–RAMP1 complex (Grace et al., 2004; 2007; Parthier et al., 2007; Pioszak et al., 2008; Koth et al., 2010; Pioszak and Xu, 2008; Sun et al., 2007; Runge et al., 2008; ter Haar et al., 2010; Underwood et al., 2010). This has resulted in confirmation of the disulphide bond pattern that had been predicted (Lisenbee et al., 2005). This structure includes a highly conserved core that includes two anti-parallel β-sheets, three disulphide bonds, and variable amino-terminal α-helix and multiple loop regions (Parthier et al., 2009). The structure also includes a conserved hydrophobic peptide-binding cleft above the stable core and between the helix and loop regions (Parthier et al., 2009).
With several of these structures including associated peptide ligands, the prediction of a peptide-binding cleft has also been realized (Grace et al., 2004; 2007; Parthier et al., 2007; Pioszak et al., 2008; Koth et al., 2010; Pioszak and Xu, 2008; Sun et al., 2007; Runge et al., 2008; ter Haar et al., 2010; Underwood et al., 2010). However, while these structures have been highly consistent, there has been less consistency in the details of the docking of the peptide ligands within these structures. This raises concern whether the hydrophobic nature of the binding cleft has resulted in non-physiological association with the hydrophobic face of the helical peptides in these complexes. Also, none of these structures have included any direct experimental insights into the orientation of the receptor amino terminus relative to the receptor core helical bundle domain. The orientation of these domains has been predicted in several of these structures; however, they have been markedly divergent and inconsistent. At this point, this relative orientation of domains must be considered as currently unknown.
The other aspect of receptor structure that must also be considered unknown is the conformation of loop regions, including both extracellular and intracellular loops. Some structure has been predicted based on nuclear magnetic resonance analysis of lipid-linked loop sequences (Pellegrini et al., 1998; Piserchio et al., 2000), but no confirmation of such structures is yet available. In fact, the ends of the intramembranous helical segments are not definitively mapped either.
Much of the current understanding of the structure of family B GPCRs has come from computational studies and the application of techniques like photoaffinity labelling that has defined spatial approximations between distinct residues within the docked ligand and residues within the receptor (Dong et al., 2004b; 2010; Chen et al., 2010). For this technique to be most meaningful, the ligand probe that includes a photolabile site of covalent attachment and a radiolabel to track through the subsequent purification must be fully biologically active. This ensures its positioning similar to the natural ligand on which it is based. The best such probes are also high affinity ligands and are efficient in establishing a covalent bond with a single distinct receptor residue. We have successfully developed and applied nine such probes that have incorporated a photolabile moiety into positions between six and 26 (Dong et al., 1999a,b; 2000; 2002; 2003; 2007; Zang et al., 2003). We have also developed amino-terminal probes with photolabile moieties in positions five, one, minus one and minus two (Dong et al., 2004a; 2008). The latter probes were developed because it was difficult to maintain high affinity binding and agonist activity with incorporation of a photolabile benzoyl-phenylalanine residue at the critical amino terminus. Of note, all of the mid-region and carboxyl-terminal probes labelled only the receptor amino terminus (Dong et al., 1999b; 2000; 2002; 2007; Zang et al., 2003). The sites of covalent attachment are described in Table 1. Only the amino-terminal probes labelled residues within the receptor core helical bundle domain (Dong et al., 2004a; 2008). This provided the first clue that the natural peptide ligand binding site was largely accommodated within the receptor amino terminus, with only the ligand amino terminus extending toward the receptor core (Figure 2).
Table 1.
Photoaffinity labelling constraints determined for the rat secretin receptor
| Secretin: site of photolabile residue | Secretin receptor residue labelled | Labelled receptor domain |
|---|---|---|
| His1 | Phe338 | EL3 |
| Thr5 | Phe349 | EL3 |
| Phe6 | Val4 | NT |
| Arg12 | Val6 | NT |
| Leu13 | Val103 | NT |
| Ser16 | Leu99 | NT |
| Arg18 | Arg14 | NT |
| Arg21 | Arg15 | NT |
| Leu22 | Leu17 | NT |
| Leu23 | Arg21 | NT |
| Leu26 | Leu36 | NT |
EL3, extracellular loop 3; NT, amino-terminal tail.
Figure 2.
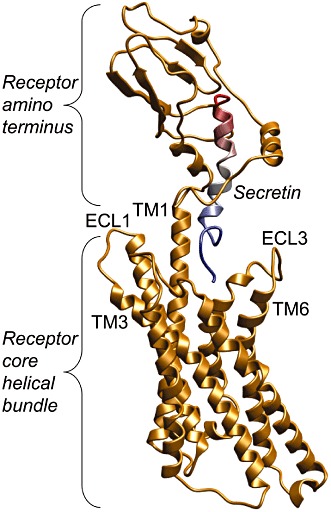
Three-dimensional molecular model of secretin-occupied secretin receptor. Shown is a working molecular model of the secretin receptor (shown in gold) occupied by natural secretin (coloured blue-to-red from its amino terminus, directed toward the receptor core helical bundle domain, to its carboxyl terminus, occupying the peptide-binding cleft within the receptor amino terminus). Selected transmembrane helices (TM) and extracellular loop regions (ECL) are labelled for orientation.
This pattern of photoaffinity labelling is shared by several members of family B GPCRs (Bisello et al., 1998; Dong et al., 2004a,b; Chen et al., 2010). This continues to support a common theme for docking natural peptide ligands for these receptors. However, despite the extensive residue-residue constraint data and the high resolution structures of the receptor amino terminus that include several of these peptides, this still does not provide adequate constraint to resolve the orientation of these major receptor structural domains. This likely reflects the high degree of flexibility of these long linear peptides and the flexibility of the receptor loop domains, with the receptor amino terminus linked to the receptor core only through the peptide backbone, with the carboxyl terminus of the receptor amino-terminal domain in contiguity with the top of transmembrane segment one.
Fluorescence studies of secretin peptides
Fluorescence studies can provide unique complementary insights to the types of studies described above. Approaches involving fluorescence have the advantage of providing not only static insights into the microdomain occupied by a fluorophore incorporated into a receptor ligand as it is normally docked at the receptor, but also dynamic insights into the agonist-induced changes in such a micro-environment. Some key insights come from collisional quenching studies using a hydrophilic molecule like potassium iodide (KI) or a hydrophobic molecule like Tempo; this provides information about the accessibility of the fluorophore to the quenching reagent. Steady-state anisotropy provides insights into the freedom of rotational movement of the fluorophore within the probe during fluorescence decay. Time-correlated single photon counting can provide information about the fluorescence lifetime of the fluorophore within the probe.
As in the photoaffinity labelling studies, these studies require a probe that binds and activates the receptor normally in order to provide meaningful data that can be extrapolated to the natural ligand. Indeed, we have developed a series of fluorescent probes that fulfil these criteria. These have involved the positioning of an alexa fluorophore at the amino terminus adjacent to position one, in the mid-region in positions 13 and 22, and at the carboxyl terminus in position 29 (Harikumar et al., 2006a) (Figure 3). All of these probes were full agonists and bound to the secretin receptor saturably and specifically, with reasonably high affinity (Harikumar et al., 2006a). Studies were performed with intact receptor-bearing cells or membranes that had been incubated with the fluorescent ligand probes. Important controls included the concurrent competition with 100-fold molar excess of non-fluorescent secretin and incubations with CHO cells not expressing the secretin receptor. Both of these provided similar low levels of signals that were subtracted from the meaningful signals reported (Harikumar and Miller, 2009).
Figure 3.
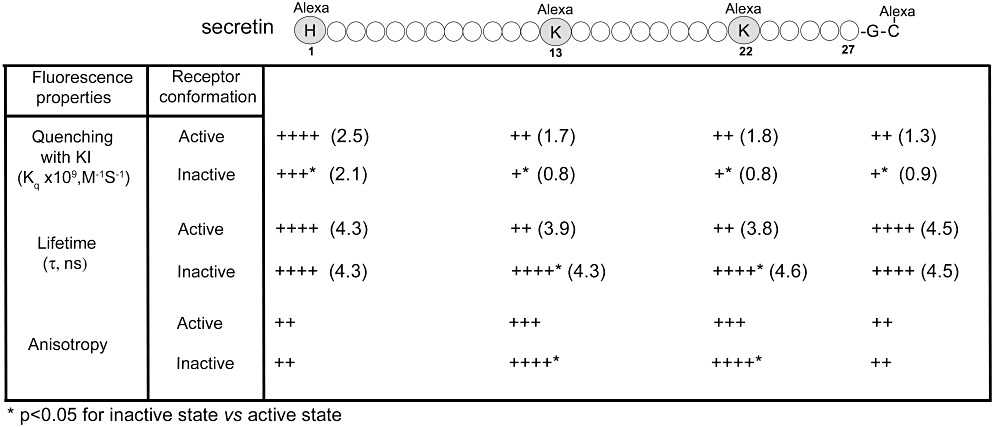
Fluorescence analysis using secretin probes bound to the secretin receptor. Shown are the positions of incorporation of fluorescent alexa488 into the amino terminus of secretin (adjacent to position 1), the carboxyl terminus of secretin (as a two-residue extension beyond position 27), and in the mid-region of secretin in positions 13 and 22. These probes were utilized in studies of the ability to quench the fluorescence with hydrophilic KI, the fluorescence lifetimes and the fluorescence anisotropy, as indications of the characteristics of the microdomains occupied by each fluorophore in the probes while bound to the secretin receptor (Harikumar et al., 2006a). The G protein-uncoupled low affinity state was achieved by incubation with GppNHp, and significant differences in each of the fluorescence characteristics relative to those when the receptor was in the high affinity state are noted. Fluorescence at the amino terminus of the peptide ligand was more readily quenched than that at any other position. The fluorescence of each of the probes was more readily quenched when the receptor was in its G protein-coupled high affinity state than in its low affinity state. For both lifetimes and anisotropy, only the mid-region probes reflected differences relative to receptor conformation, with the probe fluorescence while the receptor was in its high affinity state exhibiting shorter lifetimes and lower anisotropy than when the receptor was in its low affinity state.
Fluorescence quenching with hydrophilic KI revealed that the probe with the amino-terminal fluorophore was more accessible than the other three probes. For each of the probes, the fluorophore was quenched more easily when the receptor was in its G protein-coupled high affinity state, than when in its lower affinity state induced by incubation with the non-hydrolysable GTP analogue, guanosine-5′-(βγ-imino)triphosphate (GppNHp). Of particular interest, the micro-environments of the two mid-region probes seemed to be the most sensitive to the conformational changes, reflecting the largest changes in response to GppNHp. The fluorescence lifetimes and anisotropy of the probes varied in concert with each other. The two mid-region probes exhibited significant changes in these parameters in response to GppNHp, with the high affinity state exhibiting shorter lifetimes and lower anisotropy to reflect greater rotational dynamics. It was noteworthy that the two end probes exhibited no change in these parameters in the two states. These data support the model of the carboxyl-terminal end of the natural ligand residing in a protected peptide-binding cleft and the amino-terminal end pointing toward the helical bundle, but in a less protected environment (Harikumar et al., 2006a). No analogous data exist yet for other members of this family.
The major change upon secretin receptor coupling with its G protein and assuming its high affinity state seems to be an opening of the peptide-binding cleft, with indicator residues in that position becoming more exposed to the aqueous milieu. Also, of interest, these data do not support substantial penetration of the amino terminus of the natural ligand into the helical bundle in this state; it may be that a more subtle change in its approximation with a relevant receptor domain is involved.
Quaternary structure of the secretin receptor
In addition to being associated with their heterotrimeric G proteins, GPCRs can associate with other GPCRs and/or with RAMPs to result in complex quaternary structures. Most of the literature examining GPCR oligomerization has been directed toward family C and family A receptors, with the former having strong evidence for functionally critical dimerization that can even represent covalent complexes. The literature for family A GPCRs has been quite confusing and inconsistent, with examples of constitutive oligomerization as well as agonist-stimulated and agonist-disrupted complexes, and examples of the full spectrum of functional impact from absent to substantial (Milligan et al., 2003). No rules have yet been developed to predict the oligomerization behaviour and functional impact of oligomerization for any receptor in the A family of GPCRs.
In contrast, for family B peptide hormone-binding GPCRs, it appears that dimerization will likely be the rule, with this present constitutively, not affected by agonist binding, and facilitating G protein association and achieving the high affinity state (Gao et al., 2009). The oligomerization status of the secretin receptor has been studied more extensively than any other member of this family. This has been explored systematically to reveal structurally specific and symmetrical secretin receptor homo-dimers aligning along the lipid-exposed face of the fourth transmembrane segment that are critical for achieving the high affinity state of this receptor. These studies have utilized resonance transfer studies, both in static and saturation modes, fluorescence complementation, and establishment of covalent disulphide-bonded complexes (Harikumar et al., 2006b; 2007; 2008a; Gao et al., 2009; Lisenbee and Miller, 2006).
The simplest demonstration of receptor association was performed using bioluminescence resonance energy transfer (BRET) with receptors tagged at their carboxyl terminus with Renilla luciferase or with yellow fluorescent protein (Milligan and White, 2001). Here, the secretin receptor was shown to associate with itself in the membrane, yielding a significant BRET signal above background. This signal was independent of the presence of secretin and it was observed morphologically to start during biosynthesis in the intracellular compartments (Lisenbee and Miller, 2006). Controls established that this occurred at receptor concentrations as low as are observed physiologically and that this BRET signal was saturable (Harikumar et al., 2007). In extending these studies to determine if the BRET signal reflected a dimeric complex or a higher order oligomeric complex, bimolecular complementation with receptors tagged at their carboxyl terminus with the amino-terminal or carboxyl-terminal non-fluorescent halves of yellow fluorescent protein was utilized to yield a strong fluorescent signal. However, this was not able to act as effective donor or acceptor with a third tagged secretin receptor, as would be present in a higher order complex (Harikumar et al., 2008a).
Consistent with the homo-dimeric secretin receptor complex, competition studies were utilized to disrupt the complex and only a single region was able to do this, suggesting the presence of a single interface. These studies showed no effect of truncation of the receptor amino terminus or carboxyl terminus, and no effect of mixing with transmembrane segments except for TM4. Indeed, the relevant face of TM4 was shown to represent the lipid-exposed face in modified peptide competition studies, and this was confirmed with mutants of the intact receptor (Harikumar et al., 2007; Gao et al., 2009; Lisenbee and Miller, 2006).
After demonstrating that the lipid-exposed face of TM4 was critical for the secretin receptor dimerization, we were also successful in inducing covalent complexes through siting cysteine residues in particular positions within that interface (Gao et al., 2009). Fourteen successive residues in TM4 were mutated to cysteines, with all except for three helix-facing constructs able to traffick normally to the plasma membrane, where secretin bound and elicited cAMP responses (Gao et al., 2009). Disulphides were then induced by exposure to cuprous phenanthroline. Five constructs with lipid-exposed cysteines in positions 240, 243, 246, 247 and 250 formed covalent dimers, with three of these (243, 247 and 250) stabilized in the high affinity state (Gao et al., 2009). This provided constraints to begin to model the dimeric state of the secretin receptor (Gao et al., 2009).
Of note, every other member of family B peptide hormone-binding GPCRs has been shown to be capable of forming hetero-dimers with the secretin receptor, while family A GPCRs tested were not (Harikumar et al., 2006b; 2008b). In the few cases that have been studied, the TM4 interface for other members of this receptor family seems to be consistent (Harikumar et al., 2007; 2010).
The dimeric state of the secretin receptor has also been shown to be responsible for the negative cooperativity observed for this receptor (Gao et al., 2009). All indications suggest the structural symmetry of these dimers, yet negative cooperativity suggests an asymmetry with one protomer able to bind secretin with higher affinity than the second protomer. In another important series of studies (Dong et al., 2010), probes having two photolabile sites of covalent attachment were utilized to induce bonds to divergent regions of the secretin receptor to be certain that the bound peptide was not interacting with both receptor protomers (Dong et al., 2010). Indeed, these studies with six such pairs of dual probes confirmed that the peptide was only occupying a single protomer (Dong et al., 2010). The basis for the negative cooperativity is still unknown, but may reflect steric hindrance of the two large extracellular amino-terminal domains or selective coupling to the heterotrimeric G protein. The structural basis for the latter and the stoichiometry of receptor to G protein has not yet been established for this receptor family.
RAMPs are single transmembrane proteins that have been described to interact with several members of family B peptide hormone-binding GPCRs (Hay et al., 2006). There are three RAMPs that share approximately 30% amino acid homology and that selectively interact with receptors. These were first identified when they were found to be responsible for the translocation of the CLR from the biosynthetic intracellular organelles to the plasma membrane, where the complex bound and responded to calcitonin gene-related peptide. This was mediated by RAMP-1 association with this receptor, while its association with RAMP-2 and RAMP-3 resulted in adrenomedullin receptor phenotypes. Similarly, RAMP association with the calcitonin receptor is responsible for its amylin receptor phenotype. Recently, the secretin receptor was shown to be capable of associating with RAMP-3 (Harikumar et al., 2009). In this work, the RAMP-3 construct was translocated to the plasma membrane when co-expressed with the secretin receptor. Competition studies demonstrated that transmembrane segments TM6 and TM7 were capable of disrupting this translocation, suggesting that they contribute to the determinants of this interaction. This is clearly a distinct interface from that involved in establishing receptor homo-dimers. No clear function could be attributed to the secretin receptor–RAMP-3 complex (Harikumar et al., 2009).
Conclusion
The secretin receptor is prototypic of family B peptide hormone-binding GPCRs, following consistent themes of structure and function. It is clear that this family is distinct from the more extensively studied family A receptors, with predicted differences in the conformation of its helical bundle and in the two-step process of natural ligand binding involving the peptide-binding cleft within the disulphide-bonded receptor amino terminus and extension of the peptide toward the core helical bundle domain. Fluorescence studies suggest that the critical receptor amino-terminal domain undergoes a conformational change upon agonist binding, with this resulting in an opening of the peptide-binding cleft and some movement of the peptide ligand toward its effector domain within the receptor core. The specific structural basis for the activation event and even the relative orientation of the two key receptor domains has yet to be determined. Of note, the secretin receptor is present as a structurally specific homo-dimer in the plasma membrane, where this quaternary complex is critical to facilitate G protein association and the high affinity state of the receptor. This complex is also key for the negative cooperativity observed for this signalling system.
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK46577) and the Mayo Clinic.
Glossary
- BRET
bioluminescence resonance energy transfer
- CRF
corticotrophin-releasing factor
- GHRH
growth hormone releasing hormone
- GIP
glucose-dependent insulinotropic polypeptide
- GLP
glucagon-like peptide
- GPCR
G protein-coupled receptor
- GppNHp
guanosine-5′-(βγ-imino)triphosphate
- KI
potassium iodide
- PACAP
pituitary adenylate cyclase-activating peptide
- PTH
parathyroid hormone
- RAMP
receptor activity-modifying protein
- TM
transmembrane segment
- VIP
vasoactive intestinal polypeptide
Conflicts of interest
The authors have no conflicts of interest and no financial links with manufacturers of reagents or ligands relevant to this work.
References
- Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-activity studies of glucagon-like peptide-1. J Biol Chem. 1994;269:6275–6278. [PubMed] [Google Scholar]
- Ahn JM, Gitu PM, Medeiros M, Swift JR, Trivedi D, Hruby VJ. A new approach to search for the bioactive conformation of glucagon: positional cyclization scanning. J Med Chem. 2001a;44:3109–3116. doi: 10.1021/jm010091q. [DOI] [PubMed] [Google Scholar]
- Ahn JM, Medeiros M, Trivedi D, Hruby VJ. Development of potent truncated glucagon antagonists. J Med Chem. 2001b;44:1372–1379. doi: 10.1021/jm000453e. [DOI] [PubMed] [Google Scholar]
- Al-Sabah S, Donnelly D. The positive charge at Lys-288 of the glucagon-like peptide-1 (GLP-1) receptor is important for binding the N-terminus of peptide agonists. FEBS Lett. 2003;553:342–346. doi: 10.1016/s0014-5793(03)01043-3. [DOI] [PubMed] [Google Scholar]
- Bayliss WM, Starling EH. The mechanism of pancreatic secretion. J Physiol. 1902;28:325–353. doi: 10.1113/jphysiol.1902.sp000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisello A, Adams AE, Mierke DF, Pellegrini M, Rosenblatt M, Suva LJ, et al. Parathyroid hormone-receptor interactions identified directly by photocross-linking and molecular modeling studies. J Biol Chem. 1998;273:22498–22505. doi: 10.1074/jbc.273.35.22498. [DOI] [PubMed] [Google Scholar]
- Chen Q, Pinon DI, Miller LJ, Dong M. Spatial approximations between residues 6 and 12 in the amino-terminal region of glucagon-like peptide 1 and its receptor: a region critical for biological activity. J Biol Chem. 2010;285:24508–24518. doi: 10.1074/jbc.M110.135749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo E, De Neef P, Moguilevsky N, Petry H, Bollen A, Waelbroeck M, et al. Contribution of the second transmembrane helix of the secretin receptor to the positioning of secretin. FEBS Lett. 1998;424:207–210. doi: 10.1016/s0014-5793(98)00175-6. [DOI] [PubMed] [Google Scholar]
- Di Paolo E, Vilardaga JP, Petry H, Moguilevsky N, Bollen A, Robberecht P, et al. Role of charged amino acids conserved in the vasoactive intestinal polypeptide/secretin family of receptors on the secretin receptor functionality. Peptides. 1999;20:1187–1193. doi: 10.1016/s0196-9781(99)00122-9. [DOI] [PubMed] [Google Scholar]
- Dong M, Miller LJ. Molecular pharmacology of the secretin receptor. Receptors Channels. 2002;8:189–200. [PubMed] [Google Scholar]
- Dong M, Wang Y, Hadac EM, Pinon DI, Holicky E, Miller LJ. Identification of an interaction between residue 6 of the natural peptide ligand and a distinct residue within the amino-terminal tail of the secretin receptor. J Biol Chem. 1999a;274:19161–19167. doi: 10.1074/jbc.274.27.19161. [DOI] [PubMed] [Google Scholar]
- Dong M, Wang Y, Pinon DI, Hadac EM, Miller LJ. Demonstration of a direct interaction between residue 22 in the carboxyl-terminal half of secretin and the amino-terminal tail of the secretin receptor using photoaffinity labeling. J Biol Chem. 1999b;274:903–909. doi: 10.1074/jbc.274.2.903. [DOI] [PubMed] [Google Scholar]
- Dong M, Asmann YW, Zang M, Pinon DI, Miller LJ. Identification of two pairs of spatially approximated residues within the carboxyl terminus of secretin and its receptor. J Biol Chem. 2000;275:26032–26039. doi: 10.1074/jbc.M000612200. [DOI] [PubMed] [Google Scholar]
- Dong M, Zang M, Pinon DI, Li Z, Lybrand TP, Miller LJ. Interaction among four residues distributed through the secretin pharmacophore and a focused region of the secretin receptor amino terminus. Mol Endocrinol. 2002;16:2490–2501. doi: 10.1210/me.2002-0111. [DOI] [PubMed] [Google Scholar]
- Dong M, Li Z, Zang M, Pinon DI, Lybrand TP, Miller LJ. Spatial approximation between two residues in the mid-region of secretin and the amino terminus of its receptor. Incorporation of seven sets of such constraints into a three-dimensional model of the agonist-bound secretin receptor. J Biol Chem. 2003;278:48300–48312. doi: 10.1074/jbc.M309166200. [DOI] [PubMed] [Google Scholar]
- Dong M, Li Z, Pinon DI, Lybrand TP, Miller LJ. Spatial approximation between the amino terminus of a peptide agonist and the top of the sixth transmembrane segment of the secretin receptor. J Biol Chem. 2004a;279:2894–2903. doi: 10.1074/jbc.M310407200. [DOI] [PubMed] [Google Scholar]
- Dong M, Pinon DI, Cox RF, Miller LJ. Molecular approximation between a residue in the amino-terminal region of calcitonin and the third extracellular loop of the class B G protein-coupled calcitonin receptor. J Biol Chem. 2004b;279:31177–31182. doi: 10.1074/jbc.M404113200. [DOI] [PubMed] [Google Scholar]
- Dong M, Lam PC, Gao F, Hosohata K, Pinon DI, Sexton PM, et al. Molecular approximations between residues 21 and 23 of secretin and its receptor: development of a model for peptide docking with the amino terminus of the secretin receptor. Mol Pharmacol. 2007;72:280–290. doi: 10.1124/mol.107.035402. [DOI] [PubMed] [Google Scholar]
- Dong M, Lam PC, Pinon DI, Sexton PM, Abagyan R, Miller LJ. Spatial approximation between secretin residue five and the third extracellular loop of its receptor provides new insight into the molecular basis of natural agonist binding. Mol Pharmacol. 2008;74:413–422. doi: 10.1124/mol.108.047209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Lam PC, Pinon DI, Orry A, Sexton PM, Abagyan R, et al. Secretin occupies a single protomer of the homodimeric secretin receptor complex: insights from photoaffinity labeling studies using dual sites of covalent attachment. J Biol Chem. 2010;285:9919–9931. doi: 10.1074/jbc.M109.089730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Le A, Te JA, Pinon DI, Bordner AJ, Miller LJ. Importance of each residue within secretin for receptor binding and biological activity. Biochemistry. 2011;50:2983–2993. doi: 10.1021/bi200133u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, et al. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev. 2005;57:279–288. doi: 10.1124/pr.57.2.5. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Frimurer TM, Bywater RP. Structure of the integral membrane domain of the GLP1 receptor. Proteins. 1999;35:375–386. [PubMed] [Google Scholar]
- Gao F, Harikumar KG, Dong M, Lam PC, Sexton PM, Christopoulos A, et al. Functional importance of a structurally distinct homodimeric complex of the family B G protein-coupled secretin receptor. Mol Pharmacol. 2009;76:264–274. doi: 10.1124/mol.109.055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardella TJ, Juppner H, Wilson AK, Keutmann HT, Abou-Samra AB, Segre GV, et al. Determinants of [Arg2]PTH-(1-34) binding and signaling in the transmembrane region of the parathyroid hormone receptor. Endocrinology. 1994;135:1186–1194. doi: 10.1210/endo.135.3.8070362. [DOI] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, DiGruccio MR, Miller CL, Rivier JE, Vale WW, et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc Natl Acad Sci USA. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, Gulyas J, Digruccio MR, Cantle JP, Rivier JE, et al. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc Natl Acad Sci USA. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar E, Koth CM, Abdul-Manan N, Swenson L, Coll JT, Lippke JA, et al. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Miller LJ. Use of fluorescence indicators in receptor ligands. Methods Mol Biol. 2009;552:279–291. doi: 10.1007/978-1-60327-317-6_20. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Hosohata K, Pinon DI, Miller LJ. Use of probes with fluorescence indicator distributed throughout the pharmacophore to examine the peptide agonist-binding environment of the family B G protein-coupled secretin receptor. J Biol Chem. 2006a;281:2543–2550. doi: 10.1074/jbc.M509197200. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Morfis MM, Lisenbee CS, Sexton PM, Miller LJ. Constitutive formation of oligomeric complexes between family B G protein-coupled vasoactive intestinal polypeptide and secretin receptors. Mol Pharmacol. 2006b;69:363–373. doi: 10.1124/mol.105.015776. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Pinon DI, Miller LJ. Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J Biol Chem. 2007;282:30363–30372. doi: 10.1074/jbc.M702325200. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Happs RM, Miller LJ. Dimerization in the absence of higher-order oligomerization of the G protein-coupled secretin receptor. Biochim Biophys Acta. 2008a;1778:2555–2563. doi: 10.1016/j.bbamem.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar KG, Morfis MM, Sexton PM, Miller LJ. Pattern of intra-family hetero-oligomerization involving the G-protein-coupled secretin receptor. J Mol Neurosci. 2008b;36:279–285. doi: 10.1007/s12031-008-9060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar KG, Simms J, Christopoulos G, Sexton PM, Miller LJ. Molecular basis of association of receptor activity-modifying protein 3 with the family B G protein-coupled secretin receptor. Biochemistry. 2009;48:11773–11785. doi: 10.1021/bi901326k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar KG, Ball AM, Sexton PM, Miller LJ. Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regul Pept. 2010;164:113–119. doi: 10.1016/j.regpep.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DL, Poyner DR, Sexton PM. GPCR modulation by RAMPs. Pharmacol Ther. 2006;109:173–197. doi: 10.1016/j.pharmthera.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Holtmann MH, Hadac EM, Miller LJ. Critical contributions of amino-terminal extracellular domains in agonist binding and activation of secretin and vasoactive intestinal polypeptide receptors. Studies of chimeric receptors. J Biol Chem. 1995;270:14394–14398. doi: 10.1074/jbc.270.24.14394. [DOI] [PubMed] [Google Scholar]
- Holtmann MH, Ganguli S, Hadac EM, Dolu V, Miller LJ. Multiple extracellular loop domains contribute critical determinants for agonist binding and activation of the secretin receptor. J Biol Chem. 1996;271:14944–14949. doi: 10.1074/jbc.271.25.14944. [DOI] [PubMed] [Google Scholar]
- Igarashi H, Ito T, Hou W, Mantey SA, Pradhan TK, Ulrich CD, et al. Elucidation of vasoactive intestinal peptide pharmacophore for VPAC(1) receptors in human, rat, and guinea pig. J Pharmacol Exp Ther. 2002a;301:37–50. doi: 10.1124/jpet.301.1.37. [DOI] [PubMed] [Google Scholar]
- Igarashi H, Ito T, Pradhan TK, Mantey SA, Hou W, Coy DH, et al. Elucidation of the vasoactive intestinal peptide pharmacophore for VPAC(2) receptors in human and rat and comparison to the pharmacophore for VPAC(1) receptors. J Pharmacol Exp Ther. 2002b;303:445–460. doi: 10.1124/jpet.102.038075. [DOI] [PubMed] [Google Scholar]
- Inooka H, Ohtaki T, Kitahara O, Ikegami T, Endo S, Kitada C, et al. Conformation of a peptide ligand bound to its G-protein coupled receptor. Nat Struct Biol. 2001;8:161–165. doi: 10.1038/84159. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Nakamura S, Kaziro Y, Takahashi T, Takahashi K, Nagata S. Molecular cloning and expression of a cDNA encoding the secretin receptor. EMBO J. 1991;10:1635–1641. doi: 10.1002/j.1460-2075.1991.tb07686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koth CM, Abdul-Manan N, Lepre CA, Connolly PJ, Yoo S, Mohanty AK, et al. Refolding and characterization of a soluble ectodomain complex of the calcitonin gene-related peptide receptor. Biochemistry. 2010;49:1862–1872. doi: 10.1021/bi901848m. [DOI] [PubMed] [Google Scholar]
- Lisenbee CS, Miller LJ. Secretin receptor oligomers form intracellularly during maturation through receptor core domains. Biochemistry. 2006;45:8216–8226. doi: 10.1021/bi060494y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisenbee CS, Dong M, Miller LJ. Paired cysteine mutagenesis to establish the pattern of disulfide bonds in the functional intact secretin receptor. J Biol Chem. 2005;280:12330–12338. doi: 10.1074/jbc.M414016200. [DOI] [PubMed] [Google Scholar]
- Mann R, Nasr N, Hadden D, Sinfield J, Abidi F, Al-Sabah S, et al. Peptide binding at the GLP-1 receptor. Biochem Soc Trans. 2007;35:713–716. doi: 10.1042/BST0350713. [DOI] [PubMed] [Google Scholar]
- Mayo KE, Miller LJ, Bataille D, Dalle S, Goke B, Thorens B, et al. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- Miller LJ, Dong M, Harikumar KG, Gao F. Structural basis of natural ligand binding and activation of the Class II G-protein-coupled secretin receptor. Biochem Soc Trans. 2007;35:709–712. doi: 10.1042/BST0350709. [DOI] [PubMed] [Google Scholar]
- Milligan G, White JH. Protein-protein interactions at G-protein-coupled receptors. Trends Pharmacol Sci. 2001;22:513–518. doi: 10.1016/s0165-6147(00)01801-0. [DOI] [PubMed] [Google Scholar]
- Milligan G, Ramsay D, Pascal G, Carrillo JJ. GPCR dimerisation. Life Sci. 2003;74:181–188. doi: 10.1016/j.lfs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Murage EN, Schroeder JC, Beinborn M, Ahn JM. Search for alpha-helical propensity in the receptor-bound conformation of glucagon-like peptide-1. Bioorg Med Chem. 2008;16:10106–10112. doi: 10.1016/j.bmc.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Neumann JM, Couvineau A, Murail S, Lacapere JJ, Jamin N, Laburthe M. Class-B GPCR activation: is ligand helix-capping the key? Trends Biochem Sci. 2008;33:314–319. doi: 10.1016/j.tibs.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Nicole P, Lins L, Rouyer-Fessard C, Drouot C, Fulcrand P, Thomas A, et al. Identification of key residues for interaction of vasoactive intestinal peptide with human VPAC1 and VPAC2 receptors and development of a highly selective VPAC1 receptor agonist. Alanine scanning and molecular modeling of the peptide. J Biol Chem. 2000;275:24003–24012. doi: 10.1074/jbc.M002325200. [DOI] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D, et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci USA. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C, Reedtz-Runge S, Rudolph R, Stubbs MT. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Bisello A, Rosenblatt M, Chorev M, Mierke DF. Binding domain of human parathyroid hormone receptor: from conformation to function. Biochemistry. 1998;37:12737–12743. doi: 10.1021/bi981265h. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci USA. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piserchio A, Bisello A, Rosenblatt M, Chorev M, Mierke DF. Characterization of parathyroid hormone/receptor interactions: structure of the first extracellular loop. Biochemistry. 2000;39:8153–8160. doi: 10.1021/bi000196f. [DOI] [PubMed] [Google Scholar]
- Pozvek G, Hilton JM, Quiza M, Houssami S, Sexton PM. Structure/function relationships of calcitonin analogues as agonists, antagonists, or inverse agonists in a constitutively activated receptor cell system. Mol Pharmacol. 1997;51:658–665. doi: 10.1124/mol.51.4.658. [DOI] [PubMed] [Google Scholar]
- Robberecht P, Conlon TP, Gardner JD. Interaction of porcine vasoactive intestinal peptide with dispersed pancreatic acinar cells from the guinea pig. Structural requirements for effects of vasoactive intestinal peptide and secretin on cellular adenosine 3′:5′-monophosphate. J Biol Chem. 1976;251:4635–4639. [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge S, Wulff BS, Madsen K, Brauner-Osborne H, Knudsen LB. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br J Pharmacol. 2003;138:787–794. doi: 10.1038/sj.bjp.0705120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Solano RM, Langer I, Perret J, Vertongen P, Juarranz MG, Robberecht P, et al. Two basic residues of the h-VPAC1 receptor second transmembrane helix are essential for ligand binding and signal transduction. J Biol Chem. 2001;276:1084–1088. doi: 10.1074/jbc.M007696200. [DOI] [PubMed] [Google Scholar]
- Sun C, Song D, Davis-Taber RA, Barrett LW, Scott VE, Richardson PL, et al. Solution structure and mutational analysis of pituitary adenylate cyclase-activating polypeptide binding to the extracellular domain of PAC1-RS. Proc Natl Acad Sci USA. 2007;104:7875–7880. doi: 10.1073/pnas.0611397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW, Jin QK, Sbacchi M, Wang L, Belfiore P, Garnier M, et al. Side-chain lactam-bridge conformational constraints differentiate the activities of salmon and human calcitonins and reveal a new design concept for potent calcitonin analogues. J Med Chem. 2002;45:1108–1121. doi: 10.1021/jm010474o. [DOI] [PubMed] [Google Scholar]
- Turner JT, Jones SB, Bylund DB. A fragment of vasoactive intestinal peptide, VIP(10-28), is an antagonist of VIP in the colon carcinoma cell line, HT29. Peptides. 1986;7:849–854. doi: 10.1016/0196-9781(86)90105-1. [DOI] [PubMed] [Google Scholar]
- Ulrich CD, 2nd, Holtmann M, Miller LJ. Secretin and vasoactive intestinal peptide receptors: members of a unique family of G protein-coupled receptors. Gastroenterology. 1998;114:382–397. doi: 10.1016/s0016-5085(98)70491-3. [DOI] [PubMed] [Google Scholar]
- Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willick GE, Morley P, Whitfield JF. Constrained analogs of osteogenic peptides. Curr Med Chem. 2004;11:2867–2881. doi: 10.2174/0929867043364153. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang M, Dong M, Pinon DI, Ding XQ, Hadac EM, Li Z, et al. Spatial approximation between a photolabile residue in position 13 of secretin and the amino terminus of the secretin receptor. Mol Pharmacol. 2003;63:993–1001. doi: 10.1124/mol.63.5.993. [DOI] [PubMed] [Google Scholar]